

Abstract
Selection-amplification finds new RNA enzymes (ribozymes) among randomized RNAs with flanking unvaried sequences (primer complements). Precise removal of 3′-primer before reaction selected aminoacylation from PheAMP in 3 cycles, yielding active RNAs (kcat = 12-20 min-1) using only three conserved nucleotides, acting independently of divalent ions. This unusually simple RNA active site encouraged study of the reaction via molecular mechanics-based free energy minimization. On this basis, we suggest a chemical path for RNA-catalyzed transaminoacylation. Site modeling also predicted new features - L-stereoselectivity, 2′-regioselectivity, independence of amino acid side chain and phosphorylated activating group, that were subsequently verified. The same selection also showed that RNA aminoacylation from adenylate is simpler than from CoA thioester, potentially rationalizing translational activation by adenylates. The simplicity of this active site suggests a general route to small ribozymes.
Introduction
According to the RNA world hypothesis, RNA was once the principal bioinformational and catalytic molecule in living cells 1, 2. Substantial support for this hypothesis has been obtained by in vitro selection of previously unknown ribozymes from pools of random RNA sequences. Such newly-selected ribozymes are capable of a variety of reactions catalyzed by protein enzymes in modern cells 3. However, in view of the difficulty of RNA synthesis under primitive conditions, simpler RNA catalysts increase the plausibility of ancient RNA metabolism. Below we describe one possible route to shorter, simpler ribozymes. We use aminoacylation of RNA, a reaction required for appearance of translation at the end of the RNA world era, as our model. This reaction occurs in a variety of RNA sequence contexts 4-9, and consistent with this variability, aminoacylation should be able to respond to an uncomplicated selection protocol.
In vitro selection-amplification (SELEX) 10-12 utilizes repeated purification cycles on randomized RNAs, which possess flanking, fixed, arbitrarily chosen sequences (primer sites, ~ 20 nucleotides long) to mediate replication. To fulfill selection criteria, ribozyme activity must tolerate the fixed sequences. This necessarily decreases the number of functional sequences discovered by selection, and particularly so when the 3′- or 5′-terminus itself is a reactant. We now describe a selection requiring no 3′-fixed sequence. After only three cycles of selection, this procedure revealed a novel aminoacyl transfer center containing just three conserved nucleotides.
Encouraged by the unusual simplicity of this RNA-facilitated reaction, we attempted to understand the reaction path. Unlike affinity, which is often successfully explained by crystal structures or NOE NMR, there are no tools available to directly observe a chemical transformation. A well-validated approach is to postulate a plausible mechanism, then to test the proposed pathway against experimental data. However, for a conformationally mobile RNA molecule a mechanism can be obscure because the particular RNA fold lying on the reaction pathway is not evident. We approached this problem by comparing calculated stable active site conformations. A recurrent stable conformation was detected that also accommodated the substrate. This suggested a pathway for RNA-mediated self-aminoacylation consistent with all known properties of RNA self-aminoacylation. The mechanism predicted new features that were confirmed by experiment, suggesting that the mechanism captures essential aspects of aminoacyl transfer. Our mechanism also indicates a possibly useful generalization about the simplest, and therefore the most evolutionarily interesting RNA catalyses.
Selection scheme
The modified selection scheme is shown in Figure 1, with substrates detailed in Supplementary Figure 1. The essential step is that active randomized RNA sequences react without a 3′ constant sequence, then are ligated to a 3′ oligonucleotide (RNA-19) for amplification. The 5′-end of the ribozyme is blocked with triphosphate and RNA-19 has a 3′-ddC terminus, so ligation produces a unique product. Optimized, apparently quantitative ligation was observed at low RNA concentrations (0.1 μM), with 15-20 times excess of RNA-19.
Figure 1.

A selection cycle. Color coding: T7-promoter (17 nt) - dark red, 5'-primer (19 nt) - red, random region (50 nt) - black, HDV ribozyme (87 nt) - blue. [α 32P G] pool RNA has 69 nt; HDV ribozyme (67 nt) is distinct because it can be 3′-elongated to 87 nt without change in reactivity. Self-cleavage results in 2′,3′-cyclic phosphate at the 3′-end of transcript RNA, that must be opened and dephosphorylated.
After selected sequences are amplified by RT/PCR, the 3′-primer must be removed. We tested (not shown) RNA-19 containing a restriction site sequence, to be cleaved after PCR. But after a few cycles pooled RNAs became highly 3′-heterogeneous because T7 RNA polymerase both prematurely terminated transcription and also added extra 3′-nucleotides 13. This suggests that previous selections for aminoacylation by RNA could have been complicated by the need to acylate a varying 3′ acceptor nucleotide. Therefore we removed 3′-sequences post-transcriptionally by embedding in them the Mörl variation of the hepatitis delta virus self-cleaving ribozyme (HDV) 14. Mörl HDV RNA has no sequence requirements upstream of the cleaved internucleotide bond; therefore its use maintains 3′-terminal length but also allows any 3′-nucleotide. Self-cleavage leaves a terminal 2′,3′-cyclic phosphate ester, which we opened and hydrolyzed using T4 polynucleotide kinase to yield a 2′,3′-hydroxyl terminus for the next reaction.
Our selected reaction was 2′,3′-aminoacylation of RNA - an essential step of modern ribosome-mediated peptide synthesis. In contemporary cells, amino acids are activated as mixed anhydrides with the 5′-phosphate of AMP (aminoacyl adenylates). In reported selections of 2′,3′-RNA-aminoacylating ribozymes 3, amino acids were pre-activated as aminoacyl adenylates 5-7, cyanomethyl esters and thioesters 4, 8. Aminoacyl-CoA has also been used as a substrate, with aminoacylation of an internal 2′-OH 9.
We designed the selection as a balanced competition between two known activated amino acid substrates: aminoacyl adenylate (PheAMP) and aminoacyl-CoA (PheCoA). These were present together in the selection mixture at similar concentrations. CoA thioesters are quite stable to hydrolysis, so to maintain competition pH was decreased to 6.4, making the half-life of PheAMP ~ 54 min. CoA is arguably a prevalent cofactor in the RNA world 15 and acyl-CoA synthesis is known to be within the catalytic repertoire of modern RNA 16, 17. Thus this selection asks whether the presently universal AMP activation reaction was plausibly selected for aminoacyl-RNA biosynthesis because transacylation by an ancient RNA catalyst was simpler than transacylation from CoA-activated amino acid.
We biotinylated acylated RNA, then separated Biotin-NeutrAvidin bound self-aminoacylated biotinylated RNA from unbound, unreacted RNA 18 by column affinity chromatography (see Supplementary Online information). To release bound RNAs, the ester bond between biotinylated amino acid and the terminal ribose-OH of immobilized RNA was hydrolyzed at pH 8.3 19, providing a free 2′,3′-OH on active RNAs. We estimate that 90% of PheRNA is recovered by this assay, and have normalized for recovery throughout.
Selection results
Selection was rapid, with appearance of 2.5 % aminoacylated sequences after the third cycle of selection (Supplementary Table 1). This significantly exceeds a smaller reproducible background, mostly attributable to non-specific binding of RNA to streptavidin. We cloned after the fifth cycle. Having started with a pool of 3 × 1014 randomized machine-made ssDNAs, and assuming successful PCR of 30% of initial synthetic DNA, then 5 successive selection cycles with 0.49, 0.65, 2.5, 11.3 and 16% recoveries yield maximally 1.2×106 independent sequences, or 10-8 or fewer of the initial transcripts. Alternatively, at breakthrough in the third cycle ≤ 2.4 × 10-7 of total, or 2.4 × 107 sequences were active. Such a potentially high frequency of self-aminoacylating RNAs among randomized sequences suggests an unusually simple, or unusually varied, reactive structure, and a small active site.
Sequencing showed that active RNAs were usually derived from different initial parents, confirming the above high frequencies of active RNAs among randomized sequences. Out of 143 sequenced isolates (Figure 3, Table 1), 73.5% were two-helix junctions with one apparently non-helical 3′-nucleotide (95% U) as the aminoacyl acceptor. The junction loop was mostly a 5′ NGU 3′ triplet, with UGU the most frequent. Longer loops of 4, 5 and 6 nucleotides occur with decreasing frequency, and 4, 5 and 6 nt loops usually also end in GU 3′, like the more frequent triplet loops. Comparison of RNA-106 and RNA-113 (Figure 3, Table 2), suggests that triplet loops (RNA 106) may be more prevalent because they react faster than the CACGU junction loop in RNA 113.
Figure 3.
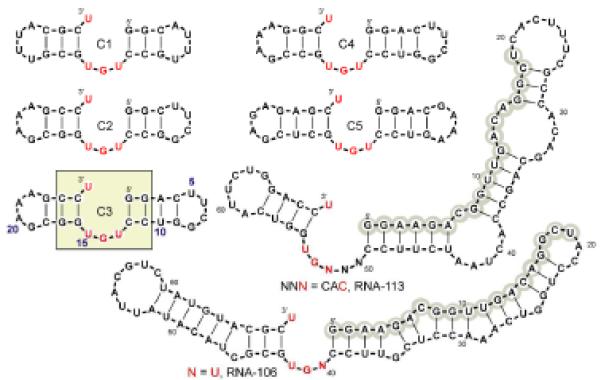
Selected and derived self-aminoacylating ribozymes. Gray circles mark 5′-constant sequence. Numbering is conventional, from 5′ end.
Table 1.
143 RNAs: selected loop and 3′ -terminal sequences.
| Triplet NGU, N | Quad N′NGY | Five/six nucleotide loops | 3′ -terminal nucleotides | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N′ | N | Y | N3' | N3'-1 | N3'-2 | N3'-3 | N3'-4 | |||
| U | 44 | 19 | 18 | 26 | ACUGU, CACGU | 136 (95%) | 21 | 30 | 44 | 36 |
| C | 9 | 2 | 12 | 13 | ACCGU | 2 | 83 (58%) | 33 | 41 | 44 |
| A | 6 | 14 | 8 | 0 | GCAAGU | 2 | 20 | 29 | 30 | 27 |
| G | 2 | 4 | 1 | 0 | GCUCGU | 3 | 19 | 51 | 28 | 36 |
| Tot | 61 (43%) | 39 (27%) | 5 (3.5%) | 143 (100%) | ||||||
Table 2.
Comparison of RNA reactivities - Reactions at 15 °C; 12 μM RNA and (in mM): 100 Pipes, 100 NaCl, 100 KCl, 5 CaCl2, 5 MgCl2, 0.1 MnCl2, final pH 6.4 (see also Supplementary Information).
| Entry | RNA name | Comment | Substrate, conc. mM | Fract PheRNA at 15 min |
|---|---|---|---|---|
| 1 | 106 | Selected RNA majority | PheAMP, 2.2 | 0.88 |
| 2 | 113 | 5 nt junction | PheAMP, 2.6 | 0.24 |
| Small Model RNAs | ||||
| 3 | C1 | 3/3 base pairs | PheAMP, 3.2 | 0.42 |
| 4 | C2 | 3/3 base pairs | PheAMP, 2.8 | 0.64 |
| 5 | C4 | 3/4 base pairs | PheAMP, 2.7 | 0.95 |
| 6 | C5 | 4/4 base pairs | PheAMP, 3.1 | 0.78 |
| 7 | C3, U/UGU | 3/4 base pairs | PheAMP, 3.3 | 0.94 |
| Altered RNA/substrate | ||||
| 8 | C3 | No divalents | PheAMP, 3.3 | 0.86 |
| 9 | C3 | 5′ OH | PheAMP, 3.0 | 0.95 |
| 10 | C3 | 5′-P, 5′-CG pair | PheAMP, 2.2 | 0.88 |
| 11 | C3 | UMP activation | PheUMP, 2.7 | 0.94 |
| 12 | C3 | Methionine | MethAMP, 3.5 | 0.65 |
| 13 | C3 | (D) amino acid | (D)PheAMP,3.2 | 0.055 |
| Loop and 3′ mutants | ||||
| 14 | G26/UGU | 3′ -end G nucleotide | PheAMP, 3.2 | 0.056 |
| 15 | C26/UGU | 3′ -end C nucleotide | PheAMP, 3.2 | 0.052 |
| 16 | A26/UGU | 3′ -end A nucleotide | PheAMP, 3.2 | 0.024 |
| 17 | U/G13GU | 5′-G loop nucl. | PheAMP, 0.60 | 0.26 |
| 18 | U/C13GU | 5′-C loop nucl. | PheAMP, 0.22 | 0.92 |
| 19 | U/A13GU | 5' -A loop nucl. | PheAMP, 0.60 | 0.033 |
| 20 | U/UC14U | Middle C loop nucl. | PheAMP, 4.0 | 0.062 |
| 21 | U/UA14U | Middle A loop nucl. | PheAMP, 2.7 | 0.27 |
| 22 | U/UU14U | Middle U loop nucl. | PheAMP, 2.3 | 0.18 |
| 23 | U/UGG15 | 3′-G loop nucl. | PheAMP, 4.0 | 0.038 |
| 24 | U/UGC15 | 3′-C loop nucl. | PheAMP, 2.4 | 0.20 |
| 25 | U/UGA15 | 3′-A loop nucl. | PheAMP, 4.0 | 0.033 |
| 26 | G26/UGU | 3′-end G nucleotide | PheAMP, 3.2 | 0.056 |
| 27 | C26/UGU | 3′-end C nucleotide | PheAMP, 3.2 | 0.052 |
| 28 | A26/UGU | 3′-end A nucleotide | PheAMP, 3.2 | 0.024 |
| 29 | A26/UGC15 | 3′-end A;3′-Gintheloop | PheAMP, 2.7 | 0.24 |
| 30 | A27/UUGC16 | 3′A; 3′-C in four nt loop | PheAMP, 2.7 | 0.19 |
The right hand bound of the active site (Figure 3) was a GC base pair mandated by the fixed 5′-primer sequence. The left-hand bound was usually a CG pair (58%), though all four base pairs occur at this helical boundary and in the four successive leftward helix positions (Table 1, Figure 3). Outside this central helix-loop-helix, the structure, even though it accommodates the mostly paired rightward primer, appeared quite varied in Bayesfold 20. These data strongly suggest that the prevalent aminoacyl-transfer center is a simple helix-loop-helix junction with a 5′ NGU or longer loop and an overhanging 3′-U acceptor, as in Figure 3 and Table 1. This simple structure possesses only three conserved nucleotides and should be frequent among randomized sequences, in agreement with rapid selection.
To compare activity of different RNAs they were aminoacylated, biotinylated and then initial reaction rates and/or degrees of transformation were found by streptavidin retardation gel assays 21 (Supplementary Figure 3). RNAs possessing cyclic 2′,3′-phosphate had less than 1% reactivity, at the level of background. Thus aminoacylation requires a 2′,3′-hydroxyl end, in all probability the site of acylation.
The population of selected sequences after the 5th cycle were reactive with PheAMP but did not show significant reactivity from PheCoA. For example, we transcribed the pool after the 5th selection and observed only background reactivity with PheCoA for these mixed sequences. Therefore an RNA active center for PheAMP is selected more easily than one that aminoacylates itself using PheCoA. RNAs that transfer aminoacyl ester from PheCoA are known 9, therefore this result suggests that an adenylate is substantially more easily used. This observation helps explain the universal choice of AMP as leaving group for amino acid activation in subsequent translation.
Kinetics of aminoacyl transfer
As shown in the accompanying figure (Figure 2A), PheRNA production shows unusual “jump” kinetics that are non-Michaelis-Menten. Initial jumps are also observed for mutated RNAs (Figure 2B), though nucleotide sequence mutations alter both the jump magnitude and rates of reaction. In Supplementary Information (Supp. Figure 4 and 5), we explain a route by which such jump kinetics may arise and be analyzed. Our preferred model, which fit data the best, supposes that fast binding of substrate yields either a reactive or unreactive complex. The essential question is how one characterizes RNAs exhibiting such complex kinetics. We wish to compare RNAs of very different activities, notably including mutants whose reaction is too slow for accurate resolution of the jump and slow phases of self-acylation. Therefore we used net reaction at an intermediate time (15 minutes; Table 2) in the presence of high (hopefully saturating) substrate adenylate concentrations. As can be appreciated from the Supplementary Online discussion, this practice combines the intrinsic reactivity of the RNA with the access of the RNA to its reactive state (expressed as jump size and the later slow rate, respectively). Nevertheless, quantitation (in Table 2) that combines intrinsic reactivity with a later reactivation of the reaction center still yields a coherent picture of the reaction.
Figure 2.

panel A - reactions of C3 RNA with PheAMP at concentrations shown.
Figure 2.

panel B - Some reactions of mutated C3 RNAs with PheAMP
The active site
To test the idea that the highly conserved RNA substructure (eg, the U26/U13G14U15 junction in RNA C3) contained all reactive elements, we made five truncated molecules preserving this active junction (see Figure 3). All these small molecules were active; in fact, most were more active than directly selected RNAs (Table 2). These constructions and selected molecules, even without other data, suggest that no structural element outside the first two nucleotide pairs rightward of the loop, nor outside the first nucleotide pair leftward of the loop, is required for rapid acylation. Given that the first leftward pair is not conserved among selected RNAs, only the loop, the one-nucleotide overhang, and the rightward structure remain as candidates, though they may require support by non-conserved structural elements.
RNAs C1 and C2 had only 24 nucleotides, but C2 has stabilizing tetraloops to aid folding and demonstrated higher reactivity. Further stabilization in 26-nucleotide RNA C3 and C4 yielded the fastest aminoacyl transfer, ≥ 2 x that of RNA-106, and maximal yield. Another base pair in RNA C5 apparently decreased the product yield, so the small derived C3 RNA was chosen as the starting point and reference in further study.
Kinetic investigations of C3 RNA showed that divalent metal ions are nonessential for activity (Table 2). Because tested RNAs came directly from EDTA electrophoretic gel purification, it seems very unlikely that active RNAs scavenge cryptic divalent ions.
All investigated ribozymes were made by T7 transcription, and so possessed 5′-G triphosphate termini. Surprisingly, removal of this large, extremely polar group close to the point of reaction made no significant difference to folding or reactivity of C3 RNA with PheAMP (Table 2).
As noted above, the GC pair at the right of the active site (Figure 3) is mandated by the initial 5′ constant sequence. Yet, when it was changed to a CG pair in synthetic C3 RNA (5′-monophosphate) the RNA was fully active (Table 2). Therefore, the rightward helix boundary is apparently not crucial, because both the base pair and the triphosphate can be changed without major effect. In addition, selection of loops with 3, 4, 5 and 6 nucleotides (Table 1) suggests that spacing of the right loop-helix junction from the aminoacyl transfer as well as the precise junction structure is free to vary.
We have followed up on the simple behavior of C3 RNA by testing mutants including all singly-mutated sequences, transcribed from altered DNA templates and processed by HDV ribozyme, as usual. Pool sequences suggested that the 3′-overhanging nucleotide should be U. We substituted 3′-U in C3 with C, A and G, and in all cases the mutated RNA's reaction was too slow for complete kinetic characterization under standard conditions (Table 2).
Selected sequences also suggest (Table 1) that, while the 5'-U in the UGU loop was favored it is not essential, with the functional order being U>C>A>G among RNAs that differ at this position. Substitution of N in NGU in mutated loops of C3 RNA decreased reactivity in the order U>C>A~G (Table 2). Thus again, rate of self-aminoacylation in model RNAs is well-correlated with selected RNA abundance.
A detailed reaction mechanism
Given the likelihood that there were only three crucial nucleotides (3′ U and loop GU 3′) in this active site, we attempted to calculate a plausible active site conformation for rapid aminoacyl transfer. We used molecular mechanics based on the parmbsc0 forcefield 22, a refined version of the more commonly used AMBER forcefield 23. The parmbsc0 forcefield corrects a tendency for anomalous backbone torsions which appear after very long simulations (approaching 100 ns) using AMBER. Recent reports show 22 that the parmbsc0 forcefield then supports the longest (>200 ns) molecular dynamic trajectories done to date for DNA and RNA oligomers. We also adopted the Ponder group's PSS method of global minimum energy search 24 rather than the more usual molecular dynamics approach. This reflects our hypothesis that, in an efficient active site, reaction is likely to occur near the most stable structure rather than in an infrequent, transient dynamic state. We also thought that the PSS method might provide a quicker search of this conformational space than usual molecular dynamic simulations. To further minimize computation we used only one explicit solvation shell containing water and monovalent ions, supplemented by a Hawkins-Cramer-Truhlar bulk solvation model 25. Because C3 RNA does not require divalent ions (Table 2), it again offered an unusually simple start point for calculation.
To test this computational implementation, we followed a 7 base pair DNA A-helix (neutralized by 14 sodium atoms) which converted into the corresponding B-form, while for the same RNA helix an initial A-form helix persisted. This realistic conformational outcome suggests that the PSS method allows accurate sampling of conformation space, and that the parmbsc0 forcefield together with a mixed solvation model provides a realistic potential for solvated DNA and RNA.
To find conformations that potentially support transaminoacylation we conducted an exhaustive set of PSS searches of the C3 reaction site (the calculated part is shown in the colored square in Figure 3). We confined investigation to conformations near the reaction path by constraining one distance between the reactive atoms (3′ or 2′ OH of terminal U26) and each of the polar groups of U15, G14 or U13 to 2-3Å. Three of the resulting computed lowest-energy conformers provided enough space to accommodate PheAMP substrate.
Introduction of PheAMP resulted in spontaneous (no constraints) hydrogen bond formation between a phosphate oxygen of PheAMP and the ring NH of U15. We explored other possibilities by restricting the amino acid's carbonyl in the proximity of all polar groups on U15 and G14 (U13 was pointed away from the substrate; Figure 4A). However, once constraints were removed and these substrate assemblies were re-optimized, in many cases, the system spontaneously reverted to the originally observed hydrogen bond between phosphate oxygen of PheAMP and the ring NH of U15. To our delight, this phosphate-NH bond-containing active site was consistent with all known experimental data and suggested new experiments. We now describe this recurrent conformation.
Figure 4A.
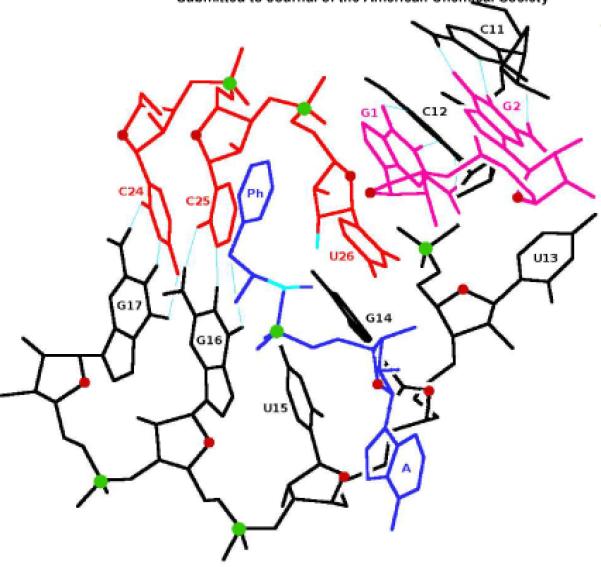
General view of the proposed C3 RNA reaction center. Color code: black - bottom strand, C11-G17; top strands: G1-G2 - magenta, C24-U26 - red; blue - (L)-5'-Phenylalanyl adenylate, (L)-PheAMP; dark red - oxygen atoms of ribose rings; green - phosphorus atoms; cyan - reactive carbon, oxygen and Watson-Crick hydrogen bonds. Adenine of (L)-PheAMP and the base of U13 are turned away from the reaction center, Acceptor U26 stacks against G1. Phenyl ring (Ph) of phenylalanine is close but perpendicular to U26; Ph is closer to the viewer than C25, so no Ph-C25 stacking interactions occur.
Phe-adenylate substrate binds to the UGU loop via five hydrogen bonds (Figure 4A, 4B):
N-H⋯O, from the ring NH of conserved loop U15 - to phosphate oxygen of PheAMP;
N-H⋯O, from NH3+ hydrogen of amino acid - to C4-carbonyl of conserved U15;
O-H⋯O, from 2′-OH of conserved terminal U26 ribose - to amino-acid carbonyl O,
N-H⋯O, from extrannular NH2 hydrogen of conserved G14- to amino acid carbonyl;
N-H⋯O, from the NH3+ hydrogen of amino acid - to phosphate oxygen of PheAMP.
Figure 4B.
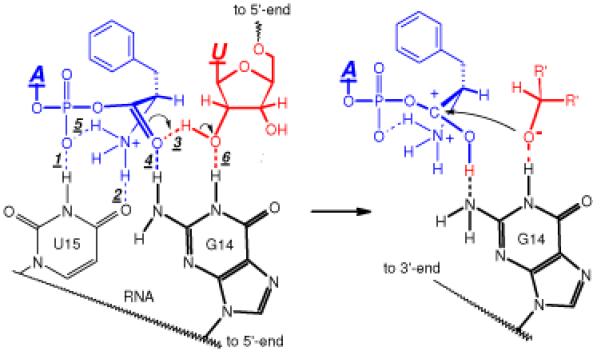
Schematic of the aminoacyl transfer reaction: proton migration followed by nucleophilic attack. Color code: black - bottom strand, U15-G14; red - top strand, U26; blue - PheAMP substrate. H-bond numbers (1-6; see text) are italicized and underlined.
The latter bond helps to orient substrate, and a sixth H-bond pulls the active site together:
-
(6)
N-H⋯O, from ring NH hydrogen of conserved loop G14 - to 2′-OH of conserved terminal U26 ribose: the bond brings the 2′-oxygen to be acylated into proximity of the amino acid carbonyl. The way these H-bonds cage and organize the reactants is further shown in Supplementary Figure 6.
The 3′-terminal U26 acceptor may contact the rightward site boundary at the first base pair (G1). But significantly, the least conserved loop nucleotide (U13), and A of PheAMP do not participate in the calculated bound state; instead pointing away from the reaction site (Figure 4A).
On the hypothetical reaction path, the 2′-OH hydrogen of U26 ribose migrates to the carbonyl of the amino acid (facilitated by H-bond #3). Migration both activates the carbonyl for the following nucleophilic attack and creates the attacking nucleophile, the 2′-oxyanion. The ring NH of conserved G14 should be essential for full reactivity (H-bond 6), stabilizing the negative charge on the nucleophilic 2′-oxygen after proton migration. The last reaction stage, ejection of the leaving AMP, is facilitated by H-bond (1).
This model explained what we knew of the reaction:
Ribozymes of the C3 RNA family are not reactive with CoA-Phe thioester because the phosphate of PheAMP is necessary for substrate binding via hydrogen bonds (1) and (5).
Divalent ions do not participate in the catalytic pathway, conformationally or chemically. They might have their observed minor effect by stabilizing the active fold of the ribozyme.
5′-Triphosphate is at some distance from the reaction site and does not interact with substrate (Figure 4A, 4B).
The substitution of G1-C12 pair with C1-G12 pair has little impact on reactivity, because it is needed only for correct folding and serves a passive role as a site boundary.
The 3'-terminal loop nucleotide must be U. We suggest that A26 or G26 would pair with U15, while C26 forms a Watson-Crick pair with G14. As a result, any nucleotide but U obstructs the binding of substrate to the UGN loop.
For proper folding of the catalytic site, N13 should not stack with G14 or N12. The decrease in reactivity U>C>A~G is consistent with the increase of N13 stacking interactions 26.
The rightward, paired, active site boundary only restricts the movement of U26. U26 can still reach its active conformation, as observed, if the boundary is moved or changed.
We further tested the following implications of the model. Overall, the properties of all single mutants of the active center seem consistent (Table 2) with the scheme (Figure 4A, 4B).
-
8
Nucleotide G14 should be required because its NH brings the reactive carbonyl and the ribose hydroxyl together. As predicted, mutation of G14 resulted in slowed reactions (Table 2).
-
9
We substituted U15 in C3 RNA by A, G and C (Table 2). While A15 and G15 mutants were extremely slow, probably because of pairing with U26, the C15 mutant reacted faster than the C26 mutant. In our proposed active site above, the major role of U15 is to provide hydrogen bonds (1) and (2). The pKa for the catalytic cytosine in the Genomic HDV ribozyme 27 reached 6.4 in conditions similar to ours. The tautomeric form of C offers the same hydrogen bonding possibilities as U and so it supports similar binding of the substrate. This may explain the observable, albeit lower, reactivity of the U26/UGC15 mutant.
-
10
We made two simultaneous substitutions in C3 RNA - A26 and C15, expecting that A26/C15 RNA should be more reactive than A26/U15. Despite introduction of a second departure from consensus, the double mutant was ≈ 10-fold more active (Table 2).
The model also suggested new properties, which were confirmed experimentally.
-
11
Adenine of PheAMP points away from the reaction center and has no discrete interactions with it, so the replacement of adenine should have little impact on the reaction. In fact, when synthetic PheUMP was used as substrate (Table 2) it was of the same order of reactivity as PheAMP.
-
12
The aromatic ring of phenylalanine interacts with U26, but is not involved in the reaction mechanism. Indeed, when PheAMP was replaced with synthetic MetAMP the reaction slowed only slightly (Table 2).
-
13
The reaction is directed to the 2′-OH of the ribose (Figure 4A). On removal of the 2'-OH it should be inhibited. Indeed, a mutant of the same sequence as C3, but having 2′-deoxyuridine instead of 3′-U26 showed no detectable reaction with PheAMP.
-
14
The model (Figure 4A-4B; Supplementary Figure 6) predicts that the stereochemistry of the aminoacyl residue is crucial, because the carbonyl of (D)-Phe is not close to U26 2′-OH. As predicted, aminoacyl transfer rate was at background levels for synthetic (D)-PheAMP substrate.
Discussion
It has been known for some time that synthesis of aminoacyl-RNA is an easy reaction for RNA having ≥ 29 residues 7, and therefore plausible for an early RNA-directed translation system. However, in this work simplified selection without arbitrary 3′-sequences yielded an unusually small self-aminoacylating RNA, ≤ 24 nucleotides. Only two of three central loop nucleotides proximal to the reactive 3′-U overhang take essential roles. The reaction tolerates different phosphorylated leaving groups and amino acids, though it is highly stereospecific for L-phenylalanine. Aminoacyl transfer requires neither RNA 5′-triphosphate nor divalent ions. This appears more flexible than the smallest previous RNA self-aminoacylators, which had 29 nucleotides and required adjacent 5′ triphosphate 28. Free choice of side chains on the phosphate leaving group and aminoacyl residue would make such a catalyst quite versatile; perhaps an adaptive quality for a primitive environment. For example, it would be predicted (Figure 4A-4B) that any nucleotide, or in fact, virtually any aminoacyl phosphate at all, might be utilized as an activated substrate by this active center.
Larger RNA active sites are expensive; with tenfold more RNA probably leading to isolation of an active site only 1.66 conserved nucleotides larger 29. Therefore, it is expeditious to ask how simple sites can be isolated. The observed structural freedom of C3 RNA, which only lightly constrains the leaving group and amino acid, in light of the calculated site structure, suggests a route by which this particular active site simplicity was attained. Essential RNA catalytic groups make interactions predicted to be tightly focused on reactive atoms, in all cases within 3 bond lengths of the point of reaction (Figure 4A, Supplemental Figure 6) and ignoring more distal, potentially substrate-specific atoms. Thus aminoacylation is accelerated without sidechain or leaving group specificity, requiring an RNA reaction apparatus of minimal size. Such RNA aminoacylation from adenylate appears much simpler and more accessible than via CoA activation using a thioester, strengthening the argument that a primordial amino acid for translation would be activated by phosphate, a phosphate ester or by a nucleotide. We wonder whether the removal of 3′-primer might lead to simplified, robust outcomes in other selections. Certainly, this seems likely when (as here) the 3′-terminus is a reactant, or is in the active center. In any case small, easily encountered, non-specific reaction modules like this one are likely participants in early molecular evolution.
Our minimal free energy reaction model for RNA-catalyzed aminoacylation agrees with experiment in so many ways that this appears unlikely to be entirely coincidental. These results therefore suggest that molecular mechanics-based free energy minimization might provide useful guidance in other RNA active sites.
Supplementary Material
Acknowledgement
Thanks to M. Illangasekare, I. Majerfeld for advice on biochemical techniques; Supported by NIH R01 GM48080 and NASA Astrobiology Institute NCC2-1052.
Footnotes
Supporting information available: Experimental methods, 6 supplementary Figures, 4 supplementary Tables, and 2 supplementary Schemes, plus a detailed explanation of non-Michaelian ribozyme jump kinetics. This information is available free of charge via the Internet at http://pubs.acs.org/. 3D information for reactive intermediates is available on request from Y. Novikov; yehor_novikov@yahoo.comm.
References
- 1.White HB., III J Mol Evol. 1976;7:101–104. doi: 10.1007/BF01732468. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert W. Nature. 1986;319:618. [Google Scholar]
- 3.Chen X, Li N, Ellington AD. Chem Biodivers. 2007;4(4):633–55. doi: 10.1002/cbdv.200790055. [DOI] [PubMed] [Google Scholar]
- 4.Saito H, Kourouklis D, Suga H. Embo J. 2001;20(7):1797–806. doi: 10.1093/emboj/20.7.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Illangasekare M, Sanchez G, Nickles T, Yarus M. Science. 1995;267(5198):643–7. doi: 10.1126/science.7530860. [DOI] [PubMed] [Google Scholar]
- 6.Illangasekare M, Yarus M. RNA. 1999;5(11):1482–9. doi: 10.1017/s1355838299991264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Illangasekare M, Yarus M. Proc Natl Acad Sci U S A. 1999;96(10):5470–5. doi: 10.1073/pnas.96.10.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee N, Bessho Y, Wei K, Szostak JW, Suga H. Nat Struct Biol. 2000;7(1):28–33. doi: 10.1038/71225. [DOI] [PubMed] [Google Scholar]
- 9.Li N, Huang F. Biochemistry. 2005;44(11):4582–90. doi: 10.1021/bi047576b. [DOI] [PubMed] [Google Scholar]
- 10.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 11.Robertson DL, Joyce GF. Nature. 1990;344:467–468. doi: 10.1038/344467a0. [DOI] [PubMed] [Google Scholar]
- 12.Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 13.Triana-Alonso FJ, Dabrowski M, Wadzack J, Nierhaus KH. J Biol Chem. 1995;270(11):6298–307. doi: 10.1074/jbc.270.11.6298. [DOI] [PubMed] [Google Scholar]
- 14.Schurer H, Lang K, Schuster J, Morl M. Nucleic Acids Res. 2002;30(12):e56. doi: 10.1093/nar/gnf055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keefe AD, Newton GL, Miller SL. Nature. 1995;373:683–685. doi: 10.1038/373683a0. [DOI] [PubMed] [Google Scholar]
- 16.Jadhav VR, Yarus M. Biochemistry. 2002;41(3):723–9. doi: 10.1021/bi011803h. [DOI] [PubMed] [Google Scholar]
- 17.Coleman TM, Huang F. Chem Biol. 2002;9(11):1227–36. doi: 10.1016/s1074-5521(02)00264-8. [DOI] [PubMed] [Google Scholar]
- 18.Putz J, Wientges J, Sissler M, Giege R, Florentz C, Schwienhorst A. Nucleic Acids Res. 1997;25(9):1862–3. doi: 10.1093/nar/25.9.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahlman RP, Dale T, Uhlenbeck OC. Mol Cell. 2004;16(5):799–805. doi: 10.1016/j.molcel.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 20.Knight R, Birmingham A, Yarus M. RNA. 2004;10(9):1323–36. doi: 10.1261/rna.5168504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theissen G, Richter A, Lukacs N. Anal Biochem. 1989;179(1):98–105. doi: 10.1016/0003-2697(89)90207-8. [DOI] [PubMed] [Google Scholar]
- 22.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA, Orozco M. Biophys J. 2007;92(11):3817–29. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. Journal of the American Chemical Society. 1995;117(19):5179–5197. [Google Scholar]
- 24.Pappu RV, Hart RK, Ponder JW. Journal of Physical Chemistry B. 1998;102(48):9725–9742. [Google Scholar]
- 25.Cramer CJ, Truhlar DG. Reviews in Computational Chemistry. 1995;6:1–72. [Google Scholar]
- 26.Saenger W. Principles of Nucleic Acid structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 27.Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. Journal of the American Chemical Society. 2007;129(43):13335–13342. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- 28.Illangasekare M, Kovalchuke O, Yarus M. J Mol Biol. 1997;274(4):519. doi: 10.1006/jmbi.1997.1414. [DOI] [PubMed] [Google Scholar]
- 29.Knight R, Yarus M. Rna. 2003;9(2):218–30. doi: 10.1261/rna.2138803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D'Ordine R, Paneth P, Anderson VE. Bioorganic Chemistry. 1995;23:169–181. [Google Scholar]
- 31.Landt O, Grunert HP, Hahn U. Gene. 1990;96(1):125–128. doi: 10.1016/0378-1119(90)90351-q. [DOI] [PubMed] [Google Scholar]
- 32.Horton RM, Cai ZL, Ho SN, Pease LR. Biotechniques. 1990;8(5):528–35. [PubMed] [Google Scholar]
- 33.Berg P. J Biol Chem. 1958;233(3):608–611. [PubMed] [Google Scholar]
- 34.Coleman TM, Li N, Huang FQ. Tetrahedron Letters. 2005;46(25):4307–4310. [Google Scholar]
- 35.Fersht A. Enzyme Structure and Mechanism. Freeman; New York: 1985. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.