

Abstract
Given activity training data from Hight-Throughput Screening (HTS) experiments, virtual High-Throughput Screening (vHTS) methods aim to predict in silico the activity of untested chemicals. We present a novel method, the Influence Relevance Voter (IRV), specifically tailored for the vHTS task. The IRV is a low-parameter neural network which refines a k-nearest neighbor classifier by non-linearly combining the influences of a chemical's neighbors in the training set. Influences are decomposed, also non-linearly, into a relevance component and a vote component.
The IRV is benchmarked using the data and rules of two large, open, competitions, and its performance compared to the performance of other participating methods, as well as of an in-house Support Vector Machine (SVM) method. On these benchmark datasets, IRV achieves state-of-the-art results, comparable to the SVM in one case, and significantly better than the SVM in the other, retrieving three times as many actives in the top 1% of its prediction-sorted list.
The IRV presents several other important advantages over SVMs and other methods: (1) the output predictions have a probabilistic semantic; (2) the underlying inferences are interpretable; (3) the training time is very short, on the order of minutes even for very large data sets; (4) the risk of overfitting is minimal, due to the small number of free parameters; and (5) additional information can easily be incorporated into the IRV architecture. Combined with its performance, these qualities make the IRV particularly well suited for vHTS.
Virtual High-Throughput Screening (vHTS) is the cost-effective, in silico complement of experimental HTS. A vHTS algorithm uses data from HTS experiments to predict the activity of new sets of compounds in silico. Although vHTS is sometimes cast as a classification task, it is more appropriately described as a ranking task, where the goal is to rank additional compounds, such that active compounds are close to the the top of the prediction-sorted list as possible. The experiments required to verify a hit are expensive, so it is critical that true actives be recognized as early as possible. Accurately ordering actives by their degree of activity, however, is not critical. The vHTS task, therefore, differs from the `ranking' task of the machine learning literature, in that the goal is not to precisely order the chemicals in relation to each other, but rather to globally rank as many actives as possible above the bulk of the inactives. Furthermore, proper vHTS training data for the ranking task, is often unavailable.
An important algorithm proposed for vHTS is the k-Nearest Neighbor (kNN) classifier, a nonparametric method which has been shown effective in a number of other problems.1,2 In the kNN approach, each new data point is classified by integrating information from its neighborhood in the training set in a very simple way. Specifically, a new data point is assigned to the class occurring most frequently among its k closest structural neighbors in the training set.
While the kNN algorithm has been applied to chemical data, it does not perform optimally3-6 because all the nearest neighbors contribute equally, regardless of their relative properties and similarities to the test chemical. Hence, important concepts—such as “the more similar a chemical is to its active neighbors, the more likely it is to be active itself” or “the closest neighbors should influence the prediction more than the furthest ones”—are not representable with a kNN. Furthermore, the kNN is usually used to classify, rather than to rank, unknown data. The kNN output can be modified to be an integer between 0 and k by counting the number of neighbors that are active, instead of taking a binary majority vote, but even with this quantization many compounds are mapped to the same integer value and therefore cannot be properly ranked. This is a critical deficiency for vHTS where economic or other reasons may dictate that only a few of the top hits be testable. kNN, even in its quantized version, does not provide a clear ranking of its top hits.
A number of researchers have attempted to rectify these deficiencies of kNN by employing alternate weighing schemes and decision rules.7-15 In many domains, including vHTS,9,10 these modifications can substantially improve classification performance. With few exceptions,10,12,13 however, these modifications are either somewhat ad hoc, untuned to the nuances of each dataset, or do not produce probabilistic predictions.
Here we propose a novel vHTS method, the Influence Relevance Voter (IRV), which can also be viewed as an extension of the kNN algorithm. The IRV uses a neural network architecture16-18 to learn how to best integrate information from the nearest structural neighbors contained in the training set. The IRV tunes itself to each dataset by a simple gradient descent learning procedure and produces continuous outputs that can be interpreted probabilistically and used to rank all the compounds. We assess the performance of IRV on two benchmark datasets from two recent open datamining competitions. For comparison purposes, we also implement two other methods: MAX-SIM and Support Vector Machines (SVM). MAX-SIM is a particularly simple algorithm, a useful baseline for comparison. In contrast, SVMs are a highly sophisticated class of methods which have been successfully applied to other chemical classification problems19,20 and are expected to yield high performances on vHTS datasets. Comparisons against the kNN method are not included because the kNN does not rank its hits and, in prior studies, has been consistently outperformed by SVMs on chemical data.
Data
The IJCNN-07 Competition and The HIV Data
In 2007, the International Joint Conference on Neural Networks (IJCNN-07)21 organized the Agnostic Learning versus Prior Knowledge Challenge (ALvPK), a blind prediction competition on five datasets from different fields. One of the datasets used in the competition, the HIV dataset, is a vHTS dataset. The dataset was derived from the Drug Therapeutics Program (DTP) AIDS Antiviral Screen made available by the National Cancer Institute (NCI)22The HIV dataset contains assay results for 42,678 chemicals experimentally tested for their ability to inhibit the replication of the Human Immunodeficiency Virus in vitro.
The conference organizers processed the raw DTP data in three steps. First, the HIV data was randomly partitioned into a training set consisting of 10% of the chemicals and a testing set composed of the remaining 90%, each labeled with their activity in the screen. Second, the `active' and `moderately active' compounds were combined into a single active class. This processing step, therefore, reduces the number of classes to two. Finally, for each chemical in the HIV dataset, the organizers of the competition provided both pre-computed feature vectors and the raw chemical structures from which they were extracted. We discard the organizers' feature vectors, preferring to extract our own features.
We constructed cross validation datasets by combining both the training and testing datasets from the competition. Active compounds were defined according to the competitions rules. The details of the data partitions for both the cross validation and competition datasets are summarized in Table 1.
Table 1.
Number of positive and negative examples in the training and testing datasets used in the IJCNN-07 and McMaster competitions, based on the HIV and DHFR screens respectively. The IJCNN-07 organizers randomly selected 10% of the data for use as a training set. In contrast, the McMaster organizers partitioned the DHFR data into two equal halves and attempted to minimize chemical similarity between each partition.
| IJCNN-07 (HIV) |
McMaster (DHFR) |
|||||
|---|---|---|---|---|---|---|
| Active | Inactive | Total | Active | Inactive | Total | |
| Train | 149 | 4,080 | 4,229 | 66 | 49,929 | 49,995 |
| Test | 1,354 | 3,7095 | 3,8449 | 94 | 49,906 | 50,000 |
| Full* | 1,503 | 41,175 | 42,678 | 160 | 99,520* | 99,680* |
The last row in the table, labeled `Full,' shows the size of the non-redundant dataset used for the additional cross-validation experiments.
The McMaster Competition and The DHFR Data
In 2005, the McMaster University organized a vHTS competition using experimental data generated in their laboratories. They screened 99,995 chemicals for inhibition activity against Dihydrofolate Reductase (DHFR). Inhibitors of DHFR are known to be effective cancer drugs, so both datasets have direct medical relevance. The 99,995 chemicals used in the screen were partitioned into a training and a testing sets of 49,995 and 50,000 chemicals, respectively, all of which are available on the web.23 For each molecule, two or three assays were performed and the results were reported as the percentage of full DHFR activity achieved in the presence of the test chemical. Lower percentages correspond to higher inhibition. Chemicals were then classified as inhibitors, i.e. active compounds, when all the experimental percentages were below 75%. According to this criterion, less than 0.2% of the data was found to be active.
It is important to note some peculiarities of the protocol used in the McMaster competition. First, the DHFR data contains 315 pairs of duplicate chemicals. These duplicates correspond to identical chemicals tested in solution with, and without, salt ions. Thus, from an experimental standpoint, these duplicate correspond to different experiments. The organizers, however, did not report which salt ions were used in each screen. Additionally, all molecules participating in duplicate tests, irrespective of the salt ions, were classified as inactive. Hence, from a computational standpoint, these duplicates are effectively redundant and should have been removed from the data to accurately assess performance. Second, rather than randomly partitioning the data, the competition organizers decided to increase the task's difficulty by minimizing structural similarity between the chemicals in the training and testing data.24 At the conclusion of the competition, this decision was criticized by the participants for artificially lowering the performance of similarity-based vHTS methods.
The submissions to this competition are evaluated in Lang et al.25 and Parker26. For comparison purposes, we first conduct tests using the same exact protocol as the one used in the competition, keeping duplicates in the data and using the same training/validation data split. However, we also conduct additional full cross-validation experiments were the competition rules do not need to be strictly followed, so we remove duplicates chemicals from the training and testing datasets. Table 1 details the characteristics of the final dataset used in this study.
Methods
As in most chemoinformatics applications, such as the search of large databases of small molecules27-29 or the prediction of their physical, chemical, and biological properties,9,10,19,20,27 all the vHTS methods we implement depend on a quantitative notion of chemical similarity to define the local geometry of chemical space. The underlying intuition, explicitly articulated as the Similar Property Principle,30 is that the more structurally similar two molecules are, the more likely they are to have similar properties. In turn, the notion of similarity is closely related to how molecules are represented. Thus, in order to precisely define our methods, we must first describe the representations and similarity measures used in this study.
Molecular Similarity and Representations
For this study, we define chemical similarity using a fairly standard fingerprint vector representation. Fingerprints are vectors recording the occurrences of particular substructures within molecular graphs. If we denote a labeled molecular graph with a calligraphic letter, 𝒜, and a fingerprint by = (Ai), each component Ai stores a 1-bit (or 0-bit) to indicate the presence (or absence) a particular substructure in 𝒜. Alternatively, each component can store the number of times the corresponding substructure appears in the graph. The resulting fingerprints are very sparse and are often compressed using a lossy algorithm.31,32 For this study, we use the lossless compression algorithm described elsewhere.33
Each component of the fingerprint corresponds to a particular labeled substructure. In terms of labeling, we consider two labelings of the molecular graph, Element (E) and Element-Connectivity (EC). In Element labeling, each vertex is labeled by the corresponding atomic symbol (e.g. C for carbon, O for oxygen, N for nitrogen). In Element-Connectivity labeling, each vertex is labeled by the atomic symbol of the corresponding atom together with the number of heavy atoms to which it is bonded (e.g. C3 for a carbon with three heavy atoms attached). In both labeling schemes, the bonds are simply labeled according to their type (single, double, triple, or aromatic). In terms of graphical substructures, we consider both paths19,20 of depth d up to 2, 5, or 8 bonds, or circular substructures34 of depth d up to 2 or 3 bonds. Thus the fingerprint components index all the labeled paths, or all the labeled trees, up to a certain depth.
Among the many possible ways of defining similarity between fingerprint vectors,35 we consider two similarity measures, known to work well with chemical data, the Jaccard-Tanimoto and MinMax measures. In the case of binary fingerprints, the Tanimoto similarity measure is defined by
| (1) |
where A∩B is the number of 1-bits in both AND , and A∪B is the number of 1-bits that appear (non-exclusively) in either OR . In the case of count fingerprints, which record the number of times each substructure occurs in a chemical, previous studies have shown that the MinMax similarity is one of the best performing similarities.20,36 MinMax is defined by
| (2) |
where the summations extend over all fingerprint components.
Maximum Similarity
One of the simplest methods for vHTS is the Maximum Similarity (MAX-SIM) algorithm,37,38 in which each test molecule is scored by its highest similarity to a known active compound. Molecules with higher scores (most similar to one of the active molecules) have greater likelihood of being active. Formally, if {𝒜1,…,𝒜|𝒜|} is the set containing all known active compounds and S is a similarity measure between two molecular fingerprints, the output of the predictor, z(𝒳), for the test chemical, 𝒳, is given by
| (3) |
MAX-SIM is trivial to implement, does not require any parameter tuning, has been well studied,37,38 and is intuitively simple. The resulting predictions yield a ranking and are somewhat interpretable; by examining the active compound the most similar to the query molecule and the corresponding similarity score, one can gain insight into the rationale behind the prediction. This method, however, takes very little information into account since, for instance, it discards all experimental information about inactive compounds. More refined methods, which consider all of the training data, can be expected to perform better.
Support Vector Machines
Alternatively, vHTS can be formulated as a machine learning classification task and solved with an SVM.4,39 As the MinMax and Tanimoto metrics both satisfy Mercer's condition,20 we can apply the standard SVM algorithm to score a compound 𝒳 according to the prediction function
| (4) |
where 𝒥 ranges over the training set. The α𝒥 weights are associated with each training example and learned from the data using a quadratic programming algorithm which maximizes the separation between active and inactive chemicals with a solution that fixes the majority of the α𝒥s to zero. The non-zero α𝒥s correspond to the support vectors which help define the decision boundary.
Two technical problems, associated with vHTS datasets, arise when attempting to apply a standard SVM implementation. First, vHTS datasets are much larger than typical SVM datasets. Using N as the number of examples in the training dataset, the standard formulation of SVM solved by quadratic programming, requires O(N2) memory usage and O(N3) processing time, quickly becoming very slow as datasets increase to the size of vHTS experiments. Fortunately, faster SVM algorithms have been developed. We use the SVMTorch online SVM implementation40 with memory-usage scaling like O(N) and training time scaling like O(N2).
Second, vHTS data is often extremely unbalanced. The HIV dataset, for instance, has about 28 times as many inactive examples as active examples. Unbalanced datasets are not always well learned by SVM algorithms, which generally assume a more balanced class distribution. The most straightforward strategy for dealing with class imbalance is to control the sensitivity, i.e. the C parameter, which limits the magnitude of each α𝒥 of the SVM.41,42 Misclassified instances of the under-represented class can be more penalized by assigning a higher sensitivity to the under-represented class. In our case, however, this method does not lead to significant improvements.
A more complex strategy, referred to as “oversampled SVMs” in the literature, adjusts for class imbalance by training ensembles of SVMs on class-balanced subsets of the training data.43,44 In this study, we build balanced subsets of the training data by splitting the inactive data into partitions of about the same size as the active data. Each of these partitions is then combined with the entire data set of active compounds to form a collection of datasets on which to train an ensemble of SVMs. At the final stage, the predicted classes (0 for inactive and 1 for active) of each SVM are summed to produce a final score. The higher this score, the more likely the test molecule is to be active. The resulting ensembles of SVMs are abbreviated as E-SVM. However, when datasets have only a small total number of instances in the under-represented class, E-SVM requires a high number of individual SVMs to be trained on a very small number of data points. For example, the McMaster dataset requires 750 individual SVMs to be trained on only 122 data points. SVMs are likely to perform poorly on such small training sets. Therefore, we are only able to apply this method to the HIV dataset.
These SVM-based methods yield accurate predictors. These predictors, however, are blackboxes, which are not easy to interpret beyond the information provided by the support vectors. The SVM decision function ignores representative examples, which are typically far from the decision boundary. Consequently, it is difficult to tease out exactly which experimental evidence suggests how a particular test compound should be classified. Furthermore, all the data's modeling power is used to learn the decision boundary; the posterior probability distribution of the data is not directly learned. Consequently, the output of an SVM, especially far away from the decision boundary, does not always correlate well with the confidence of a prediction.45 This deficiency is problematic in vHTS, where ranking active compounds at the top of the prediction list is more important than learning a single decision boundary between classes.
The Influence Relevance Voter
The Influence Relevance Voter (IRV) uses a neural network to combine information from the neighbors of a chemical to estimate its probability of being active. The input to the network is the list of labeled nearest neighbors and their similarities to the chemical, possibly with some variations described below. While many neural network architecture are possible, here we propose a specific implementation which uses the concept of weight-sharing to reduce the number of free parameters, hence avoiding overfitting.16,17,46 This algorithm addresses deficiencies of the SVM-based algorithms by directly modeling the posterior probability distribution of the data. Furthermore, the IRV naturally exposes the data used to classify each test instance in an interpretable way.
At a high-level, the IRV is defined by a preprocessing step, during which all the neighbors of a test molecule are identified, and a processing step, during which information from each neighbor is fed into a neural network to produce a prediction. The prediction is computed from the “influence” of each neighbor, which in turn is computed as the product of each neighbor's “relevance,” encoding how much each neighbor should affect the prediction, and “vote,” encoding the direction towards which the prediction should be pushed. Figure 1 shows the architecture of the IRV, using a notation similar to the plate notation used for graphical models, which displays the local dependencies that are replicated throughout the network, illustrating its structure and exposing the extensive weight-sharing across the network.
Figure 1.
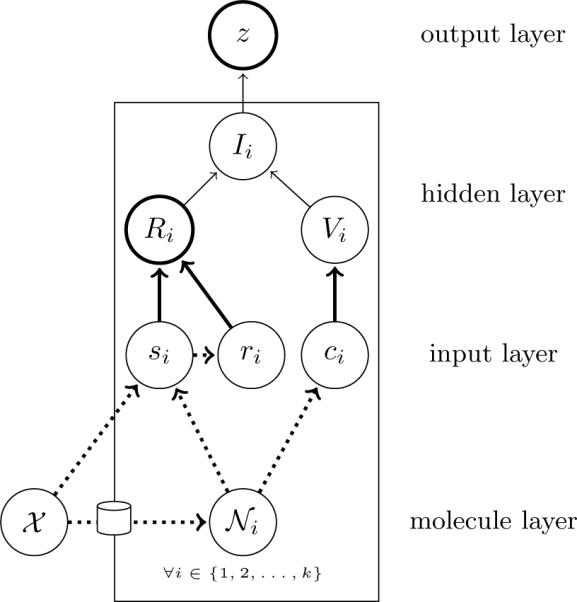
The neural network architecture of the IRV using a notation similar to the plate notation of graphical models. The structure and weights depicted inside the plate are replicated for and shared with each neighbor i. Thick lines depicts elements of the graph learned from the data. Dotted lines depict portions of the network computed during the preprocessing step, during which the test chemical(𝒳 is used to retrieve a list of neighbors, {𝒩1,…,𝒩k} from all the chemicals in the training data. The influences Ii of each neighbor are summed up to into a logistic output node to produce the final prediction, z(𝒳).
Formally, we compute the IRV's output as,
| (5) |
where 𝒳 is the test molecule, i ranges from 1 to k over all the k nearest neighbors, Ii is the “influence” of the ith neighbor on the output, wz is the bias of the output node, and σ (·) is the logistic function 1/(1+e-x). These influences indicate exactly how much, and in which direction, each training example contributes to the prediction and can be used to interpret each final prediction. The influence of the ith node is defined by
| (6) |
where Ri is the relevance and Vi is the vote of the ith neighbor. For this study, the relevance is defined as
| (7) |
where si is the similarity S(𝒳, 𝒩i) of the ith closest neighbor to the test compound, ri is the rank of the ith neighbor in the similarity-sorted list of neighbors, ws and wr are parameters tuning the importance of different inputs, and wy is the bias of the logistic unit. In order to facilitate learning, all inputs are normalized into standard units during the preprocessing step, by subtracting the corresponding mean and dividing by the corresponding standard deviation. For this study, we define the vote by
| (8) |
where w0 is the weight associated with inactive neighbors, w1 is the weight associated with active neighbors, and ci ∈ {0,1} is the class of the ith neighbor.
The logistic output of the IRV can be interpreted as a probability and directly encodes the confidence of each prediction18,47
| (9) |
In other words, the output of the network on a test molecule is approximately equal to the probability of the test molecule being active given its structure and the training data. This is enforced by training the network to minimize the relative-entropy or Kullback-Leibler divergence between the true target distribution t(𝒥) and the predicted distribution z(𝒥) across all molecules 𝒥 in the training set.18 Thus the IRV is trained by gradient descent to minimize the error or, equivalently, the negative log-likelihood given by
| (10) |
It is possible to add weight decay terms to the error function, equivalent to defining a gaussian prior on the weights, to prevent over-fitting during training. In practice, overfitting is unlikely because so few parameters are optimized during training. Three parameters, ws, wr, and wy, are shared across all k neighbors. The model requires just three additional parameters, w1, w0, and wz. Therefore, a total of only six parameters are learnt from the data. Nevertheless, weight decay is included in this implementation because it seems to further decrease the training time. The corresponding penalized negative log-likelihood is given by
| (11) |
where the first summation is over all six weights.
Each influence, Ii, encodes how much, and in what direction, its associated neighbor pushes the prediction. Influences with the greatest absolute values affect the output the most, their associated neighbors are the data upon which the prediction is made. Those with negative values push the output towards an inactive prediction, while those with positive values push the output towards an active prediction. Thus the influences and the prediction are readily understandable by direct inspection and visualization.
Variations on the IRV
The IRV's performance can be fine-tuned by several variations. In this study, we particularly consider: (1) the number of neighbors selected; (2) the class-balance of the neighbors; and (3) the weighing of different classes during training. More complex variations are explored in the discussion.
The network can be presented different total numbers k of neighbors. The network's performance is not sensitive to the exact value of k above a certain threshold, but sometimes small performance improvements can be realized. For the competitions, we fixed k at 20 and were able to obtain good results with low computational cost.
Rather than presenting the IRV network with nearest neighbors irrespective of their class, it is possible instead to present an equal number of neighbors from each class. For example, instead of selecting the 20 nearest neighbors, one can select both the 10 nearest active and the 10 nearest inactive compounds. The latter is most effective in situations where the number of active compounds is particularly small (e.g. the DHFR dataset).
During training, it is also possible to overweigh the log-likelihood of one of the classes, so as to increase its contribution to the training process. This can be used to address the class imbalance in most vHTS datasets. For example, if there are 20 times more inactive chemicals than active ones, then the error associated with each of the active compounds can be multiplied by 20 during training to restore the balance. Initial experiments indicate that this variation does not appreciably improve performance, so this variation is excluded from the results.
In any case, the IRV comes with a relatively small number of hyperparameters, primarily the number k of neighbors and their composition, and inferences regarding their values can be made from the statistical properties of the data, as in the case of other machine learning methods.
Performance Measures
Each of the vHTS algorithms we describe, MAX-SIM, SVM, and IRV, outputs a number for each chemical in the uknown dataset. This number is used to rank the dataset and it is the ranks of the true actives that are used by different metrics to quantify performance and assess each method.
The IJCNN-07 competition determines winners according to the Balanced Error Rate (BER) metric. This metric requires to set a threshold, learned during the training stage, to separate predicted actives from predicted inactives. Once this is done, the BER is defined as the average of the proportion of actives mistakenly predicted as inactives and the proportion of inactives mistakenly predicted as actives. The lower the BER score, the better the method is at separating classes.
Whenever the participants associate a list of prediction values, rather than binary classes, with their submission, the IJCNN-07 competition also reports the area under the ROC curve (AUC). The ROC curve plots the True Positive Rate (TPR), which is the proportion of correctly predicted actives, against the False Positive Rate (FPR), which is the proportion of inactives incorrectly predicted as actives, for every possible threshold. The higher the AUC, the better the method is at separating classes.
In the McMaster competition, performance is assessed according to the number of active compounds retrieved in the top 1% and top 5% of the prediction-ranked test data. This is equivalent to looking at the Enrichment Factor (EFf%), which is the proportion of active compounds retrieved in the first f percent of the ranked list of chemicals.
In addition to these metrics, we also report for each dataset the value of the Boltzmann-Enhanced Discrimination of the Receiver Operating Characteristic (BEDROC) metric with α fixed at 20.0. The BEDROC metric, which quantifies the ability of a method to rank active compounds early at the top of the prediction-sorted test data, is particularly suitable to the evaluation of vHTS methods. Additional details about assessing early recognition and the mathematical derivation of the BEDROC metric can be found in Truchon and Bayly48.
Results
We report the cross-validation performance of the MAX-SIM, SVM and IRV methods on both the HIV and DHFR datasets. In order to directly compare our methods with others, we also present the performance of each method obtained while following the IJCNN-07 and McMaster competition protocols. Table 2 reports the hyperparameters (the similarity scheme and neighbor selection protocol) which yields the best performance for each experiment. The best similarity scheme is consistent across all methods and all folds of the cross-validation within each experiment.
Table 2.
Each experiment's optimal hyperparameters, in terms of similarity scheme (labeling, feature type, and metric) and whether or not the class-balance of IRV's nearest neighbors is enforced. If class-balance is enforced, the IRV is presented the 10 nearest active neighbors and the 10 nearest inactive neighbors (10+10); otherwise, the IRV is presented 20 nearest neighbors (irrespective of class). The optimal hyperparameters are always consistent across all methods and across all folds of the cross-validation experiments.
| Competition Rules |
Cross-Validation |
|||
|---|---|---|---|---|
| HIV | DHFR | HIV | DHFR | |
| Label | E | EC | EC | EC |
| Feature | Trees (d ≤ 2) | Paths (d ≤ 8) | Trees (d ≤ 2) | Paths (d ≤ 8) |
| Metric | MinMax | Tanimoto | Tanimoto | MinMax |
| Balanced | No (20) | Yes (10+10) | No (20) | Yes (10+10) |
HIV Cross-Validation
The various vHTS methods are evaluated by cross-validation on the HIV data. The performance is computed by averaging 10 random 10-fold cross-validated performances, using for each fold 90% of the data for training and 10% of the data for validation. Detailed results are described in Table 3. When feasible, we also report the Leave-One-Out (LOO) cross-validation performance. The SVM and E-SVM require about 1,000 seconds to train each fold, which amounts to approximately 500 CPU-days to LOO cross-validate the full dataset. Therefore, the SVM LOO performances are excluded.
Table 3.
Performance of several methods on the HIV data used in the IJCNN-07 competition. The IJCNN-07 row shows the best performance submitted to the competition not including results submitted by our laboratory. The “RANDOM” row shows the performance of a random classifier. Leave-one-out cross-validation performances are displayed between parenthesis, all other cross-validation performances are 10-fold cross-validations. A value that is not available is denoted by “-”. Best performances are in bold, and second best performances are in italics.
| Competition Rules |
Cross-Validation |
|||||
|---|---|---|---|---|---|---|
| BEDROC | BER | AUC | BEDROC | BER | AUC | |
| IJCNN-07 | - | 0.283 | 0.771 | - | - | - |
| RANDOM | 0.035 | 0.500 | 0.500 | 0.035 | 0.5000 | 0.5000 |
| IRV | 0.500 | 0.271 | 0.762 | 0.630 (0.633) | 0.210 (0.208) | 0.845 (0.839) |
| E-SVM | 0.465 | 0.269 | 0.764 | 0.573 | 0.199 | 0.845 |
| SVM | 0.507 | 0.280 | 0.758 | 0.616 | 0.217 | 0.836 |
| MAX-SIM | 0.439 | 0.283 | 0.739 | 0.526 (0.515) | 0.225 (0.222) | 0.806 (0.811) |
The IRV achieves the highest BEDROC of 0.630. The best BER of 0.1993, however, is obtained by the E-SVM. Both SVM and E-SVM are clearly behind the IRV in term of BEDROC performance, with values of 0.616 and 0.530 respectively. The ROC curves for the HIV data are plotted in Figure 3. These curves demonstrate that the SVM and IRV are approximately equivalent for this task and that the E-SVM does not perform as well in the critical early part of the ROC curve.
Figure 3.

The 10-fold cross-validated performances of various algorithms on the full HIV data are displayed using (1) a standard ROC curve; and (2) a pROC curve. The pROC curve plots the False Positive Rate on a logarithmic axis to emphasize the crucial initial portion of the ROC curve.49
Figure 2 illustrates how easy it is to intepret the IRV prediction, by tracking and visualizing the underlying influences corresponding to the experimental evidence used in order to make a prediction. Visualizing the IRV influence's is much more informative than explanations derived from an SVM, which prunes most of the experimental data from the final model.
Figure 2.

The influences on an accurately predicted hit from the HIV dataset, obtained in the cross-validation experiment, are displayed as a bar graph. The IRV used to compute these influences was trained using the 20 nearest neighbors. The experimental data both supporting and countering the prediction is readily apparent. The structures of four neighbors, two actives and two inactives, are shown. Compounds on the right are structurally similar and active. Compounds on the left are structurally similar and inactive.
The IJCNN-07 Competition
We participated in the IJCNN-07 competition by entering our E-SVM and IRV predictions for the HIV dataset in the Prior Knowledge track. As the competition was judged by BER, the ESVM won first place with a BER of 0.2693 and the IRV took second place with a BER of 0.2782. The IRV's performance was not fully optimized at the time, and we now report a BER of 0.2705, slightly above the winning entry. Thus again on this dataaset the performance of the IRV is at least equal to the performance of the SVM-based methods. Table 3 reports the performance of the MAX-SIM, SVM, and IRV methods in comparison with both a random classifier and the best participants in the IJCNN-07 experiment.
Although the BER was used to select the winners of the competition, the BEDROC metric better evaluates the suitability of methods for the vHTS problem. The best BEDROC of 0.507 is obtained with the SVM instead of the E-SVM. The IRV still ranks just behind the SVM, with a BEDROC of 0.500.
In the case of the SVM-based methods, there may be a trade-off between BER and BEDROC performance. For this dataset, the E-SVM optimizes the BER, but ranks third in BEDROC performance. The SVM optimizes the BEDROC, but ranks third in BER performance. The IRV, in contrast, is a close second in both BER and BEDROC performance, suggesting that it may optimize both performances simultaneously. Altogether, these results suggest the IRV method is slightly more suitable for vHTS, but E-SVM can better optimize the global class separation, quantified by the BER in the IJCNN-07 competition.
DHFR Cross-Validation
We also cross-validate our methods on the DHFR dataset. Performances are computed by averaging the results of ten random 10-fold cross-validations. We also report the LOO cross-validation performance of both the MAX-SIM and IRV methods.
The performance of our methods on the full, non-redundant data is reported in Table 4. The optimal performances are reached using the relaxed criteria for assigning actives and presenting the IRV with the 10 nearest active neighbors and 10 nearest inactive neighbors. Retrieving 20.6% of the actives in the top 5% of the ranked list, the SVM performs much better than both the random classifier and MAX-SIM, which retrieves only 8.8% of the actives in the same fraction. The IRV performs much better, retrieving 25% of the active compounds in the top 5% of the prediction-sorted data. The superiority of the IRV over the SVM is also apparent in its BEDROC performance of 0.251 for the IRV versus 0.200 for the SVM. Furthermore, of the top 20 IRV predictions, nine were true actives. The top twelve hits are listed in Figure 4. The nearly ideal ranking of the top hits was not duplicated by the SVM.
Table 4.
Results from the DHFR dataset used in the McMaster competition. The “RANDOM” row shows the performance of a random classifier. Leave-one-out cross validation performances are displayed between parenthesis, all other cross validation performances are 10-fold cross-validations. A value that is not available is denoted by '-'. Best performances are in bold and second best performance are in italics.
| Competition Rules |
Cross-Validation |
|||||
|---|---|---|---|---|---|---|
| BEDROC | 1% | 5% | BEDROC | 1% | 5% | |
| McMaster | - | 2 | 13 | - | - | - |
| RANDOM | 0.002 | 0.9 | 4.7 | 0.002 | 1.6 | 8.0 |
| IRV | 0.100 | 3 | 13 | 0.251 (0.258) | 20 (23) | 40 (44) |
| SVM | 0.084 | 1 | 4 | 0.200 | 22 | 33 |
| MAX-SIM | 0.045 | 0 | 3 | 0.062 (0.069) | 4 (4) | 13 (14) |
Figure 4.

Top twelve accurately predicted hits by an IRV cross-validated on the McMaster dataset. Prediction-sorted rankings are in bold and the IRV outputs are in parenthesis. Strikingly, the top hits are ranked at the very top of the prediction-sorted list. Although they exhibit a high degree of similarity, the top hits have several different scaffolds.
The cross-validated ROC curves for the DHFR data are displayed in Figure 5. Consistently with the performance metrics, these curves clearly show the superiority of the IRV on this particularly challenging dataset.
Figure 5.

The 10-fold cross-validated performances on the DHFR data. (a) ROC (b) pROC curve. The pROC curve plots the FPR on a logarithmic axis to emphasize the crucial first portion of the ROC curve.49
The McMaster Competition
We did not enter any of our predictors in the McMaster competition, which took place before the beginning of this work, but we do retrospectively compare our results, following the competition rules, to published results. The winners were picked based on the number of actives ranked in the top 1% and 5% of the predictions. This is equivalent to looking at the Enrichment Factors (EF) calculated at 1% and 5%. For this competition, even the winner was only able to correctly retrieve a small fraction of the active chemicals in the test set: 2 in the top 1% and 13 in the top 5% of the prediction-ranked output. A detailed comparison of the performances obtained while following the competition rules are presented in Table 4.
For the IRV, optimal results are achieved by presenting the 10 nearest active neighbors and 10 nearest inactive neighbors to the network. Furthermore, for all the classifiers, optimal results are obtained using the relaxed criterion to label actives during training. For the relaxed criterion, instead of labeling active compounds when both experimental scores are less than 75%, we label compounds active if at least one score is below 75%. This increases the pool of positive examples during training, feeding the classifiers additional information. Actives are assigned by the relaxed criterion only during training; test performance are computed using the more stringent competition criterion to ensure our results are comparable with the literature.
Both the SVM and the MAX-SIM method perform worse than random, retrieving only 4.2% and 3.1% of the actives respectively, in the top 5% of their prediction-ranked lists. This performance is similar to most submissions to the competition, as 29 out of 32 participants also retrieved 4.2% of hits in the top 5% of their ranked list, and only one of them also found more than 1.1% of hits in the top 1%.
The IRV, in contrast, detects almost 14% (13 out of 94) of the actives in the top 5% of the ranked list, which is as good as the best entry in the competition. Moreover, the IRV detects one more active in the top 1% of the prediction-sorted data than this best entry. Altogether, these results show that the IRV performs much better than the SVM and slightly better than the best methods in the community on this challenging dataset.
Although the results from the McMaster competition provide a useful basis for performance comparison, the organizers did not maintain any information regarding the exact methods used by each participant. As a result, information regarding the methods used to generate the competition's best entry, for instance, is not readily available.
Discussion
According to metrics best suited to evaluate vHTS, the IRV compares favorably with SVMs and other state-of-the-art methods. On the HIV dataset, the IRV performance is comparable to the best SVM performance, while on the DHFR dataset the IRV performance is clearly superior to SVMs. In addition to outperforming SVMs, the IRV is preferable because: (1) it is trained much more quickly; (2) it provides a framework which easily allows the incorporation of additional information, beyond the chemical structures; and (3) its predictions are interpretable.
IRV Training Time
A first advantage of the IRV is that it is quickly trained, once the nearest neighbors have been computed. Each chemical is represented using only a small number of informative features, allowing the neural network to be trained in minutes rather than hours or days, and making leave-one-out cross-validation possible, even on very large datasets. Learning the weights is efficient, with both space and time complexity scaling approximately as O(N · k). This corresponds to the fact that, under typical conditions, neural network training converges in a constant number of epochs (one epoch corresponding to one training pass through the training set). Thus the IRV training complexity scales like the complexity of computing its full gradient.
Finding the nearest neighbors requires much more time than learning the weights. Although computing the neighbors is computationally expensive, it is trivially parallelizable and less expensive than training an SVM. The naive algorithm, using a full pairwise comparison of the training data, requires O(N2 logk) time. The pruning search algorithms described in Swamidass and Baldi29 can reduce the complexity to approximately O(N1.6 logk). Furthermore, finding the nearest neighbors is trivially distributed across an arbitrary number C of computers, with negligible overhead. The complexity of the parallelized algorithm for finding neighbors, therefore, can be approximated as O(N1.6/C · logk).
Alternatively, Locality-Sensitive Hashing (LSH) could be used to find most of the nearest neighbors in only O(N · logN) time, enabling predictions in O(logN) time, further reducing the complexity.50 LSH, however, is an approximate algorithm, and is not guaranteed to find all the neighbors exactly. In principle, the IRV may be able to tolerate the noise introduced by inexact nearest-neighbor algorithms, but this possibility is beyond the scope of this study and currently untested.
Although the weights can be trained quickly by gradient descent, the IRV requires the manual selection of a few hyperparameters, such as the number of nearest neighbors and the way they are pooled and ordered. The IRV, however, does not appear to be highly sensitive to these parameters. Selecting the 20 nearest neighbors seems to be about right for the two datasets studied. There still remains some reason to believe information may be hidden among distant neighbors, especially when one class is particularly rare.8 In principle, both the information from distant neighbors can be included and the arbitrariness inherent in picking k can be removed by setting k = ∞ and presenting the network with all instances in the training set instead of just the nearest few neighbors. Unfortunately, this does increase the time and space complexity of training and classification. This variation is also beyond the scope of this study and left for future work.
IRV Incorporation of Additional Information
A second advantage of the IRV is that it can naturally be extended to cohesively integrate additional types of information. Several of these extensions are visually depicted in Figure 6. In the case of the HIV dataset, compounds classified as active for IJCNN-07 are in fact known to be either “active” or “moderately active”, the distinction between which is ignored. We can, however, learn the predictive value of the chemicals belonging to the “moderately active” class by adding another class of neighbors to the IRV model. This amounts to redefining Vi as
| (12) |
where, for the neighbors, w0, w1 and w2, corresponding to `inactive', “moderately active” or “active” compounds. Of course, if the aim is to find active compounds as defined by the competition, the likelihood function used during training would remain unchanged, with the “moderately active” compounds labeled as active compounds. The IRV can be modified in similar ways to use the raw inhibition data, ai, reported in the DHFR dataset, by using
| (13) |
where wa and wv are learned from the data. More generally, one could define
| (14) |
where f (·) is a parameterized function-with parameters to be learned from the data-of both the inhibition data and the assigned class of each neighbor. Initial experiments indicate that these modifications can further optimize performance.
Figure 6.
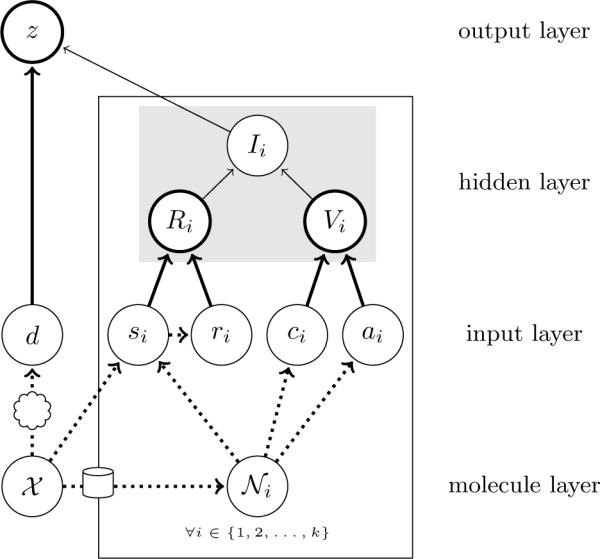
Examples of IRV architectural extensions. The variable d denotes the docking score derived by fitting each test chemical, 𝒳, into the binding pocket of a known protein target, depicted as a cloud in the figure. The grayed out rectangle delimits a portion of the network which could be trivially replaced with an arbitrarily complex neural network to generate an IRV with increased modeling power; additional replaceable portions can be imagined. The variable ai denotes the real-valued activity of each neighbor, 𝒩i, in the HTS screen.
Instance-level data, also, can easily be integrated in the IRV framework. For example, solubility is known to be an important property that can affect the ability of an HTS experiment to measure the activity of a compound. Feeding the test compound's predicted solubility into the output node could improve prediction accuracy. Likewise, in situations where the structure of the target protein is available, such as for the McMasters competition, performance gains could be realized by feeding the docking energy of the test compound directly into the output node, naturally integrating docking and similarity data into the same predictor.
Similarly, it is possible to replace portions of the IRV with neural networks of arbitrary complexity, thereby increasing the modeling power. This comes of course at a cost of increasing the number of parameters which must be learned, requiring the model to be trained on increasing amounts of data.
IRV Interpretability
A third advantage of the IRV is that its predictions are transparent. The exact experimental data used to make a prediction can be extracted from the network by examining each prediction's influences. Thus, although the IRV is composed of a neural network, it should be considered a “white-box” method. In contrast, the predictions of an SVM are more difficult to interpret. Along the same lines, the IRV's output is probabilistic and directly encodes each prediction's uncertainty, enabling reasonably accurate estimation of the number of hits in an arbitrary set of test molecules. Although an SVM output can be rescaled onto a probability scale,51-53 SVMs can exhibit a tendency to predict with overly high confidence in regions where the training data is sparse.
Often, there is a trade-off between understandable models and high performing models,1,54 requiring researchers to choose if interpretability is more important than accuracy. The IRV seems to solve this dilemma by yielding high performance on HTS data using an easily understandable framework.
Acknowledgement
Work supported by NIH Biomedical Informatics Training grant (LM-07443-01), NSF MRI grant (EIA- 0321390), NSF grant 0513376, and a Microsoft Research Award to PB. We would like also to acknowledge the OpenBabel project and OpenEye Scientific Software for their free software academic licenses.
References
- (1).Karakoc E, Cherkasov A, Sahinalp SC. Distance Based Algorithms For Small Biomolecule Classification And Structural Similarity Search. Bioinformatics. 2006;22:243. doi: 10.1093/bioinformatics/btl259. [DOI] [PubMed] [Google Scholar]
- (2).Zheng W, Tropsha A. Novel Variable Selection Quantitative Structure-Property Relationship Approach Based on the k-Nearest-Neighbor Principle. J. Chem. Inf. Comput. Sci. 2000;40:185–194. doi: 10.1021/ci980033m. [DOI] [PubMed] [Google Scholar]
- (3).Cannon EO, Bender A, Palmer DS, Mitchell JBO. Chemoinformatics-Based Classification of Prohibited Substances Employed for Doping in Sport. J. Chem. Inf. Model. 2006;46:2369–2380. doi: 10.1021/ci0601160. [DOI] [PubMed] [Google Scholar]
- (4).Geppert H, Horvath T, Gartner T, Wrobel S, Bajorath J. Support-Vector-Machine-Based Ranking Significantly Improves the Effectiveness of Similarity Searching Using 2D Fingerprints and Multiple Reference Compounds. J. Chem. Inf. Model. 2008;48:742–746. doi: 10.1021/ci700461s. [DOI] [PubMed] [Google Scholar]
- (5).Plewczynski D, Spieser SAH, Koch U. Assessing Different Classification Methods for Virtual Screening. J. Chem. Inf. Model. 2006;46:1098–1106. doi: 10.1021/ci050519k. [DOI] [PubMed] [Google Scholar]
- (6).Simmons K, Kinney J, Owens A, Kleier D, Bloch K, Argentar D, Walsh A, Vaidyanathan G. Comparative Study of Machine-Learning and Chemometric Tools for Analysis of In-Vivo High-Throughput Screening Data. J. Chem. Inf. Model. 2008 doi: 10.1021/ci800142d. [DOI] [PubMed] [Google Scholar]
- (7).Hastie T, Tibshirani R. Discriminant Adaptive Nearest Neighbor Classification and Regression. Adv. Neural. Inform. Process. Syst. 1996:409–415. [Google Scholar]
- (8).Holmes C, Adams N. Likelihood inference in nearest-neighbour classification models. Biometrika. 2003;90(14):99–112. [Google Scholar]
- (9).Kozak K, Kozak M, Stapor K. Weighted k-Nearest-Neighbor Techniques for High Throughput Screening Data. Int. J. Biomed. Sci. 2006;01:155–160. [Google Scholar]
- (10).Niwa T. Using General Regression and Probabilistic Neural Networks To Predict Human Intestinal Absorption with Topological Descriptors Derived from Two-Dimensional Chemical Structures. J. Chem. Inf. Comput. Sci. 2003;43:113–119. doi: 10.1021/ci020013r. [DOI] [PubMed] [Google Scholar]
- (11).Shang W, Qu Y, Zhu H, Huang H, Lin Y, Dong H. An Adaptive Fuzzy kNN Text Classifier Based on Gini Index Weight. ISCC '06: Proceedings of the 11th IEEE Symposium on Computers and Communications.2006. pp. 448–453. [Google Scholar]
- (12).Specht D. Probabilistic neural networks for classification, mapping, or associative memory. IEEE International Conference on Neural Networks, 1988.1988. pp. 525–532. [Google Scholar]
- (13).Specht D. A general regression neural network. IEEE Trans. Neural Netw. 1991;2:568–576. doi: 10.1109/72.97934. [DOI] [PubMed] [Google Scholar]
- (14).Tan S. Neighbor-weighted K-nearest neighbor for unbalanced text corpus. Expert Syst. Appl. 2005;28:667–671. [Google Scholar]
- (15).Zhang H, Berg AC, Maire M, Malik J. SVM-KNN: Discriminative Nearest Neighbor Classification for Visual Category Recognition. Proc. CVPR 2006. 2006;02:2126–2136. [Google Scholar]
- (16).Bishop C. Neural Networks for Pattern Recognition. Chapter 1 Oxford University Press, Inc; New York: 1995. [Google Scholar]
- (17).Baldi P. Gradient descent learning algorithm overview: a general dynamical systems perspective. IEEE Trans. Neural Netw. 1995;6:182–195. doi: 10.1109/72.363438. [DOI] [PubMed] [Google Scholar]
- (18).Baldi P, Brunak S. Bioinformatics: the machine learning approach. Second edition MIT Press; Cambridge, MA: 2001. [Google Scholar]
- (19).Azencott CA, Ksikes A, Swamidass SJ, Chen JH, Ralaivola L, Baldi P. One- to Four-Dimensional Kernels for Virtual Screening and the Prediction of Physical, Chemical, and Biological Properties. J. Chem. Inf. Model. 2007 doi: 10.1021/ci600397p. [DOI] [PubMed] [Google Scholar]
- (20).Swamidass SJ, Chen JH, Bruand J, Phung P, Ralaivola L, Baldi P. Kernels for Small Molecules and the Predicition of Mutagenicity, Toxicity, and Anti-Cancer Activity. Bioinformatics; Proceedings of the 2005 ISMB Conference; 2005. p. 359. [DOI] [PubMed] [Google Scholar]
- (21).Agnostic Learning vs. Prior Knowledge Challenge. http://www.agnostic.inf.ethz.ch/index.php (accessed Jan 15, 2009)
- (22).DTP - AIDS Antiviral Screen Data. http://dtp.nci.nih.gov/docs/aids/aids_data.html (accessed Jan 15, 2009)
- (23).HTS Data Mining and Docking Competition. http://hts.mcmaster.ca/HTSDataMiningandDockingCompetition.html (accessed Jan 15, 2009)
- (24).Bender A, Mussa HY, Glen RC. Screening for Dihydrofolate Reductase Inhibitors Using MOLPRINT 2D, a Fast Fragment-Based Method Employing the Naïve Bayesian Classifier: Limitations of the Descriptor and the Importance of Balanced Chemistry in Training and Test Sets. J. Biomol. Screen. 2005;10:658–666. doi: 10.1177/1087057105281048. [DOI] [PubMed] [Google Scholar]
- (25).Lang PT, Kuntz ID, Maggiora GM, Bajorath J. Evaluating the high-throughput screening computations. J. Biomol. Screen. 2005;10:649–652. doi: 10.1177/1087057105281269. [DOI] [PubMed] [Google Scholar]
- (26).Parker CN. McMaster University data-mining and docking competition: computational models on the catwalk. J. Biomol. Screen. 2005;10:647–648. doi: 10.1177/1087057105281268. [DOI] [PubMed] [Google Scholar]
- (27).Leach AR, Gillet VJ. An Introduction to Chemoinformatics. Springer; Dordrecht, The Netherlands: 2005. [Google Scholar]
- (28).Chen J, Swamidass SJ, Dou Y, Bruand J, Baldi P. ChemDB: A Public Database Of Small Molecules And Related Chemoinformatics Resources. Bioinformatics. 2005;21:4133–4139. doi: 10.1093/bioinformatics/bti683. [DOI] [PubMed] [Google Scholar]
- (29).Swamidass SJ, Baldi P. Bounds and Algorithms for Exact Searches of Chemical Fingerprints in Linear and Sub-Linear Time. J. Chem. Inf. Model. 2007;47:302–317. doi: 10.1021/ci600358f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Johnson MA, Maggiora GM, editors. Concepts and Applications of Molecular Similarity. John Wiley; 1990. [Google Scholar]
- (31).Flower DR. On the Properties of Bit String-Based Measures of Chemical Similarity. J. Chem. Inf. Comput. Sci. 1998;38:378–386. [Google Scholar]
- (32).Raymond JW, Willett P. Effectiveness of Graph-Based and Fingerprint-Based Similarity Measures for Virtual Screening of 2D Chemical Structure Databases. J. Comput. Aided Mol. Des. 2001;16:59–71. doi: 10.1023/a:1016387816342. [DOI] [PubMed] [Google Scholar]
- (33).Baldi P, Benz R, Hirschberg D, Swamidass S. Lossless compression of chemical finger-prints using integer entropy codes improves storage and retrieval. J. Chem. Inf. Model. 2007;47:2098–2109. doi: 10.1021/ci700200n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hassan M, Brown RD, Varma-O'Brien S, Rogers D. Cheminformatics analysis and learning in a data pipelining environment. Mol. Divers. 2006;10:283–299. doi: 10.1007/s11030-006-9041-5. [DOI] [PubMed] [Google Scholar]
- (35).Holliday JD, Hu CY, Willett P. Grouping of coefficients for the calculation of intermolecular similarity and dissimilarity using 2D fragment bit-strings. Comb. Chem. High Throughput Screen. 2002;5:155–66. doi: 10.2174/1386207024607338. [DOI] [PubMed] [Google Scholar]
- (36).Ralaivola L, Swamidass SJ, Saigo H, Baldi P. Graph Kernels for Chemical Informatics. Neural Netw. 2005;18:1093–1110. doi: 10.1016/j.neunet.2005.07.009. [DOI] [PubMed] [Google Scholar]
- (37).Hert J, Willett P, Wilton DJ, Acklin P, Azzaoui K, Jacoby E, Schuffenhauer A. Comparison of Fingerprint-Based Methods for Virtual Screening Using Multiple Bioactive Reference Structures. J. Chem. Inf. Model. 2004;44:1177–1185. doi: 10.1021/ci034231b. [DOI] [PubMed] [Google Scholar]
- (38).Hert J, Willett P, Wilton DJ, Acklin P, Azzaoui K, Jacoby E, Schuffenhauer A. Enhancing the effectiveness of similarity-based virtual screening using nearest-neighbor information. J. Med. Chem. 2005;48:7049–54. doi: 10.1021/jm050316n. [DOI] [PubMed] [Google Scholar]
- (39).Mahé P, Ralaivola L, Stoven V, Vert JP. The Pharmacophore Kernel for Virtual Screening with Support Vector Machines. J. Chem. Inf. Model. 2006;46:2003–2014. doi: 10.1021/ci060138m. [DOI] [PubMed] [Google Scholar]
- (40).Collobert R, Bengio S. SVMTorch: Support Vector Machines for Large-Scale Regression Problems. J. Mach. Learn. Res. 2001;1:143–160. http://www.idiap.ch/learning/SVMTorch.html. [Google Scholar]
- (41).Veropoulos K, Campbell C, Cristianini N. Controlling the Sensitivity of Support Vector Machines. Proceedings of the International Joint Conference on AI.1999. pp. 55–60. [Google Scholar]
- (42).Shin H, Cho S. How to Deal with Large Dataset, Class Imbalance and Binary Output in SVM based Response Model. Proceedings of the Korean Data Mining Conference; 2003. pp. 93–107. Best Paper Award. [Google Scholar]
- (43).Estabrooks A, Jo T, Japkowicz N. A Multiple Resampling Method for Learning From Imbalanced Data Set. Comput. Intell. 2004;20 year. [Google Scholar]
- (44).Orriols A, Bernad-Mansilla E. The Class Imbalance Problem in Learning Classifier Systems: A Preliminary Study. Proceedings of the 2005 Workshops on Genetic and Evolutionary Computation; Washington, D.C.. June 25 - 26, 2005; New York, NY: 2005. pp. 74–78. [Google Scholar]
- (45).Tipping ME. Sparse Bayesian Learning and the Relevance Vector Machine. J. Mach. Learn. Res. 2001;1:211–244. [Google Scholar]
- (46).Baldi P, Pollastri G. The principled design of large-scale recursive neural network architectures DAG-RNNS and the protein structure prediction problem. J. Mach. Learn. Res. 2003;4:575–602. [Google Scholar]
- (47).Dybowski R, Roberts SJ. Confidence intervals and prediction intervals for feed-forward neural networks. Clinical Applications of Artificial Neural Networks. 2001:298–326. [Google Scholar]
- (48).Truchon JF, Bayly CI. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007;47:488–508. doi: 10.1021/ci600426e. [DOI] [PubMed] [Google Scholar]
- (49).Clark RD, Webster-Clark DJ. Managing Bias in ROC Curves. J. Comput. Aided Mol. Des. 2008;22:141–146. doi: 10.1007/s10822-008-9181-z. [DOI] [PubMed] [Google Scholar]
- (50).Dutta D, Guha R, Jurs P, Chen T. Scalable partitioning and exploration of chemical spaces using geometric hashing. J. Chem. Inf. Model. 2006;46:321–333. doi: 10.1021/ci050403o. [DOI] [PubMed] [Google Scholar]
- (51).Smola AJ, Bartlett P, Scholkopf B, Schuur-mans D, editors. Probabilities for Support Vector Machines. MIT Press; 1999. pp. 61–74. [Google Scholar]
- (52).Sollich P. Probabilistic interpretations and Bayesian methods for support vector machines. Artificial Neural Networks, 1999. ICANN 99. Ninth International Conference on (Conf. Publ. No. 470).1999. pp. 91–96. [Google Scholar]
- (53).Kwok JTY. Moderating the outputs of support vector machine classifiers. IEEE Trans. Neural Netw. 1999;10:1018–1031. doi: 10.1109/72.788642. [DOI] [PubMed] [Google Scholar]
- (54).Szafron D, Lu P, Greiner R, Wishart DS, Poulin B, Eisner R, Lu Z, Anvik J, Macdonell C, Fyshe A, Meeuwis D. Proteome Analyst: Custom Predictions with Explanations in a Web-based Tool for High-throughput Proteome Annotations. Nucleic Acids Res. 2004;32:365. doi: 10.1093/nar/gkh485. [DOI] [PMC free article] [PubMed] [Google Scholar]