

Abstract
OBJECTIVE
Smoking decreases insulin action and increases the risk of type 2 diabetes in humans. Mechanisms responsible for smoking-induced insulin resistance are unclear. We hypothesized smokers would have increased intramuscular triglyceride (IMTG) and diacylglycerol (DAG) concentration and decreased fractional synthesis rate (FSR) compared with nonsmokers.
RESEARCH DESIGN AND METHODS
Nonsmokers (n = 18, aged 20 ± 0.5 years, BMI 22 ± 0.4 kg/m2, body fat 20 ± 2%, 0 cigarettes per day) and smokers (n = 14, aged 21 ± 0.7 years, BMI 23 ± 0.4 kg/m2, body fat 20 ± 3%, 18 ± 0.7 cigarettes per day) were studied in a fasted condition after a standardized diet. [U-13C]palmitate was infused during 4 h of rest followed by a skeletal muscle biopsy and intravenous glucose tolerance test.
RESULTS
Smokers were less insulin sensitive (Si) compared with nonsmokers (Si 5.28 ± 0.5 nonsmokers vs. 3.74 ± 0.3 smokers 10−4 · μU−1 · ml−1, P = 0.03). There were no differences in IMTG or DAG concentration (IMTG 24.2 ± 3.4 nonsmokers vs. 27.2 ± 5.9 smokers μg/mg dry wt, DAG 0.34 ± 0.02 nonsmokers vs. 0.35 ± 0.02 smokers μg/mg dry wt) or IMTG FSR between groups (0.66 ± 0.1 nonsmokers vs. 0.55 ± 0.09 smokers %/hr). Intramuscular lipid composition was different, with increased percent saturation of IMTG (32.1 ± 1.2 nonsmokers vs. 35.2 ± 1.0 smokers %, P = 0.05) and DAG (52.8 ± 1.7 nonsmokers vs. 58.8 ± 2.2 smokers %, P = 0.04) in smokers. Smokers had significantly decreased peroxisome proliferator–activated receptor-γ (1.76 ± 0.1 nonsmokers vs. 1.42 ± 0.11 smokers arbitrary units [AU], P = 0.03) and increased monocyte chemotactic protein-1 (3.11 ± 0.41 nonsmokers vs. 4.83 ± 0.54 smokers AU, P = 0.02) mRNA expression compared with nonsmokers. We also found increased insulin receptor substrate-1 Ser636 phosphorylation in smokers compared with nonsmokers (0.73 ± 0.08 nonsmokers vs. 1.14 ± 0.09 smokers AU, P = 0.002).
CONCLUSIONS
These data suggest: 1) IMTG concentration and turnover are not related to alterations in insulin action in smokers compared to nonsmokers, 2) increased saturation of IMTG and DAG in skeletal muscle may be related to insulin action, and 3) basal inhibition of insulin receptor substrate-1 may decrease insulin action in smokers.
The latest data from the Centers for Disease Control and Prevention indicate 19.8% of the U.S. population smokes cigarettes (1). Cigarette smoking is an independent risk factor for the development of type 2 diabetes (2,3), likely because overwhelming evidence indicates smoking promotes insulin resistance (4–9). Nevertheless, the mechanistic link between smoking and insulin resistance is not clear. Understanding the mechanisms of how smoking promotes insulin resistance may provide drug targets for diabetes treatment or prevention in people for whom smoking cessation cannot be achieved.
Intramuscular triglyceride (IMTG) concentration may be an important marker of skeletal muscle insulin resistance as the content of IMTG is negatively associated with insulin-stimulated glucose disposal in healthy humans (10), individuals with type 1 diabetes (11) and type 2 diabetes (12), and offspring of individuals with type 2 diabetes (13). Others (14,15) speculated that in addition to total content, low rates of IMTG synthesis and degradation, rather than concentration, may more closely relate to insulin action. There are several mechanisms that may increase IMTG storage in smokers, including increased free fatty acid (FFA) appearance rate, concentration, and delivery to skeletal muscle (16,17) as well as decreased adipocyte but increased skeletal muscle lipoprotein lipase activity (16,18), with similar fat oxidation compared with nonsmokers (17). These data suggest smoking may increase trafficking of fat from adipose tissue to skeletal muscle, which may enhance IMTG storage and help explain insulin resistance in smokers compared with nonsmokers.
Although there have been many investigations on metabolic alterations with smoking, the relationship between smoking, insulin resistance, and intramuscular lipid metabolism has not been explored. Investigating IMTG metabolism in smokers compared with nonsmokers may allow a more thorough understanding of insulin action in smokers to better target therapies to prevent and treat type 2 diabetes. Insight may also be obtained for mechanisms promoting insulin resistance induced by second-hand smoke exposure (19). We performed this study to test the hypothesis that IMTG concentration would be greater and fractional synthesis rate lower in cigarette smokers compared with nonsmokers. We predicted decreased insulin action in smokers would be related to decreased IMTG synthesis compared with control subjects.
RESEARCH DESIGN AND METHODS
Eighteen healthy sedentary nonsmokers (11 men, 7 women) and 14 smokers (12 men, 2 women) were recruited for this study (Table 1). Subjects gave written informed consent and were excluded if they had a BMI < 20 kg/m2 or >25 kg/m2; diabetes; hyperlipidemia; liver, kidney, thyroid or lung disease; or were taking medications that can affect glucose or lipid metabolism. Smokers had been smoking cigarettes for an average of 3.3 ± 0.7 years. All subjects were sedentary and engaged in moderate to vigorous exercise <3 h/week. Subjects were weight stable in the 6 months before the study. This study was approved by the Colorado Multiple Institution Review Board at the University of Colorado Denver.
TABLE 1.
Anthropometric and metabolic characteristics of the study population
| Nonsmokers | Smokers | P | |
|---|---|---|---|
| N (men/women) | 18 (11/7) | 14 (12/2) | |
| Age (years) | 20.1 ± 0.4 | 21.2 ± 0.7 | 0.17 |
| BMI (kg/m2) | 22.6 ± 0.4 | 22.9 ± 0.4 | 0.40 |
| Body fat (%) | 20.2 ± 2.1 | 19.7 ± 2.2 | 0.86 |
| Fasting glucose (mg/dl) | 82.0 ± 1.8 | 85.2 ± 0.9 | 0.16 |
| FFAs (μmol/l) | 738 ± 66 | 716 ± 45 | 0.79 |
| Lactate (mmol/l) | 0.75 ± 0.1 | 0.84 ± 0.1 | 0.35 |
| Insulin (μU/ml) | 4.4 ± 0.4 | 5.5 ± 0.4 | 0.06 |
| Glucagon (pg/ml) | 63 ± 4 | 53 ± 4 | 0.10 |
| Triglycerides (mg/dl) | 67 ± 9 | 117 ± 13§ | 0.001 |
| Norepinephrine (pg/ml) | 220 ± 12 | 280 ± 59 | 0.39 |
| Epinephrine (pg/ml) | 27 ± 2 | 33 ± 5.1 | 0.46 |
| Adiponectin (pg/ml) | 12.5 ± 1.4 | 11.9 ± 0.9 | 0.97 |
| TNF-α (pg/ml) | 1.4 ± 0.1 | 1.6 ± 0.2 | 0.38 |
| IL-6 (pg/ml) | 1.67 ± 0.3 | 4.61 ± 1.1 | 0.007 |
| Cigarettes per day (n) | 0 | 18.0 ± 0.8§ | <0.001 |
| Breath CO (ppm) | 3.1 ± 0.5 | 27.2 ± 2.4§ | <0.001 |
| Urinary cotinine (ng/ml) | 3.8 ± 1.2 | 342.6 ± 51§ | <0.001 |
| Glucose Ra (mg · kg−1 · min−1) | 1.75 ± 0.08 | 1.75 ± 0.08 | 0.98 |
Values are means ± SE.
§ = significantly different between nonsmokers and smokers, P < 0.05. Ra, rate of appearance.
Preliminary testing.
Subjects reported to the General Clinical Research Center (GCRC) for screening procedures after a 12-h overnight fast, where they were given a health and physical exam followed by a fasting blood draw. Body composition was determined using dual-energy X-ray absorptiometry (DEXA) analysis (Lunar DPX-IQ; Lunar, Madison, WI).
Diet and exercise control.
All subjects were given a prescribed diet for 3 days before admission to the GCRC. Daily caloric requirement was estimated from the DEXA measurement of fat-free mass using the equation 1.4 × [372 + (23.9 × fat-free mass)] and analysis of dietary records. Composition of this diet was 55% carbohydrate, 30% fat, and 15% protein. The fat content of the diet was controlled with the composition of saturated, monounsaturated, and polyunsaturated fat in a 1:1:1 ratio. Subjects were asked to refrain from planned physical activity for 48 h before the tracer infusion study.
Tracer infusion study.
Subjects arrived to the GCRC after an overnight fast at 7 a.m., when an antecubital vein in one arm was cannulated for isotope infusion and a retrograde dorsal hand vein in the contralateral side was catheterized for blood sampling via the heated hand technique. At 7:30 a.m., blood and breath samples were taken for background isotope enrichment. Starting at 8 a.m., a primed (4.5 mg/kg) constant (0.03 mg · kg−1 · min−1) infusion of [6,6-2H2]glucose and a continuous infusion of [U-13C] palmitate (Isotec, Miamisburg, OH) (0.0174 μmol · kg−1 · min−1) were initiated and continued for 4 h and throughout the muscle biopsy procedure. Subjects remained fasting during the tracer infusion. During the 4-h infusion, only smokers smoked one cigarette (Camel Lights; R.J. Reynolds, Winston-Salem, NC) every 30 min until the start of the final blood sample, for a total of eight cigarettes. No further smoking occurred throughout the study. Blood sampling was performed during minutes 210, 220, 230, and 240 of infusion for metabolite and hormone concentrations. A percutaneous needle biopsy was performed after 240 min of isotope infusion for determination of IMTG and diacylglycerol (DAG) concentration, composition, and turnover (20). Muscle biopsies were taken from midway between the greater trochanter of the femur and patella. The anatomic location and depth of the biopsy was as similar as possible among subjects to minimize variance in muscle fiber composition that varies with depth and length in the vastus lateralis (21). Muscle was immediately flash frozen in liquid nitrogen and stored at −80°C until dissection and analysis.
After the tracer infusion, insulin sensitivity was determined via an insulin-modified frequently sampled intravenous glucose tolerance test using standard methods (22). Briefly, after baseline samples, intravenous glucose (0.3 g/kg) was infused over 1 min, followed by insulin at 0.03 units/kg 20 min after glucose administration. Blood samples were then frequently sampled over 3 h and whole-body insulin sensitivity (Si) calculated using the Bergman minimal model (22).
Metabolite and hormone analyses.
Standard enzymatic assays were used to measure glucose (Olympus AU400e Chemistry analyzer, Olympus America, Center Valley, PA), lactate (Sigma Kit #826, St. Louis, MO), and FFA (NEFA Kit, Wako, TX). Insulin, glucagon, and adiponectin were measured using an RIA (Diagnostic Systems Laboratories, Webster, TX) and catecholamines using high-performance liquid chromatography. Plasma cotinine (Calbiotech, Spring Valley, CA) as well as interleukin (IL)-6 and tumor necrosis factor (TNF)-α (R&D Systems HS600B and HSTA00C, respectively, Minneapolis, MN) were measured using an ELISA kit. Breath CO was measured using an EC50-Micro Smokerlyzer (Bedfont Scientific, Medford, NJ).
Muscle lipid analysis.
Skeletal muscle samples were dissected free of extramuscular fat on ice as described by Guo et al. (23). Then, a sample of muscle (∼70 mg) was lyophilized, weighed, added to 1 ml iced MeOH along with internal standards of tripentadecanoic acid and dipentadencanoic acid, and homogenized for 1 min on ice (Omni TH; Omni International, Marietta, GA). Total lipids were then extracted as previously described (24). Samples were shaken on a rotational mixer for 1.5 h at 4°C, then spun at 3,000 rcf for 15′ to separate phases. The nonpolar bottom layer was dried down under N2 at 40°C, resuspended in chloroform, and added to aminopropyl solid-phase extraction columns (Supelclean LC-NH2, 3 ml, Supelco Analytical). FFAs, phospholipids, IMTGs, and DAGs were isolated using solid-phase extraction based on the methods originally described by Kaluzny et al. (25). The FFA fraction was methylated by adding 0.5 ml 2% sulfuric acid, capping, and heating at 100°C for 1.5 h. Phospholipid, IMTG, and DAG fractions were converted to fatty acid methyl esters by transmethylation using sodium methoxide. Stable isotope ratios of 13C in fatty acid methyl esters were measured at the Stable Isotope Laboratory at the University of California at Davis using a GC-combustion isotope ratio mass spectrometer (GC/C-IRMS) system composed of a Trace GC Ultra gas chromatograph (Thermo Electron, Milan, Italy) with a SGE BPX70 column (30 m × 0.25 mm internal diameter, 0.25 micron film thickness) coupled to a Delta Plus Advantage isotope ratio mass spectrometer through a GC/C-III interface (Thermo Electron, Bremen, Germany). Enrichment was calculated based on a standard curve of known enrichments and corrected for variations in abundance (26). Concentration and composition analysis was performed on an HP 6890 GC with a 30-m DB-23 capillary column connected to an HP 5973 MS. Peak identities were determined by retention time and mass spectra compared with standards of known composition.
Western blotting.
Frozen skeletal muscle samples were weighed and homogenized on ice using a Kontes glass homogenizer (Kimble/Kontes, Vineland, NJ) in a previously described buffer (27) containing protease (Roche Applied Science, Indianapolis, IN) and phosphatase inhibitors (Sigma, St. Louis, MO). Samples were agitated at 4°C for 2 h, then spun at 16,000 g for 15′ to pellet insoluble protein. Supernatant was saved and used to determine protein concentration (Calbiochem, San Diego, CA).
Forty micrograms of sample protein and internal standard were run on an SDS-PAGE 8% Bis-Tris gel (Invitrogen, Carlsbad, CA), transferred to a polyvinylidene fluoride membrane, and blocked with 5% BSA for 1 h at room temperature. Primary antibody incubations were performed in 5% BSA overnight at 4°C, and a horseradish peroxidase–conjugated secondary antibody was incubated for 1 h at room temperature. Enhanced chemiluminescence was used to visualize protein bands of interest. Intensity of protein bands was captured using an AlphaImager 3300 and quantified using FluorChem software (Alpha Innotech, San Leandro, CA). Protein bands were normalized to β-actin as a loading control and then normalized to an internal standard that was run on each gel. Rabbit anti–4-hydroxynonenal antibody was a generous gift from Dr. Dennis Peterson. Other antibodies were purchased from cell signaling (Danvers, MA) and Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies were from Bio-Rad (Bio-Rad, Hercules, CA).
RT-PCR.
Total RNA was extracted from homogenized muscle biopsies using the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA was analyzed and quantified using the Experion System (Bio-Rad). Reverse transcription was performed using 45 ng total RNA with iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was performed using primer sets for genes of interest or two reference genes (all spanned exon-exon boundaries) and ABsolute Blue QPCR SYBR Green Fluorescein Mix (Thermo Fisher Scientific, Waltham, MA) following manufacturer's protocol. Reactions were run in duplicate on an iQ5 Real-Time PCR Detection System (Bio-Rad) along with a no-template control per gene. We performed RT-PCR to determine differences in gene expression that may explain the insulin resistant phenotype of smokers. We determined differences in the expression of genes for desaturation of long-chain acyl-CoA (stearoyl CoA desaturase 1 [SCD1]), IMTG synthesis (diacylglycerol acyltransferase 1 [DGAT1]), IMTG degradation (adipose triglyceride lipase [ATGL]), a transcription factor regulating genes controlling lipogenesis (sterol regulatory element binding protein 1c [SREBP1c]), a nuclear transcription factor influencing fatty acid oxidation (peroxisome proliferator–activated receptor α [PPAR-α]) and synthesis (PPAR-γ), a nuclear binding partner to PPAR (retinoid X receptor γ [RXR-γ]), a transcription factor regulating the PPAR/RXR dimer (forkhead box class 01 [FOXO1]), and markers of macrophage infiltration (monocyte chemoattractant protein 1 [MCP-1]) and local inflammation (IL-6). RNA expression data were normalized to levels of ribosomal protein L13a (RPL13A) and ubiquitin C (UBC) using the comparative threshold cycle method. The following forward primers were used in this analysis:
SCD1 (AACTGGTGATGTTCCAGAGGAGGT)
FOXO1 (TCCCACACAGTGTCAAGACAACGA)
DGAT1 (ATGTCTTTGCTGTGGCTGCATTCC)
ATGL (ATCCCACTTCAACTCCAAGGACGA)
SREBP-1c (GGAGCCATGGATTGCACTTT)
PPAR-α (TGCACTGGAACTGGATGACAGTGA)
PPAR-γ (TCATCCTCTCAGGAAAGGCCAGTA)
RXR-γ (AGTCTCCCAGTGGAGCATTGAACA)
IL-6 (AAATTCGGTACATCCTCGACGGCA)
RPL13A (CCTGGAGGAGAAGAGGAAAGAGA)
UBC (ATTTGGGTCGCGGTTCTTG)
Plasma palmitate and glucose isotope analysis.
Plasma glucose derivatization and analysis was determined as previously described (27). Methylation and extraction of plasma palmitate was performed as previously described (28). Samples were run on an HP 6890 GC with a 30-m DB-23 capillary column connected to an HP 5973 MS. Enrichments were calculated based on a standard curve of known enrichments and corrected for variations in abundance (26).
Isotope ratio mass spectrometry.
Two milliliters of breath CO2 was transferred into a 20-ml exetainer for measurement of 13CO2/12CO2 with continuous flow isotope ratio mass spectrometry (IRMS) (Delta V, Thermo Electron). Each sample was injected (1.2 μl per injection) in duplicate for isotope ratio analyses, with an average standard deviation for all injections of 0.0001 atom percent.
Calculations.
IMTG fractional synthesis rate was calculated as previously described (29). The average enrichment of the skeletal muscle FFA pool during the 4-h infusion was used to represent the precursor pool from which IMTG was synthesized.
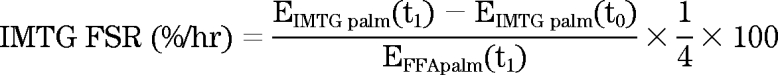 |
DAG fractional synthesis rates were calculated in a similar way.
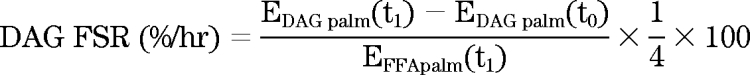 |
Where EIMTG palm(t1) is the enrichment of palmitate in the IMTG pool after 4 h of infusion, EIMTG palm(t0) is the enrichment of background palmitate in the IMTG pool, EDAG palm(t1) is the enrichment of palmitate in the muscle DAG pool after 4 h of infusion, and EDAG palm(t0) is the enrichment of background palmitate in the muscle DAG pool. EFFApalm(t1) is the enrichment of skeletal muscle palmitate in the FFA pool. The enrichment of stearate in IMTG and DAG during the 4-h muscle biopsy was used to represent the background enrichment of palmitate before isotope infusion without having to take a background biopsy as previously described (29). Skeletal muscle FFA was assumed to be the precursor pool for IMTG and DAG synthesis (29).
Where FFA represents concentration of individual FFA species in IMTG and DAG after transmethylation.
Palmitate and glucose rate of disappearance and palmitate rate of oxidation were calculated using steady-state kinetics and a whole-body estimate of carbon label retention as previously described (30). Palmitate incorporation rate into IMTG was calculated as the product of IMTG fractional synthesis rate (FSR) and palmitate pool size in IMTG. Fat-free mass was used to extrapolate skeletal muscle palmitate IMTG storage to the whole body.
Statistics.
Data are presented as means ± SE. Differences in normally distributed data between smokers and nonsmokers were analyzed using a one-way ANOVA (SPSS, Chicago, IL). Non-normally distributed data were log transformed before analysis using a one-way ANOVA. P values were corrected for multiple comparisons using the Bonferroni method. Differences between IMTG and DAG fractional synthesis rates within individuals were determined using a paired t test. An α level of 0.05 was used throughout.
RESULTS
Subject characteristics and demographic information are shown in Table 1. All subjects were college-aged men and women with similar BMI, percent body fat, and blood metabolites. Smokers reported smoking almost one pack of cigarettes per day. There was an approximate doubling of fasting blood triglyceride concentration in smokers compared with nonsmokers, as well as significantly greater IL-6, urinary cotinine, and breath CO concentrations.
Insulin sensitivity was significantly lower in smokers compared with nonsmokers (Fig. 1, P = 0.03). Contrary to our hypothesis, we did not find a difference in resting concentration or FSR of IMTG in smokers compared with nonsmokers (Fig. 2A and B). However, composition of IMTG differed between groups, with significantly greater proportion of saturated fatty acid species in smokers compared with nonsmokers (Fig. 2C, P = 0.02). There was also a significant inverse relationship between IMTG content and FSR (Fig. 2D).
FIG. 1.

Insulin sensitivity measured using the Bergman minimal model in nonsmokers (□) and smokers (■). Values are means ± SE. § = significantly different than nonsmokers, P < 0.05.
FIG. 2.

Intramuscular triglyceride concentration (A), fractional synthesis rate (B), saturation (C) in nonsmokers (□) and smokers (■). Values are means ± SE. § = significantly different than nonsmokers, P < 0.05. The significant relationship between IMTG concentration and turnover in all subjects is shown in panel (D).
Skeletal muscle DAG concentration was also not different between groups, suggesting alterations in DAG-stimulated protein kinase C activation cannot explain the insulin resistance in smokers (Fig. 3A). The FSR of DAG was also not different between groups (Fig. 3B). The FSR of DAG was significantly greater than IMTG in both groups (P < 0.001). Similar to IMTG, saturation of DAG was significantly greater in smokers compared with nonsmokers (Fig. 3C, P = 0.03).
FIG. 3.

Intramuscular DAG concentration (A), fractional synthesis rate (B), and saturation (C) in nonsmokers (□) and smokers (■). D: Skeletal muscle DAG composition in nonsmokers and smokers. Values are means ± SE. § = significantly different than nonsmokers, P < 0.05.
We found significantly lower expression of PPAR-γ and significantly higher expression of MCP-1 in smokers compared with nonsmokers (Fig. 4, P < 0.05). There were trends (P < 0.10) toward higher expression of FOXO1 and lower DGAT1 and RXR-γ in smokers compared with nonsmokers.
FIG. 4.

RT-PCR summary of skeletal muscle biopsy data comparing nonsmokers (□) with smokers (■). Values are means ± SE. § = significantly different than nonsmokers, P < 0.05; † = different than nonsmokers, P < 0.10.
Table 2 shows a summary of Western blot data, which was not significantly different between groups, including total IRS-1 content. The only significant difference was significantly greater content of insulin receptor substrate (IRS)-1 Ser636 phosphorylation, which is inhibitory to insulin signaling in smokers compared with nonsmokers (Fig. 5, P = 0.002). This difference was significant even after correcting for multiple comparisons using Bonferroni.
TABLE 2.
Summary of Western blot data from resting skeletal muscle biopsies from nonsmokers and smokers
| Nonsmokers | Smokers | P | |
|---|---|---|---|
| GRP78 | 0.93 ± 0.12 | 0.85 ± 0.10 | 0.65 |
| ADRP | 0.74 ± 0.08 | 0.76 ± 0.07 | 0.84 |
| MAP4K4 | 1.43 ± 0.20 | 1.84 ± 0.30 | 0.18 |
| 4-HNE | 0.85 ± 0.13 | 0.78 ± 0.12 | 0.87 |
| SDH | 1.23 ± 0.08 | 1.16 ± 0.08 | 0.52 |
| PGC1α | 0.60 ± 0.05 | 0.67 ± 0.06 | 0.36 |
| TLR2 | 0.98 ± 0.11 | 1.3 ± 0.16 | 0.12 |
| SOCS1 | 0.76 ± 0.05 | 0.74 ± 0.04 | 0.48 |
| PKCζ | 0.74 ± 0.13 | 1.02 ± 0.33 | 0.78 |
| PKCθ | 0.42 ± 0.04 | 0.52 ± 0.05 | 0.15 |
| Phospho-p44 MAPK/total | 0.51 ± 0.07 | 0.38 ± 0.08 | 0.15 |
| Phospho-ser307/IRS1 total | 0.67 ± 0.16 | 0.81 ± 0.17 | 0.54 |
| Phospho-AMPK/total | 1.04 ± 0.23 | 0.86 ± 0.18 | 0.56 |
| Phospho IKKalpha/total | 0.84 ± 0.15 | 0.77 ± 0.22 | 0.85 |
| Phospho-JNK/total | 0.63 ± 0.05 | 0.68 ± 0.06 | 0.50 |
| Phospho-p70S6k/total | 1.10 ± 0.34 | 0.80 ± 0.10 | 0.96 |
Values are means of arbitrary units ± SE. GRP78, glucose-regulated protein 78; ADRP, adipose differentiation–related protein; MAP4K4, mitogen-activated protein kinase kinase kinase kinase 4; 4-HNE, 4-hydroxynonenal; SDH, succinate dehydrogenase; PGC1a, proliferator-activated receptor γ coactivator-1a; TLR2, toll-like receptor 2; SOCS1, suppressor of cytokine signaling 1; AMPK, AMP-activated protein kinase; IKK, IκB kinase; JNK, c-Jun N-terminal kinase.
FIG. 5.

Ser636 phosphorylation of IRS-1 in nonsmokers (□) and smokers (■). There was not a significant difference in total IRS-1 content between groups. Values are means ± SE. Nonsmokers = 0.73 ± 0.08, smokers = 1.14 ± 0.9, AU. § = significantly different than nonsmokers, P < 0.05.
Whole-body fat oxidation was not significantly different between groups (control subjects = 1.4 ± 0.1 μmol · kg−1 · min−1; smokers = 1.3 ± 0.1 μmol · kg−1 · min−1). Palmitate turnover was significantly higher in smokers compared with nonsmokers (Fig. 6A, P < 0.001). Breath 13CO2 enrichment was significantly greater in smokers compared with nonsmokers (smokers = 1.117 ± 0.001 Atom%, control subjects = 1.112 ± 0.001 Atom%; P = 0.001). The rate of oxidation of plasma palmitate was also significantly higher in smokers compared with nonsmokers (Fig. 6B, P = 0.01). However, the rate of palmitate incorporation into IMTG was not significantly different between groups (Fig. 6C).
FIG. 6.

Whole-body palmitate rate of appearance (A), oxidation (B), and incorporation (C) into IMTG in nonsmokers (□) and smokers (■). Values are means ± SE. § = significantly different than nonsmokers, P < 0.05.
DISCUSSION
Many studies have demonstrated a detrimental effect of smoking on insulin action. Data from epidemiologic (7–9) as well as cross-sectional studies report decreased insulin sensitivity in middle-aged smokers (4–6). We are the first to report chronic cigarette smoking promotes insulin resistance in young, otherwise healthy college-aged men and women. Major findings from this study suggest saturation of intramuscular lipids, independent of concentration or turnover, may be related to insulin action. Further, smoking resulted in basal inhibition of insulin signaling. The induction of such metabolic defects at a young age will surely result in profound and premature untoward health effects.
In addition to lower insulin action, smokers have greater delivery of plasma FFA (16,17) and lipoproteins (16,18) to skeletal muscle. Therefore, we hypothesized smoking may promote IMTG and DAG accumulation. However, we found no differences in IMTG or DAG concentration between groups. These data add to other studies finding a dissociation between IMTG concentration and insulin action (31,32) and support the notion that IMTG may only be a marker of insulin resistance rather than a cause. Similar DAG concentrations were surprising considering data linking DAG accumulation with insulin resistance (33). Supporting a lack of DAG-induced insulin resistance were similar PCKζ and θ content between groups. However, these data are from whole-cell lysates, and it is possible that differences in cytosolic versus sarcolemmal localization went unnoticed. Together, our data suggest increased skeletal muscle IMTG and DAG concentrations are not related to insulin resistance in college-aged chronic smokers.
We hypothesized IMTG and DAG FSR would be positively related to insulin sensitivity (14,15). Increased synthesis rates of IMTG may enhance FFA clearance and decrease ceramide and long-chain acyl-CoA synthesis, which may increase insulin action (34,35). In support of this, recent evidence indicates increasing IMTG synthesis in rodents and humans protects against fat-induced insulin resistance (31,32). In the current study, however, we found no differences in IMTG and DAG FSR between smokers and nonsmokers, suggesting rates of IMTG and DAG synthesis may not be related to insulin sensitivity in this population. Further, we found no change in whole-body glucose kinetics between groups similar to some (17) but not all previous studies (36). Smokers had increased plasma palmitate turnover as has been shown before in older smokers (17). Our data on breath V13CO2 and 13C palmitate incorporation into IMTG suggests that the increased plasma palmitate disposal in smokers is oxidized and not stored as IMTG. These data suggest decreased mitochondrial FFA oxidation and mitochondrial dysfunction does not play a major role in smoking-induced insulin resistance.
Interestingly, IMTG and DAG were significantly more saturated in smokers compared with nonsmokers. Initially, we thought this could be explained by increased habitual consumption of saturated fat in smokers. However, there were no differences in overall phospholipid composition, which has been used as a surrogate measure of dietary lipid intake, between groups (37). Less muscle lipid desaturation in smokers is unlikely, as the expression and protein content of SCD1, which converts saturated palmitoyl-CoA and stearoyl-CoA to monounsaturated palmitoleoyl-CoA and oleoyl-CoA, respectively, was not different between groups. Although indirect, a common surrogate for SCD1 activity are ratios of 16:1 to 16:0 and 18:1 to 18:0 (38). None of these ratios were significantly different for IMTG and DAG pools between smokers and nonsmokers. Therefore, it is unlikely that changes in SCD1 content or activity explain alterations in muscle lipid saturation, but the exact mechanism by which smoking changes neutral lipid storage is not known. The importance of saturated lipids on tissue insulin action was highlighted by a study that developed a transgenic Elovl6 (elongation of long-chain fatty acids family member 6) knockout mouse (39). They found no change in hepatic triglyceride content but a significant change in triglyceride composition and increased hepatic insulin action. We extend these data to humans and suggest IMTG and DAG composition may influence insulin sensitivity in skeletal muscle.
We analyzed many potential mediators of intracellular insulin resistance to explain why smokers were insulin resistant. There was no indication of plasma inflammation, lipid toxicity, lipid peroxidation, or endoplasmic reticulum stress. The only measure significantly different between groups was greater IRS-1 Ser636 phosphorylation in smokers compared with nonsmokers. This difference may explain decreased insulin action in smokers because of basal inhibition of insulin signaling.
There are several known mechanisms that lead to phosphorylation of Ser636 on IRS-1. Increased mammalian target of rapamycin (mTOR) and/or p44/42 mitogen-activated protein kinase (MAPK, a.k.a. ERK1/2) activity promotes insulin resistance by phosphorylating this specific site on IRS-1. We measured the phosphorylation state of the downstream kinase for mTOR/p70s6k and found no differences between groups. Further, we measured ERK1/2 phosphorylation and found no differences between groups. Therefore, these data imply chronic signaling through mTOR or ERK1/2 cannot explain increased IRS-1 Ser636 phosphorylation in smokers compared with nonsmokers. However, our data are consistent with transitory nicotine-stimulated ERK1/2 activity that could result in basal IRS-1 Ser636 phosphorylation.
The effect of smoking to decrease insulin action is likely because of nicotine, which promotes insulin resistance following direct infusion in humans (40). However, the mechanism behind smoking or nicotine-induced insulin resistance is not known. Recent evidence indicates nicotine increases ERK1/2 phosphorylation in a wide variety of cells (41–43). Further, the increase in ERK1/2 phosphorylation is transient, starting after 2 min of nicotine exposure, peaking after ∼10 min, and decreasing to basal values after 15–30 min of continued exposure (41–43). Ser636 phosphorylation of IRS-1 induced by ERK1/2 activity has been reported in primary human muscle cell cultures from individuals with type 2 diabetes (44) and following TNFα-induced insulin resistance in 3T3L-1 adipocytes (45). Interestingly, ERK1/2 phosphorylation can decrease to basal levels before measuring increased serine phosphorylation of IRS-1 in 3T3L1 adipocytes (45). The current data are consistent with nicotine exposure during tobacco smoking promoting insulin resistance via Ser636 phosphorylation of IRS-1, induced via transient ERK1/2 stimulation.
Skeletal muscle RT-PCR analysis supports other mechanisms may also be working to promote insulin resistance in smokers. Expression of MCP-1 was increased in skeletal muscle from smokers compared with nonsmokers. MCP-1 is a chemokine produced by adipocytes, vascular cells, and skeletal muscle, which promotes macrophage entry into tissues and itself may promote insulin resistance (46). These data suggest smoking may promote macrophage infiltration in skeletal muscle, possibly promoted by smoking-induced endothelial damage (47), which may result in local cytokine production and insulin resistance (48). PPAR-γ expression was also decreased in smokers compared with nonsmokers. We found a trend for decreased RXR-γ expression, which dimerizes with PPAR-γ in the nucleus, and a nonsignificant increase in FOXO1 expression in smokers, which negatively regulates PPAR-γ/RXR promoter activity (49). Muscle-specific knockout of PPAR-γ results in an insulin-resistant phenotype (50). Therefore, these changes point to alterations in downstream targets of PPAR-γ as potential mechanisms in smoking-induced insulin resistance. Abbasi et al. (51) treated insulin-resistant smokers for 12 weeks with pioglitazone and found increased insulin sensitivity and decreased plasma triglyceride concentrations. These data imply increased PPAR-γ stimulation may reverse part of the insulin resistance in smokers and suggest decreased muscle PPAR-γ may be important in insulin resistance in smokers.
There are several limitations to this study. The sample size studied was small, which may have limited our ability to measure small differences between groups. We chose subjects who were sedentary and engaged in planned physical activity less than 3 h/week. Even though succinate dehydrogenase content was similar between groups, it is possible that differences in physical activity may have influenced our results.
In conclusion, chronic cigarette smokers were less insulin sensitive compared with control subjects that did not smoke. There were no differences in IMTG or DAG content or synthesis rates, but smokers had increased saturation of skeletal muscle lipids. Insulin resistance in smokers may be because of increased basal inhibition of insulin signaling via Ser636 phosphorylation of IRS-1.
Acknowledgments
This work was partially supported by the National Institutes of Health General Clinical Research Center Grant RR-00036, National Institute of Diabetes and Digestive and Kidney Diseases Grants to L.P. (DK-064811), R.H.E. (DK-26356), and B.C.B. (DK-059739), and the Colorado Tobacco Research Program grant awarded to B.C.B. (3K-027).
No potential conflicts of interest relevant to this article were reported.
We thank Dr. Dennis Peterson at the University of Colorado Health Sciences Center for the generous donation of the 4-hydroxynonenal antibody.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Centers for Disease Control. Cigarette smoking among Adults—United States, 2007. MMWR 2008;57:1221–1226 [PubMed] [Google Scholar]
- 2.Feskens EJ, Kromhout D: Cardiovascular risk factors and the 25-year incidence of diabetes mellitus in middle-aged men. The Zutphen Study. Am J Epidemiol 1989;130:1101–1108 [DOI] [PubMed] [Google Scholar]
- 3.Perry IJ, Wannamethee SG, Walker MK, Thomson AG, Whincup PH, Shaper AG: Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men. Br Med J 1995;310:560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attvall S, Fowelin J, Lager I, Von Schenck H, Smith U: Smoking induces insulin resistance: a potential link with the insulin resistance syndrome. J Intern Med 1993;233:327–332 [DOI] [PubMed] [Google Scholar]
- 5.Frati AC, Iniestra F, Ariza CR: Acute effect of cigarette smoking on glucose tolerance and other cardiovascular risk factors. Diabetes Care 1996;19:112–118 [DOI] [PubMed] [Google Scholar]
- 6.Eliasson B, Attvall S, Taskinen MR, Smith U: The insulin resistance syndrome in smokers is related to smoking habits. Arterioscler Thromb 1994;14:1946–1950 [DOI] [PubMed] [Google Scholar]
- 7.Manson JE, Ajani UA, Liu S, Nathan DM, Hennekens CH: A prospective study of cigarette smoking and the incidence of diabetes mellitus among US male physicians. Am J Med 2000;109:538–542 [DOI] [PubMed] [Google Scholar]
- 8.Persson PG, Carlsson S, Svanstrom L, Ostenson CG, Efendic S, Grill V: Cigarette smoking, oral moist snuff use and glucose intolerance. J Intern Med 2000;248:103–110 [DOI] [PubMed] [Google Scholar]
- 9.Uchimoto S, Tsumura K, Hayashi T, Suematsu C, Endo G, Fujii S, Okada K: Impact of cigarette smoking on the incidence of type 2 diabetes mellitus in middle-aged Japanese men: the Osaka Health Survey. Diabet Med 1999;16:951–955 [DOI] [PubMed] [Google Scholar]
- 10.Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB, Storlien LH: Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 1997;46:983–988 [DOI] [PubMed] [Google Scholar]
- 11.Ebeling P, Essen-Gustavsson B, Tuominen JA, Koivisto VA: Intramuscular triglyceride content is increased in IDDM. Diabetologia 1998;41:111–115 [DOI] [PubMed] [Google Scholar]
- 12.Goodpaster BH, Theriault R, Watkins SC, Kelley DE: Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism 2000;49:467–472 [DOI] [PubMed] [Google Scholar]
- 13.Perseghin G, Scifo P, De Cobelli F, Pagliato E, Battezzati A, Arcelloni C, Vanzulli A, Testolin G, Pozza G, Del Maschio A, Luzi L: Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H–13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999;48:1600–1606 [DOI] [PubMed] [Google Scholar]
- 14.Goodpaster BH, He J, Watkins S, Kelley DE: Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 2001;86:5755–5761 [DOI] [PubMed] [Google Scholar]
- 15.Russell AP: Lipotoxicity: the obese and endurance-trained paradox. Int J Obes Relat Metab Disord 2004;28(Suppl.):S66–S71 [DOI] [PubMed] [Google Scholar]
- 16.Sztalryd C, Hamilton J, Horwitz BA, Johnson P, Kraemer FB: Alterations of lipolysis and lipoprotein lipase in chronically nicotine-treated rats. Am J Physiol 1996;270:E215–E223 [DOI] [PubMed] [Google Scholar]
- 17.Hellerstein MK, Benowitz NL, Neese RA, Schwartz JM, Hoh R, Jacob P, 3rd, Hsieh J, Faix D: Effects of cigarette smoking and its cessation on lipid metabolism and energy expenditure in heavy smokers. J Clin Invest 1994;93:265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chajek-Shaul T, Berry EM, Ziv E, Friedman G, Stein O, Scherer G, Stein Y: Smoking depresses adipose lipoprotein lipase response to oral glucose. Eur J Clin Invest 1990;20:299–304 [DOI] [PubMed] [Google Scholar]
- 19.Barnoya J, Glantz SA: Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation 2005;111:2684–2698 [DOI] [PubMed] [Google Scholar]
- 20.Bergstrom J, Hermansen L, Hultman E, Saltin B: Diet, muscle glycogen and physical performance. Acta Phys Scand 1967;71:140–150 [DOI] [PubMed] [Google Scholar]
- 21.Lexell J, Henriksson-Larsen K, Winblad B, Sjostrom M: Distribution of different fiber types in human skeletal muscles: effects of aging studied in whole muscle cross sections. Muscle Nerve 1983;6:588–595 [DOI] [PubMed] [Google Scholar]
- 22.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN: MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther 2003;5:1003–1015 [DOI] [PubMed] [Google Scholar]
- 23.Guo Z, Mishra P, Macura S: Sampling the intramyocellular triglycerides from skeletal muscle. J Lipid Res 2001;42:1041–1048 [PubMed] [Google Scholar]
- 24.Rosendal J, Knudsen J: A fast and versatile method for extraction and quantitation of long-chain acyl-CoA esters from tissue: content of individual long-chain acyl-CoA esters in various tissues from fed rat. Anal Biochem 1992;207:63–67 [DOI] [PubMed] [Google Scholar]
- 25.Kaluzny MA, Duncan LA, Merritt MV, Epps DE: Rapid separation of lipid classes in high yield and purity using bonded phase columns. J Lipid Res 1985;26:135–140 [PubMed] [Google Scholar]
- 26.Patterson BW, Zhao G, Klein S: Improved accuracy and precision of gas chromatography/mass spectrometry measurements for metabolic tracers. Metabolism 1998;47:706–712 [DOI] [PubMed] [Google Scholar]
- 27.Bergman BC, Cornier MA, Horton TJ, Bessesen D: Effects of fasting on insulin action and glucose kinetics in lean and obese men and women. Am J Physiol 2007;293:E1103–E1111 [DOI] [PubMed] [Google Scholar]
- 28.Patterson BW, Zhao G, Elias N, Hachey DL, Klein S: Validation of a new procedure to determine plasma fatty acid concentration and isotopic enrichment. J Lipid Res 1999;40:2118–2124 [PubMed] [Google Scholar]
- 29.Guo Z, Jensen MD: Determination of skeletal muscle triglyceride synthesis using a single muscle biopsy. Metabolism 2002;51:1198–1205 [DOI] [PubMed] [Google Scholar]
- 30.Wolfe R: Radioactive and Stable Isotope Tracers in Biomedicine: Principles and Practice of Kinetic Analysis New York, Wiley-Liss, 1992 [Google Scholar]
- 31.Liu L, Zhang Y, Chen N, Shi X, Tsang B, Yu YH: Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest 2007;117:1679–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schenk S, Horowitz JF: Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest 2007;117:1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itani SI, Ruderman NB, Schmieder F, Boden G: Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes 2002;51:2005–2011 [DOI] [PubMed] [Google Scholar]
- 34.Ellis BA, Poynten A, Lowy AJ, Furler SM, Chisholm DJ, Kraegen EW, Cooney GJ: Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am J Physiol Endocrinol Metab 2000;279:E554–E560 [DOI] [PubMed] [Google Scholar]
- 35.Houmard JA, Tanner CJ, Yu C, Cunningham PG, Pories WJ, MacDonald KG, Shulman GI: Effect of weight loss on insulin sensitivity and intramuscular long-chain fatty acyl-CoAs in morbidly obese subjects. Diabetes 2002;51:2959–2963 [DOI] [PubMed] [Google Scholar]
- 36.Huie MJ, Casazza GA, Horning MA, Brooks GA: Smoking increases conversion of lactate to glucose during submaximal exercise. J Appl Physiol 1996;80:1554–1559 [DOI] [PubMed] [Google Scholar]
- 37.Andersson A, Nalsen C, Tengblad S, Vessby B: Fatty acid composition of skeletal muscle reflects dietary fat composition in humans. Am J Clin Nutr 2002;76:1222–1229 [DOI] [PubMed] [Google Scholar]
- 38.Ntambi JM, Miyazaki M: Regulation of stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res 2004;43:91–104 [DOI] [PubMed] [Google Scholar]
- 39.Matsuzaka T, Shimano H, Yahagi N, Kato T, Atsumi A, Yamamoto T, Inoue N, Ishikawa M, Okada S, Ishigaki N, Iwasaki H, Iwasaki Y, Karasawa T, Kumadaki S, Matsui T, Sekiya M, Ohashi K, Hasty AH, Nakagawa Y, Takahashi A, Suzuki H, Yatoh S, Sone H, Toyoshima H, Osuga J, Yamada N: Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat Med 2007;13:1193–1202 [DOI] [PubMed] [Google Scholar]
- 40.Axelsson T, Jansson PA, Smith U, Eliasson B: Nicotine infusion acutely impairs insulin sensitivity in type 2 diabetic patients but not in healthy subjects. J Intern Med 2001;249:539–544 [DOI] [PubMed] [Google Scholar]
- 41.Ueno H, Pradhan S, Schlessel D, Hirasawa H, Sumpio BE: Nicotine enhances human vascular endothelial cell expression of ICAM-1 and VCAM-1 via protein kinase C, p38 mitogen-activated protein kinase, NF-κB, and AP-1. Cardiovasc Toxicol 2006;6:39–50 [DOI] [PubMed] [Google Scholar]
- 42.Chowdhury P, Bose C, Udupa KB: Nicotine-induced proliferation of isolated rat pancreatic acinar cells: effect on cell signalling and function. Cell Prolif 2007;40:125–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wada T, Naito M, Kenmochi H, Tsuneki H, Sasaoka T: Chronic nicotine exposure enhances insulin-induced mitogenic signaling via up-regulation of α7 nicotinic receptors in isolated rat aortic smooth muscle cells. Endocrinology 2007;148:790–799 [DOI] [PubMed] [Google Scholar]
- 44.Bouzakri K, Roques M, Gual P, Espinosa S, Guebre-Egziabher F, Riou JP, Laville M, Le Marchand-Brustel Y, Tanti JF, Vidal H: Reduced activation of phosphatidylinositol-3 kinase and increased Ser636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes 2003;52:1319–1325 [DOI] [PubMed] [Google Scholar]
- 45.Engelman JA, Berg AH, Lewis RY, Lisanti MP, Scherer PE: Tumor necrosis factor α-mediated insulin resistance, but not dedifferentiation, is abrogated by MEK1/2 inhibitors in 3T3–L1 adipocytes. Mol Endocrinol 2000;14:1557–1569 [DOI] [PubMed] [Google Scholar]
- 46.Sell H, Kaiser U, Eckel J: Expression of chemokine receptors in insulin-resistant human skeletal muscle cells. Horm Metab Res 2007;39:244–249 [DOI] [PubMed] [Google Scholar]
- 47.Henderson B, Csordas A, Backovic A, Kind M, Bernhard D, Wick G: Cigarette smoke is an endothelial stressor and leads to cell cycle arrest. Atherosclerosis 2008;201:298–305 [DOI] [PubMed] [Google Scholar]
- 48.Sartipy P, Loskutoff DJ: Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A 2003;100:7265–7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamei Y, Miura S, Suganami T, Akaike F, Kanai S, Sugita S, Katsumata A, Aburatani H, Unterman TG, Ezaki O, Ogawa Y: Regulation of SREBP1c gene expression in skeletal muscle: role of retinoid X receptor/liver X receptor and forkhead-O1 transcription factor. Endocrinology 2008;149:2293–2305 [DOI] [PubMed] [Google Scholar]
- 50.Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC, Hirshman MF, Rosen ED, Goodyear LJ, Gonzalez FJ, Spiegelman BM, Kahn CR: Muscle-specific PPARγ-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest 2003;112:608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abbasi F, Farin HM, Lamendola C, McGraw L, McLaughlin T, Reaven GM: Pioglitazone administration decreases cardiovascular disease risk factors in insulin-resistant smokers. Metabolism 2008;57:1108–1114 [DOI] [PubMed] [Google Scholar]