

Summary
Most bacterial exported proteins cross the cytoplasmic membrane as unfolded polypeptides. However, little is known about how they fold during or after this process due to the difficulty in detecting folding intermediates. Here we identify co-translational and post-translational folding intermediates of a periplasmic protein in which the protein and DsbA, a periplasmic disulfide bond-forming enzyme, are covalently-linked by a disulfide bond. The co-translational mixed-disulfide intermediate is, upon further chain elongation, resolved, releasing the oxidized polypeptide, thus allowing us to follow the folding process. This analysis reveals that two cysteines that are joined to form a structural disulfide can play different roles during the folding reaction and that the mode of translocation (co-translational verse post-translational) can affect the folding process of a protein in the periplasm. The latter finding leads us to propose that the activity of the ribosome (translation) can modulate protein folding even in an extra-cytosolic compartment.
Introduction
In living organisms, most exported proteins cross the cytoplasmic membrane through the Sec channel (Wickner and Schekman, 2005). In order for it to go through the channel, a protein must be maintained in an unfolded state (Liu et al., 1988; Osborne and Rapoport, 2007). However, poorly understood is the nature of the folding process of a protein during and after its passage through the channel (Akiyama and Ito, 1993; Ureta et al., 2007).
A critical step in the folding of many exported proteins is the formation of disulfide bonds (Sevier and Kaiser, 2006). Thus, one convenient way to study aspects of folding of an exported protein in vivo is by following the appearance of disulfide bonds in that protein (Jansens et al., 2002).
Disulfide bonds are generally introduced into exported proteins by members of the thioredoxin superfamily in both prokaryotes and eukaryotes (Kadokura et al., 2003; Sevier and Kaiser, 2006). In the periplasm of Escherichia coli, the formation of disulfide bonds is catalyzed by the protein DsbA, a thioredoxin superfamily member, which oxidizes substrates by donating its disulfide bond to a pair of cysteines on a substrate (Bardwell et al., 1991; Kamitani et al., 1992). The process likely begins with the attack by a deprotonated cysteine of the substrate protein on the disulfide bond of DsbA, leading to a short-lived complex of DsbA and substrate linked together by an intermolecular disulfide bond (Figure 1A) (Kadokura et al., 2004). Next, a deprotonated second cysteine of the substrate attacks the mixed-disulfide to resolve the complex, releasing oxidized substrate and reduced DsbA. Results obtained in vitro are consistent with this model (Darby and Creighton, 1995; Frech et al., 1996). However, due to the difficulty of detecting the disulfide-linked enzyme-substrate complex, the process leading to formation of a disulfide bond in a folding protein via this complex has not been followed in vivo. Additionally, many questions remain about the mechanism of disulfide bond formation in vivo. We do not know how formation of disulfide bonds is coordinated with protein translocation and other folding processes. We also do not know which cysteines in substrates are used to form the intermediate mixed-disulfide complex with DsbA.
Figure 1.

Oxidative folding of PhoA in vivo. (A) Substrate oxidation by DsbA. (B) Use of the dsbA P151T mutant (lanes 7 to 18) and wild-type strain (1 to 6) to study the oxidative folding of PhoA. The strains expressing PhoA were pulsed for 30 s with [35S]-methionine and then chased with cold methionine for 10 min in the presence (lanes 13 to 18) or absence (1 to 12) of chloramphenicol (Cm). The samples were AMS-alkylated, immunoprecipitated with anti-PhoA antibody, and analyzed by SDS-PAGE and autoradiography. Relative molecular masses are shown in kDa. The positions of the oxidized (oxi) and reduced (red) forms of PhoA are indicated. See Figure S7 for the identity of these bands. Open arrowheads, non-specific bands; asterisks, DsbA-PhoA mixed-disulfide complexes; double asterisks, the “65-kDa” complex; upward arrow, the elongating chains of PhoA. (C) The PhoA sample as loaded in panel B, lane 13 was subjected to the second immunoprecipitation with the indicated antibodies. (D) The dsbA P151T mutant expressing PhoAss-FLAG-PhoA or PhoA-FLAG was used to discriminate the co-translational folding intermediates from those of the post-translational. The samples were processed as panel B but using anti-FLAG antibody and chase being done in the presence of chloramphenicol.
We previously reported a mutation that generated a Pro151 to Thr change in DsbA, which slows down the resolution of covalent mixed-disulfide complexes between DsbA and its substrates and, thus, allows the detection of these intermediates (Kadokura et al., 2004). Here we use this mutant and wild-type cells to follow the oxidative folding of a periplasmic protein in vivo. This approach enables us to 1)delineate the two steps of electron transfer that lead to the formation of a disulfide bond in a folding protein by a thioredoxin-superfamily member (Figure 1A) and 2)specify the substrate cysteine that is used to form a disulfide-linked enzyme-substrate intermediate. Our results show that the two cysteines that form a structural disulfide bond can play different roles in the oxidative protein folding reaction catalyzed by a thioredoxin superfamily member in vivo. Moreover, we provide evidence that the mode of translocation can affect the protein folding process in this compartment. Our detection and characterization of the intermediates reveal novel features of protein folding in an extra-cytosolic compartment.
Figure 2.

Use of non-reducing, reducing 2D SDS-PAGE to follow the oxidative folding of the elongating chains of PhoA. (A and B) The samples, prepared as in Figure 1D, from the wild-type strain expressing PhoAss-FLAG-PhoA that was pulsed and then chased for the indicated times in the presence of chloramphenicol, were separated on the 2D gels. (C and D) 2D gel analysis of the folding of PhoAss-FLAG-PhoA with the [CCAA] (C) or [AACC] (D) mutation. Signals on the diagonal line above the PhoA precursor are those of backgrounds, which become visible after long exposure required to show the faint signals from the intermediate complexes. See Results for details.
Results
Detection of disulfide-linked complexes formed between DsbA and a newly synthesized PhoA
To better understand the folding process in vivo, we used pulse-chase experiments to follow the time course and nature of the intermediates in disulfide bond formation in alkaline phosphatase (PhoA). PhoA is a homodimeric periplasmic protein with two intra-chain disulfide bonds, one between Cys168 and Cys178 and the other between Cys286 and Cys336 (Bradshaw et al., 1981). PhoA is translocated across the cytoplasmic membrane by a mixed co-and post-translational mechanism in which a substantial portion of the polypeptide chain is synthesized before protein translocation begins (Josefsson and Randall, 1981; see also Figure S1 for co-translational and post-translational translocation). To determine the oxidation (disulfide-bonded) status of PhoA at each time point, cultures were directly treated with acid to prevent any post-harvest oxidation of cysteines, and the free cysteines were alkylated with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS, 0.5 kDa). This modification retards the mobility of the reduced form of proteins on gels. Since the presumed intermediates of oxidative folding that form between DsbA and its substrates have never been detected before in wild-type cells, we first utilized the DsbA P151T mutant which allows detection of such covalent intermediates (Kadokura et al., 2004). To follow the oxidation of PhoA, cellular proteins were pulse-labeled with [35S]-methionine for 30 s at 30°C, chased for up to 10 min and immunoprecipitated with anti-PhoA antibody. Immediately after the pulse, a small fraction (∼8%) of PhoA is in the oxidized form (Figure 1B, lane 7, oxi; see also Figure S2, lane 7). The remainder runs at 1) the positions of the reduced forms (red), 2) five positions representing complexes of apparently higher molecular weights than PhoA (asterisks), and 3) positions of possible nascent chains (upward arrow). Note that the reduced forms are observed at the positions of the reduced PhoA precursor still containing its signal sequence (faint upper band) and the reduced mature PhoA (lower band). After a 10 min chase, PhoA is largely converted to the oxidized form (Figure S2, lanes 7 to 12). We suggest that the five higher molecular weight bands (Figure 1B, lane 7, asterisks) represent disulfide-linked intermediates containing both DsbA and PhoA, since these bands disappear when the samples are treated with reductant before electrophoresis (not shown, see also Figure 2AB and related discussion on it), and both anti-DsbA and anti-PhoA antibodies precipitate them (Figure 1C). Importantly, these complexes were also detected in wild-type cells, although not appearing as prominently on gels because they were present in reduced amounts (Figure 1B, lanes 1 to 6; see also Figure 2B). Detection in vivo of a disulfide-linked complex between a thioredoxin superfamily member and its folding substrate is difficult (Appenzeller-Herzog and Ellgaard, 2008). Indeed, the amounts of the complexes in wild-type cells indicated by the single asterisks are so small that it was necessary to slightly overexpose the film in Figure 1B for their detection. Nevertheless, these results show that the bands seen with the P151T mutant are not artifacts generated by the alteration of DsbA, confirming the mutant's utility in allowing their detection. This result represents the first detection, in wild-type cells, of disulfide-bonded complexes between DsbA and a substrate.
An intermediate complex formed between the periplasmic catalyst DsbA and the PhoA polypeptide elongating from the ribosome
We noticed that the smallest DsbA-PhoA complex, a diffuse “65-kDa” band (Figure 1B, double asterisk), behaves differently from the other complexes (single asterisk). Note that this complex appears to have a molecular weight lower than predicted for that of a complex containing both full-length DsbA and full-length PhoA. The latter should run at a position corresponding to a molecular weight of about 71-kDa. In the presence of chloramphenicol, a peptide elongation inhibitor that freezes the polysome (Ennis, 1972), the four high molecular weight complexes disappeared during chase accompanied by a concomitant increase in the amount of oxidized PhoA (Figure 1B, lanes 13 to 18; Figure S2, lanes 13 to 18). However, the “65-kDa” complex persisted after the chase in the presence of the inhibitor (Figure 1B, lanes 13 to 18), indicating that ongoing peptide elongation is needed to resolve this complex. Surprisingly, during the chase in the presence of chloramphenicol, we observed an increase in the amount of the “65-kDa complex (see also Figure 1D, lanes 1 to 5). The implication of this finding will be discussed later.
To further characterize what portion of PhoA was present in the intermediates, we inserted a FLAG tag either at the N-terminus of PhoA after the signal sequence cleavage site (PhoAss-FLAG-PhoA) or after the C-terminus of the protein (PhoA-FLAG) (Figure S3). Anti-FLAG antibody can pull down nascent polypeptides from the N-terminally tagged but not the C-terminally tagged construct, because nascent polypeptides have not completed synthesis of the C-terminus. We found that the “65-kDa” complex was precipitated by anti-FLAG antibody only when we used the PhoAss-FLAG-PhoA (Figure 1D). This result, together with the previous finding that ongoing peptide elongation is needed to resolve the “65-kDa” complex, indicates that it is a complex between DsbA and PhoA nascent chains. In contrast, the other four higher molecular weight complexes were pulled down from both FLAG-tagged constructs (Figure 1D), indicating that they consisted of DsbA and full-length PhoA.
To assess the nature of the PhoA in the intermediates, immunoprecipitates were separated by two-dimensional (2D) gel electrophoresis in which the first dimension was non-reducing and the second reducing (Note that, in Figures 2, 3, 5 and 6, and Figures S5, and S6, we used the strain with wild-type DsbA). Each of the four high molecular weight complexes yielded a spot that represents the full-length mature PhoA (Figure 2ABD, arrowhead), supporting the conclusion that they contained DsbA and full-length PhoA. In contrast, the reduced “65-kDa” complex yielded a faint but reproducible short streak around 43 kDa (Figure 2ABD, marked as “65-kDa” complex; see also Figure 5B). The apparent molecular mass of the complex (∼65-kDa) agrees well with the sum of the molecular masses of DsbA (21 kDa) and ∼43 kDa. Thus, PhoA forms a disulfide-linked complex with DsbA when the polypeptide has elongated to an approximate molecular mass of 43 kDa.
Figure 3.
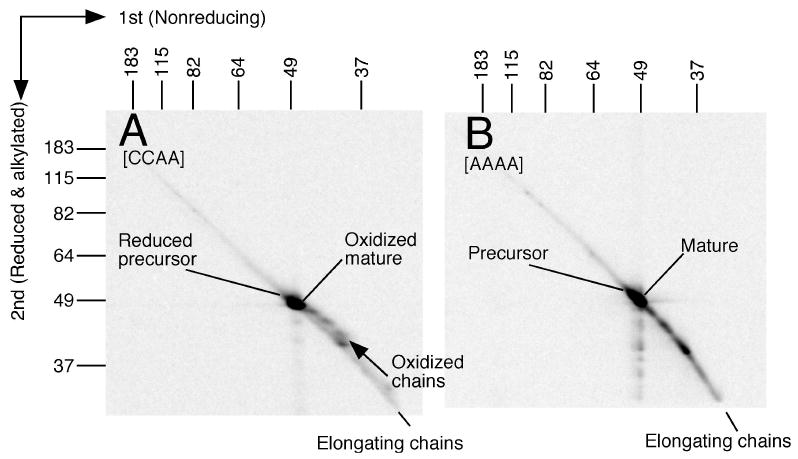
Two dimensional gel electrophoresis and in-gel AMS-alkylation to detect the oxidation of the Cys168-Cys178 pair in the elongating chains of PhoA. (A and B) HK353 (wild-type strain) expressing PhoAss-FLAG-PhoA[CCAA] (A), or PhoAss-FLAG-PhoA[AAAA] (B) was subjected to radio-labeling, AMS-alkylation and immunoprecipitation with anti-FLAG antibody. The latter variant served as a negative control for the N-terminal disulfide bond formation. After separation on a non-reducing gel, the AMS-alkylated PhoA molecules were subjected to reduction, in-gel AMS alkylation and separation on the second dimension gels. Note that, to achieve better separation of the samples, the gels were run for longer time in both dimensions.
Figure 5.

The SfmC signal sequence drives co-translational export and folding of PhoA. (A) The export of PhoA was studied by SDS-PAGE using the samples processed from the wild-type strain expressing PhoAss-FLAG-PhoA (lanes 1 to 4) or SfmCss-FLAG-PhoA (5 to 8) as described in Figure 1D. (B) Oxidative folding of PhoA from SfmCss-FLAG-PhoA was studied by 2D separation of the sample from the 0-min chase time-point. The gels were run for longer time in both dimensions
Figure 6.

The signal sequence affects protein folding in the periplasm. (A) To study the effect of the signal sequence on the folding of PhoA lacking Cys178, expression of PhoAss-FLAG-PhoA (lanes 1 to 6) or SfmCss-FLAG-PhoA (lanes 7 to 12) with the indicated cysteine to alanine mutations was induced with 10 μ M IPTG at 30°C for 50 min in the wild-type strain (lanes 1, 3, 5, 7, 9 and 11) or a ΔdsbC strain (lanes 2, 4, 6, 8, 10, and 12). The samples were subjected to AMS-alkylation, SDS-PAGE and western blotting with anti-FLAG antibody. (B) The activity of the ribosome can affect protein folding in the periplasm. See discussion for details.
Oxidation of the polypeptide chain in the periplasm as it elongates from the ribosome
The long diagonal line on the same 2D gel, ending at the position of the PhoA precursor (marked as “Reduced precursor”), represents elongating chains of PhoA (Figure 2), since no corresponding line is present when anti-FLAG antibody is used to precipitate PhoA from PhoA-FLAG (compare lane 1 with lane 6 in Figure 1D). From about 44 to 48 kDa, a streak appears above the diagonal line (Figure 2ABD, arrow) resulting in a doubling of the line at this position. Our evidence indicates that this streak consists of nascent chains that contain a disulfide bond in the first non-reducing dimension causing them to move at a faster rate in the first dimension than non-disulfide bonded PhoA chains of the same molecular weight. These chains then moved more slowly in the second dimension after they had been reduced. The greater gel mobility in the first dimension could be due to the compactness of the protein generated by the disulfide bond. This conclusion is supported by the following analysis of the effect of each of its two disulfide bonds on gel mobility of PhoA species.
To determine which cysteine pair of PhoA (Cys168-Cys178 or Cys286-Cys336) is involved in the formation of the nascent intra-chain disulfide bond that generates the more slowly moving streak, we asked which of the two disulfide bonds affects mobility on gels in this way. Each of the two cysteine pairs was mutated separately to convert both of the cysteines to alanine and the oxidation of the nascent chains in these two species of PhoA was studied. We found that the appearance of the off-diagonal streak depended on the presence of the Cys286-Cys336 pair but not the Cys168-Cys178 pair (Figure 2CD). This finding indicates that the formation of the intra-chain disulfide bond in nascent PhoA polypeptides affecting mobility on gels is due to the oxidation of the Cys286-Cys336 pair. These results also indicate that the Cys286-Cys336 disulfide bond starts to form when the polypeptide is elongated to 44 kDa. We also found that the absence of the Cys286-Cys336 pair led to the disappearance of the spot derived from the reduction of the “65-kDa” complex indicating that this complex is one between DsbA and one cysteine of this pair of cysteines (Figure 2CD). These results also suggest that the “65-kDa” complex is an intermediate that precedes the formation of the Cys286-Cys336 disulfide bond.
We note that, when the chase was done in the presence of chloramphenicol, the disulfide bond seen in the streak above the diagonal line formed at slightly earlier stages of translation and did so more efficiently (Figure 2AB). The formation of the “65-kDa” complex, as mentioned above, was also enhanced during the chase (Figure 1B, lanes 13 to 18 and Figure 1D, lanes 1 to 5). These findings can be most readily explained by assuming that the nascent chains could still undergo further translocation even though protein synthesis was blocked during the chase. In this way, the appropriate cysteines were exposed in the periplasm allowing formation of DsbA-PhoA complexes and of the carboxyl-terminal disulfide bond. In fact, the enhanced accumulation of the “65-kDa” complex observed during the chase was largely diminished by azide, a translocation inhibitor (Oliver et al., 1990) (Figure S4). Thus, the nascent chains underwent translation-uncoupled translocation into the periplasm during the chase period.
Use of 2D, in-gel alkylation to study the oxidation of a closely-located cysteine pair
The reduction of the Cys168-Cys178 disulfide did not change the migration of PhoA on gels (Figure S5A, lanes 5-6), consistent with a previous report (Sone et al., 1997b). This observation can explain why oxidation of this cysteine pair did not lead to a streak comparable to that seen with the Cys286-Cys336 pair on the 2D gel (Figure 2C). To allow visualization of nascent chains containing the Cys168-Cys-178 disulfide bond, we caused alteration of its mobility on gels by performing an in-gel AMS alkylation step after AMS-alkylated proteins had been run on the first gel and treated with reductant. This step adds ∼0.5 kDa to each cysteine that had been involved in a disulfide bond, causing the originally oxidized chains to migrate more slowly on the second gel. With this new approach we were able to detect the oxidation of the first cysteine pair (Figure 3A). This oxidation occurred when the polypeptides were elongated to approximately 39 kDa (Figure 3A), not surprisingly an earlier point than that seen for the formation of the C-terminal disulfide bond, but at a considerably longer time after the cysteines had been incorporated into the protein. This finding is consistent with previous studies which showed that a considerable length of the PhoA polypeptide is made before translocation across the membrane begins (Josefsson and Randall, 1981).
We were unable to detect a DsbA-PhoA mixed-disulfide intermediate that leads to the formation of the Cys168-Cys178 disulfide bond (Figure 2C, Figure 3A). We suppose that this intermediate is quite short-lived as the close proximity of these two cysteines should result in the mixed-disulfide with DsbA being immediately attacked by the closely following cysteine to resolve this complex and form the disulfide bond. This situation contrasts with that of the “65-kDa” complex where the two cysteines (Cys286 and Cys336) are separated by 49 amino acids.
Identification of the substrate cysteine that is used to form the nascent chain mixed-disulfide intermediate
To further study the mechanism of formation of the C286-Cys336 disulfide bond, we analyzed the structure of the “65-kDa” DsbA-PhoA complex, using cysteine to alanine mutants of PhoA. Introduction of a C286A mutation into PhoA (Figure 4A, lanes 9-12), but not of a C336A mutation (lanes 5-8), prevented the formation of this intermediate. Thus, Cys286 of PhoA is the cysteine that attacks DsbA to form the “65-kDa” complex (Figure 4B). However, the C286A mutant also formed a complex, but it moved to around a 69-kDa position 4 kDa higher than the “65-kDa” complex. The 69-kDa complex is not seen when both Cys286 and 336 are present in PhoA. This finding indicates that Cys336 can attack DsbA in the absence of Cys286, but only when the polypeptide is elongated further to around 47 kDa. However, the predominance of the “65-kDa” complex when both Cys286 and Cys336 are present indicates that, with the native PhoA, Cys286 is normally used to attack DsbA to form the intermediate during translocation (see, for contrasting conditions, the section following the next one).
Figure 4.
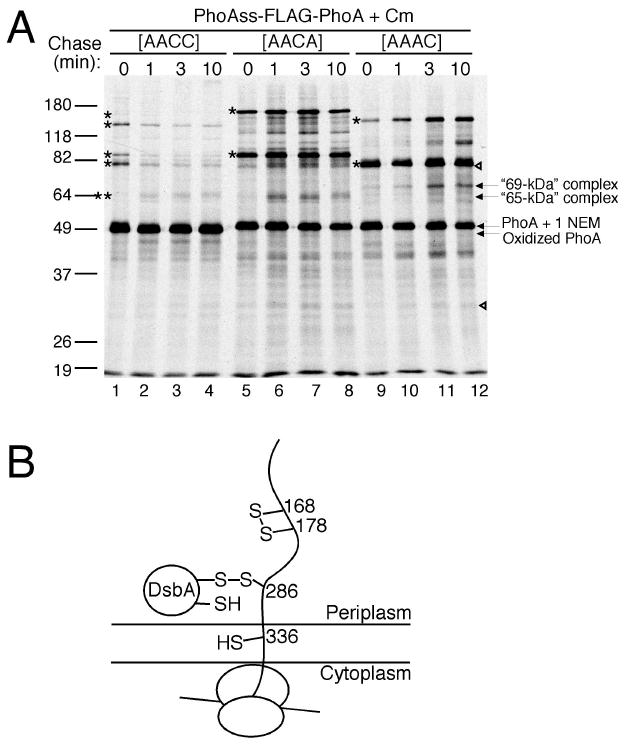
Analysis of the structure of the DsbA-PhoA complexes. (A) Use of cysteine to alanine mutations to determine the cysteine on PhoA that is used to form the “65-kDa” complex (double asterisks) and the other DsbA-PhoA complexes (asterisk). The dsbA P151T mutant strain, expressing PhoAss-FLAG-PhoA with the indicated cysteine to alanine mutations, was labeled, chased, and processed as described in Figure 1D except that 20 mM N-ethylmaleimide was used instead of AMS. See Results for the “69-kDa” complex which appeared with the C286A mutation. Note that some of the high molecular weight complexes appeared very strongly when the resolving cysteine of the pair was mutated. See Supplemental Text for discussion on this finding. (B) Model for the “65-kDa” complex.
SRP-dependent signal sequences drive co-translational folding
In our pulse-chase experiments, a portion of PhoA is found as full-length precursor at early time points (e.g. “Reduced precursor” in Figure 2A and Figure 5A). Since this precursor chases into mature PhoA, this species must have crossed the membrane post-translationally (Figure S1a). This finding is consistent with previous reports that the processing of many secretory proteins, including PhoA, occurs both co-translationally and post-translationally in vivo (Josefsson and Randall, 1981). To determine further whether the folding of a protein might be influenced by the mechanism of its export, we also caused PhoA to be exported predominantly by the co-translational pathway (Tian and Bernstein, 2009). We did this by replacing the signal sequence of PhoA with the signal sequences of SfmC and TorT (Figure S3), which utilize the signal recognition particle (SRP) pathway to direct co-translational targeting of the protein to the translocation channel in contrast to the native PhoA signal sequence (Huber et al., 2005). In these experiments, consistent with a co-translational initiation of protein export, we found no accumulation of the signal sequence-containing precursor of PhoA in the pulses (compare Figure 5A, lanes 5 to 8 and Figure S6A, lanes 1 to 4 with Figure 5A, lanes 1 to 4). Separation by 2D gel of the PhoA products from these variants shows that disulfide bond formation in these constructs occurs co-translationally (Figure 5B and Figure S6B), indicating the coupling of protein export and oxidative folding.
Interestingly, we found that the continuation of protein export and oxidative folding seen with the PhoA signal sequence during the chase in the presence of chloramphenicol were absent when the SRP signal sequences were used (e.g. compare the “65-kDa” complex formation in Figure 1D, lanes 1 to 5 and Figure 5A, lanes 1 to 4 with that of Figure 5A, lanes 5 to 8). This finding suggests that when the SRP pathway is used, protein export and folding are tightly coupled to peptide elongation at least at late stages of translation. This finding is unexpected because, unlike mammalian SRP, bacterial SRP failed to exhibit “elongation arrest”, a mechanism to couple peptide elongation to protein translocation (Powers and Walter, 1997).
Finally, we point out that, when we use SRP-dependent signal sequences for PhoA export, we observe the efficient formation of the “65-kDa” complex, which is followed by chain oxidation (Figure 5B).
Studies on the high molecular weight complexes
Up to this point, the high molecular weight disulfide-linked complexes formed between DsbA and the completed chains of PhoA remained a mystery. To gather further insight into the nature of these complexes, we determined their disulfide bond connectivity using the same cysteine to alanine mutations described above. In contrast to the nascent-chain mixed-disulfide intermediate which was exclusively formed between DsbA and Cys286, the high molecular weight mixed-disulfide complexes included ones that used either Cys286 or Cs336 for the linkage with DsbA (Figure 4A, asterisks). To analyze the structure of these high molecular weight complexes, we purified them using a hexahistidine tag fused to DsbA (Kadokura et al., 2004) and a FLAG tag fused to PhoA. Upon analysis of the purified complexes by mass spectrometry, we detected tryptic fragments from DsbA and PhoA but not of any other protein (not shown), showing that they consist only of these two proteins. Since Cys33, the C-terminal cysteine of the CXXC active site of DsbA, is inaccessible to the surface of the protein, Cys30 should be the only cysteine available from DsbA for the mixed-disulfide formation with PhoA. On the other hand, from the PhoA mutant variants, only one cysteine (either Cys286 or Cys336) is available. Accordingly, the high molecular weight complexes are likely to include hetero-dimers of DsbA and the completed chain of PhoA linked by Cys30 of DsbA and either Cys286 or Cys336 of PhoA.
It should be noted that the high molecular weight complexes migrated in gels differently in two ways. First, the high molecular weight complexes formed between DsbA and Cys336 of PhoA migrated faster than those formed between DsbA and Cys286 of PhoA (Figure 4A). This behavior can be explained by the fact that the rate of migration of denatured covalent complexes in gels does not depend only on the sum of the molecular weights of the components, but also depends on the branch structure (Cho et al., 2007). Second, the electrophoretic mobilities of two of the high molecular weight complexes (corresponding to calculated molecular weight of 130-kDa and 150-kDa) are much slower than those expected from the sum of the mass of DsbA and the full length-PhoA (∼71 k-Da). Since such slowly-migrating molecules were not observed for the nascent-chain mixed-disulfide intermediate, their appearance likely requires a step that occurs only after the synthesis of the C-terminal portion of PhoA. However, without further analysis, we cannot speculate on the nature of these unusual complexes.
For study of the folding of PhoA that takes place during translation (co-translational folding), it is possible to use the chain-length as a time scale for the progression of event. However, the folding that takes place after translation is completed (post-translational folding) lacks such a clear scale. This fact, in conjunction with the presence of the several high molecular weight mixed-disulfide complexes and the very transient nature of the mixed-disulfide intermediate formation, prevents us from establishing the precursor-product relationship for these larger potential intermediates. Thus, it is not clear whether these high molecular complexes are the intermediates of the same or different pathways or whether they are on-pathway or off-pathway intermediates. However, importantly, these results indicate a difference in the kinds of the intermediate complexes formed between the co-translational folding and post-translational folding. We observe the presence of multiple complexes of this sort occurring apparently after translation has been completed, in contrast to the one mixed-disulfide intermediate, the “65-kDa” complex, that occurred with co-translational folding (e.g. Figure 5B). Strikingly, the post-translational high molecular weight mixed-disulfide complexes used either Cys286 or Cys336 of PhoA for their formation in contrast to the co-translational intermediate (“65-kDa”) which used only Cys286. These findings lead us to consider it likely that differences between translocation of proteins exported in a co-translational versus post-translational mechanism result in differences in the folding process of proteins in the periplasm.
In particular, since the intermediate formed during the co-translational export used only Cys286 for complex formation, the more N-terminal cysteine of the Cys286-Cys336 pair, it appears that oxidative folding of this protein proceeds more vectorially from the N-terminus to the C-terminus during co-translational folding than it does during post-translational folding in the periplasm.
Signal sequences can affect protein folding in the periplasm
To test whether the mode of export affects the folding process, we constructed PhoA [CACC], a PhoA variant which lacks the second cysteine, and exported it either co-translationally with the SfmC signal sequence or mainly post-translationally with the PhoA signal sequence. A mutant of this sort, lacking one of the cysteines of the N-terminal cysteine pair has been shown to cause misfolding of this protein (Sone et al., 1997a). We reasoned that co-translational export causes rather tight vectorial folding and, as a result, disulfide bonds form in order as each pair of cysteines appears in the periplasm. If this is the case, then the elimination of the second cysteine from the construct would result in formation of an incorrect disulfide bond between Cys168 and Cys286. This bond would seriously compromise the folding of this protein, possibly resulting in decreased levels of the protein in the cell.
Consistent with this expectation, the export of the PhoA [CACC] variant by the co-translational SfmC signal sequence caused a stronger defect in the accumulation of this protein than that of the same variant with its native PhoA signal sequence (Figure 6A, lanes 3, and 9). This finding indicates that the biosynthesis of PhoA with the C178A mutation is compromised when this protein is exported with the co-translational signal sequence. Since there is a periplasmic disulfide bond isomerase, DsbC, which acts to repair wrongly-formed disulfide bonds (Kadokura et al., 2003), we asked whether eliminating DsbC from cells would affect the level of PhoA protein expressed from these constructs. Strikingly, most of the PhoA [CACC] exported with the SfmC signal sequence failed to accumulate in the oxidized state in the dsbC mutant (lane 10), suggesting that the majority of the protein did not fold properly in the absence of the disulfide isomerase. Furthermore, when both the first and second cysteines, Cys168 and Cys178, were mutated to alanine, the levels of this protein were restored to the levels of wild-type PhoA (Figure 6A, lanes 11, and 12). The latter result establishes that the decreased accumulation of PhoA in the PhoA [CACC] variant exported by the SfmC signal sequence is not merely due to the lack of the second cysteine (Cys178) itself, but to the presence of the unpaired cysteine (Cys168) in PhoA. Thus, when exported by the co-translational signal sequence, the unpaired cysteine caused more severe misfolding of this protein than can be fully repaired by periplasmic DsbC. These results show that the nature of a signal sequence can affect the folding of the reporter protein in the periplasm.
Discussion
In this study, we followed the steps of DsbA-catalyzed oxidative folding of PhoA using pulse-chase experiments. Our analysis of the process, in particular the detection and characterization of the mixed-disulfide intermediate complexes formed between PhoA and DsbA, provides new insights into protein folding in an extra-cytosolic compartment. Our data point to the following picture for the co-translational formation of the Cys286-Cys336 disulfide bond: The process begins with Cys286 attacking DsbA when the PhoA polypeptide elongates to about 43 kDa in length. This reaction leads to the formation of a mixed-disulfide between DsbA and the nascent chain of PhoA. Further elongation of the polypeptide allows Cys336 to attack the mixed-disulfide, resulting in the resolution of the complex and oxidation of the nascent chain. Our detection of the “65-kDa” complex and demonstration of nascent chain oxidation establish that protein folding in the periplasm can occur as the polypeptide elongates from the ribosome.
Mixed-disulfide complexes between thioredoxin superfamily members and their folding substrates have been detected in both prokaryotes and eukaryotes, but the detailed mechanism of the formation of a specific disulfide bond via such a complex have not been studied (Di Jeso et al., 2005; Kadokura et al., 2004; Morinari and Helenius, 1999). One problem with the analysis of the complexes formed between a thioredoxin superfamily member in eukaryotes and its substrates is that the substrates often have many disulfide bonds, which makes analysis of the process difficult. Furthermore, the very transient nature of the mixed-disulfide complexes makes it hard to detect the intermediate complexes themselves (Appenzeller-Herzog and Ellgaard, 2008). Using a simple substrate (PhoA) with only two disulfide bonds and both the wild-type cells and the DsbA P151T mutant enabled us to delineate the steps of electron transfer leading to the formation of the C-terminal disulfide bond of PhoA. Our visualization of the steps of co-translational formation of the disulfide bond provides direct evidence that disulfide bond formation by a thioredoxin superfamily member proceeds through a mixed-disulfide complex between the enzyme and its folding substrate in vivo.
Here we showed that, during co-translational folding of PhoA, Cys286 attacks DsbA to initiate the formation of the Cys286-Cys336 disulfide bond. This step is followed by an attack of Cys336 on the mixed-disulfide bond formed between DsbA and Cys286, thus completing the formation of the disulfide bond. These results provide direct evidence that two cysteines of a folding protein can play different roles in the formation of their disulfide bond in a process catalyzed by a thioredoxin family member. The differentiation of the role of individual cysteines in the formation of a disulfide bond may be important for coordinating the steps involved in proper protein folding. This may be the case particularly for proteins with complex disulfide patterns.
We suggest that at least two factors may contribute to determining which cysteines attack DsbA during the process of disulfide bond formation. First, evidently, the cysteine on the substrate must be sterically available to DsbA for the reaction to take place. Second, since the deprotonation of a sulfhydryl group is required for a cysteine to attack a disulfide bond, the reactivity of a substrate cysteine may also be influenced by its pKa. In the case of PhoA that is co-translationally crossing the membrane, apparently the availability (first appearance in the periplasm) of the cysteine determines which cysteine attacks DsbA. Further studies that identify and characterize attacking cysteines in proteins with complicated disulfide pattern by detecting disulfide-linked intermediates with the P151T mutant should help to reveal the relative roles of these properties in determining oxidative protein folding pathways.
We have observed a difference in the types of mixed-disulfide intermediates formed when we compare co-translational and post-translational folding of PhoA. Using SRP-dependent (co-translational) and non-SRP-dependent (post-translational and co-translational) signal sequences to promote PhoA export, we detect one mixed-disulfide intermediate between DsbA and PhoA during co-translational folding (the “65-kDa” DsbA-PhoA complex), and four mixed-disulfide complexes during post-translational folding. We showed that while Cys286, the N-terminal cysteine of the Cys286-Cys336 pair, is used to form the co-translational intermediate, the post-translational complexes could use either Cys286 or Cys336 to form an intermolecular disulfide bond with DsbA. Further, we showed, using the C178A mutant, that, in contrast to post-translational folding, co-translational folding of PhoA is very sensitive to the presence of an unpaired cysteine in the protein. These findings are consistent with the proposal that oxidative protein folding in the periplasm occurs more vectorially from the N-terminus to the C-terminus during protein synthesis than after completion of protein synthesis and indicate that the mode of translocation (co-translational verse post-translational) can affect the folding process of a protein in the periplasm.
The apparent vectorial nature of disulfide bond formation, as shown here for PhoA, especially during its co-translational folding, may be a shared property of many bacterial exported proteins (Berkmen et al. 2005; Jansens et al., 2002). If so, disulfide bonds would tend to be formed between cysteines that appear sequentially in the polypeptide, leading to the formation of incorrect disulfide bonds in those proteins that should include non-consecutive disulfide bonds. In fact, efficient folding of certain E. coli proteins with non-consecutive disulfide bonds requires the bacterial disulfide bond isomerase DsbC for the repair of incorrect disulfide bonds (Berkmen et al., 2005; Hiniker and Bardwell, 2004). In addition, for those organisms that make disulfide bonds, the majority of bacterial cell envelope proteins appear to have evolved to avoid an unpaired cysteine, perhaps to prevent the formation of inappropriate disulfide bonds (Dutton et al., 2008). Nevertheless, our results on the behavior of PhoA during the post-translational folding together with the findings of others (Messens et al., 2007) indicate that oxidative folding does not necessarily proceed vectorially in this organism.
How might the mode of translocation affect the folding process of a protein? There is reason to think that the steps in protein folding in the periplasm may depend on the rate at which a protein is translocated across the cytoplasmic membrane and appears in the periplasm. In a co-translational mechanism of translocation, the rate of appearance of the protein in the periplasm is dependent on protein synthesis. In post-translational translocation it depends on the efficiency of the Sec apparatus to drive proteins through the membrane. That these processes do result in different rates of protein translocation is supported by studies which estimate that protein translocation through the SecYEG channel is more than 10 times faster than that of protein translation (Matsuyama et al, 1992; Pugsley, 1993). To calculate the rate of protein translocation, these reports use a) the estimated number of channels in a cell, b) the estimated number of polypeptides a single cell needs to export per generation, and c) the estimated average size of exported polypeptides. These calculations would indicate that, during co-translational export, the rate of translocation is limited by the rate of peptide elongation from the ribosome and, thus, a polypeptide chain appears in the periplasm more slowly than when it is exported in a post-translational manner. Therefore, when the polypeptide is crossing the channel in a co-translational manner, it may be the slowness in the appearance in the periplasm that results in polypeptide folding more vectorially from the N-terminus to the C-terminus as we have observed with the mutant variant (C178A) of PhoA. In addition, the presence of the ribosome attached to the nascent chain itself might influence the way a protein passes through the channel or its rate of translocation (e.g. by altering the interaction between SecA and the nascent chain), thereby affecting protein folding in the periplasm.
It is well established that the ribosome plays an important role in protein folding in the cytosol of organisms (Albanese et al., 2006; Evans et al., 2008; Fedorov and Baldwin, 1997; Rutkowska et al., 2008). Here, we propose, that the activity of the ribosome (translation) can also affect extracytosolic protein folding in a compartment such as the periplasm (Figure 6B). The significance of this concept should apply to eukaryotes as well as bacteria, since both co-translational and post-translational protein translocation into the endoplasmic reticulum have been demonstrated (Wickner & Schekman, 2005).
Interestingly, the “65-kDa” complex, formed between DsbA and Cys286 of the nascent-chain of PhoA, persisted after the chase in the presence of chloramphenicol both in the wild-type cells and in the P151T mutant (Figure 1B and Figure 5A) indicating that this complex is highly stable. The stability can be explained in part by the unavailability of the partner cysteine (Cys336) at the time when the complex forms. However, one might still have imagined that the resolving cysteine of DsbA (Cys33) could attack the intermolecular disulfide bond, reversing the reaction and generating oxidized DsbA and PhoA with the Cys286 in the reduced state. However, the stable nature of this complex suggests that DsbA or its substrate may have evolved to stabilize intermediate complexes thus promoting forward reactions. Perhaps, one of the features of DsbA that contributes to the stability of this complex is its high redox potential.
The methods we have developed for this work provide useful tools for the studies of the coordination between protein export and folding processes in E. coli and in other organisms. These methods include positional tagging to discriminate co- and post-translational folding, in-gel AMS alkylation to detect oxidation of closely-located cysteines, use of the dsbA P151T mutant to facilitate the detection of the mixed-disulfides, and use of SRP-dependent signal peptides to coordinate peptide elongation with protein export.
In this paper, we have shown, by detailed analysis of the formation of disulfide bonds, that it is possible to follow in vivo aspects of the folding of proteins exported to the bacterial cell envelope. Our demonstration that we can observe co-translational, co-translocational oxidative folding intermediates as well as post-translational steps encourages us to believe that it should be possible to assess the role of many cellular factors in protein folding. Future studies will determine how periplasmic molecular chaperones (Harms et al., 2001), peptidyl-prolyl cis-trans isomerases (Duguay and Silhavy, 2004), export machinery proteins such as SecD and SecF (Ureta et al., 2007), and the ribosome cooperate with the disulfide catalyst to drive protein folding.
Experimental Procedures
Strains and growth conditions
Strains and plasmids are listed in Table S1. Unless otherwise noted, cultures were grown at 30°C in M63 glucose medium (Kadokura et al., 2004) supplemented with appropriate antibiotics and amino acids.
Pulse-chase, alkylation and immunoprecipitations
To follow the oxidative folding of PhoA, cells harboring a plasmid encoding each PhoA construct were grown at 30°C until saturation in M63 glucose medium supplemented with 50 μg/ml of each of 18 amino acids (all except for methionine and cysteine) and 200 μg/ml of ampicillin, and then, were subcultured 1:100. When the culture was grown to an optical density at 600 nm (OD600) of 0.3, expression of the PhoA construct from the plasmid was induced by addition of isopropyl-β-D-thiogalactopyranoside to a final concentration of 10 μM. Note that the expression level of PhoA is critical for the success of the experiments of this sort as the expression of this protein above a certain level causes inefficient export or folding of the protein, which becomes a problem especially when co-translational export and folding are studied. When the culture reached an OD600 of 0.5, the cells were pulse-labeled for 30 sec with 40 μCi of [35S]-methionine per 1 ml, and chased with a final concentration of 0.1% methionine. When used to prevent peptide elongation, chloramphenicol was added at the beginning of the chase to a final concentration of 100 μg per ml. After chase, a portion of the culture (800 μl) was harvested and immediately mixed with 100 μl of 100 % trichloroacetic acid and kept on ice for 20 min to stop peptide elongation and further export of proteins and to inhibit the reactivity of cysteines. To analyze the formation of mixed-disulfides and to determine the in vivo redox states of proteins, the free cysteines in the sample were subjected to alkylation, unless otherwise noted, with 10 mM AMS (4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid, Invitrogen-Molecular Probes, Eugene, OR) as described (Kadokura et al., 2004). After alkylation, the PhoA product was collected from the samples by immunoprecipitation essentially as described (Ito et al., 1981) using an appropriate antibody. The precipitated proteins were separated on a 10% SDS-polyacrylamide gel and visualized by a Biomax MR film (Kodak, Rochester, NY) or a Typhoon 9400 Imager (Amersham Biosciences, Sunnyvale, CA).
Non-reducing, reducing two-dimensional gel electrophoresis and in-gel alkylation of proteins with AMS
The immunoprecipitates from the pulse-chased samples were first separated on a non-reducing gel. The gel lanes were cut out, incubated in Laemmli SDS sample buffer containing 5% β-mercaptoethanol at 90°C for 5 min. Then, the proteins in the gel was separated on the second dimension gel. When in-gel alkylation of proteins with AMS was needed, the gel lane from the first dimension was treated with a reductant, TCEP (tris-(2-carboxyethyl)phosphine, hydrochloride) (Invitrogen, Eugene, OR), rinsed three times with water, and then treated with AMS, prior to electrophoresis on the second dimension gel. Details are provided in the Supplemental Data.
Supplementary Material
Acknowledgments
We thank members of J.B. laboratory especially Markus Eser, and Seung-Hyun Cho for discussions. H.K. thanks Kenji Kohno, in whose laboratory part of this work was done. J.B. is an American Cancer Society Professor. This work was supported by NIH grant GM41883 (to J.B.) and in part by an international research fellowship from the Global COE program in NAIST and Grant-in-Aid for Scientific Research C (21580092) (to H.K.).
Footnotes
Supplemental Data: Supplemental Data include two tables, seven figures, experimental procedures, and text, and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama Y, Ito K. Folding and assembly of bacterial alkaline phosphatase in vitro and in vivo. J Biol Chem. 1993;268:8146–8150. [PubMed] [Google Scholar]
- Albanese V, Yam AY, Baughman J, Parnot C, Frydman J. Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell. 2006;124:75–88. doi: 10.1016/j.cell.2005.11.039. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Ellgaard L. In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid Redox Signal. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- Berkmen M, Boyd D, Beckwith J. The nonconsecutive disulfide bond of Escherichia coli phytase (AppA) renders it dependent on the protein-disulfide isomerase, DsbC. J Biol Chem. 2005;280:11387–11394. doi: 10.1074/jbc.M411774200. [DOI] [PubMed] [Google Scholar]
- Bradshaw RA, Cancedda F, Ericsson LH, Neumann PA, Piccoli SP, Schlesinger MJ, Shriefer K, Walsh KA. Amino acid sequence of Escherichia coli alkaline phosphatase. Proc Natl Acad Sci USA. 1981;78:3473–3477. doi: 10.1073/pnas.78.6.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Porat A, Ye J, Beckwith J. Redox-active cysteines of a membrane electron transporter DsbD show dual compartment accessibility. EMBO J. 2007;26:3509–3520. doi: 10.1038/sj.emboj.7601799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby NJ, Creighton TE. Catalytic mechanism of DsbA and its comparison with that of protein disulfide isomerase. Biochemistry. 1995;34:3576–3587. doi: 10.1021/bi00011a012. [DOI] [PubMed] [Google Scholar]
- Di Jeso B, Park YN, Ulianich L, Treglia AS, Urbanas ML, High S, Arvan P. Mixed-disulfide folding intermediates between thyroglobulin and endoplasmic reticulum resident oxidoreductases ERp57 and protein disulfide isomerase. Mol Cell Biol. 2005;25:9793–9805. doi: 10.1128/MCB.25.22.9793-9805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguay AR, Silhavy TJ. Quality control in the bacterial periplasm. Biochim Biophys Acta. 2004;1694:121–134. doi: 10.1016/j.bbamcr.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci USA. 2008;105:11933–11938. doi: 10.1073/pnas.0804621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis HL. Polysome metabolism in Escherichia coli: effect of antibiotics on polysome stability. Antimicrob Agents Chemother. 1972;1:197–203. doi: 10.1128/aac.1.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MS, Sander IM, Clark PL. Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo. J Mol Biol. 2008;383:683–692. doi: 10.1016/j.jmb.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov AN, Baldwin TO. Cotranslational protein folding. J Biol Chem. 1997;272:32715–32718. doi: 10.1074/jbc.272.52.32715. [DOI] [PubMed] [Google Scholar]
- Frech C, Wunderlich M, Glockshuber R, Schmid FX. Preferential binding of an unfolded protein to DsbA. EMBO J. 1996;15:392–398. [PMC free article] [PubMed] [Google Scholar]
- Harms N, Koningstein G, Dontje W, Muller M, Oudega B, Luirink J, de Cock H. The early interaction of the outer membrane protein phoe with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J Biol Chem. 2001;276:18804–18811. doi: 10.1074/jbc.M011194200. [DOI] [PubMed] [Google Scholar]
- Hiniker A, Bardwell JC. In vivo substrate specificity of periplasmic disulfide oxidoreductases. J Biol Chem. 2004;279:12967–12973. doi: 10.1074/jbc.M311391200. [DOI] [PubMed] [Google Scholar]
- Huber D, Boyd D, Xia Y, Olma MH, Gerstein M, Beckwith J. Use of thioredoxin as a reporter to identify a subset of Escherichia coli signal sequences that promote signal recognition particle-dependent translocation. J Bacteriol. 2005;187:2983–2991. doi: 10.1128/JB.187.9.2983-2991.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Bassford PJ, Jr, Beckwith J. Protein localization in E. coli: is there a common step in the secretion of periplasmic and outer-membrane proteins? Cell. 1981;24:707–717. doi: 10.1016/0092-8674(81)90097-0. [DOI] [PubMed] [Google Scholar]
- Jansens A, van Duijn E, Braakman I. Coordinated nonvectorial folding in a newly synthesized multidomain protein. Science. 2002;298:2401–2403. doi: 10.1126/science.1078376. [DOI] [PubMed] [Google Scholar]
- Josefsson LG, Randall LL. Different exported proteins in E. coli show differences in the temporal mode of processing in vivo. Cell. 1981;25:151–157. doi: 10.1016/0092-8674(81)90239-7. [DOI] [PubMed] [Google Scholar]
- Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes. Annu Rev Biochem. 2003;72:111–135. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science. 2004;303:534–537. doi: 10.1126/science.1091724. [DOI] [PubMed] [Google Scholar]
- Kamitani S, Akiyama Y, Ito K. Identification and characterization of an Escherichia coli gene required for the formation of correctly folded alkaline phosphatase, a periplasmic enzyme. EMBO J. 1992;11:57–62. doi: 10.1002/j.1460-2075.1992.tb05027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GP, Topping TB, Cover WH, Randall LL. Retardation of folding as a possible means of suppression of a mutation in the leader sequence of an exported protein. J Biol Chem. 1988;263:14790–14793. [PubMed] [Google Scholar]
- Matsuyama S, Fujita Y, Sagara K, Mizushima S. Overproduction, purification and characterization of SecD and SecF, integral membrane components of the protein translocation machinery of Escherichia coli. Biochim Biophys Acta. 1992;1122:77–84. doi: 10.1016/0167-4838(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Messens J, Collet JF, Van Belle K, Brosens E, Loris R, Wyns L. The oxidase DsbA folds a protein with a nonconsecutive disulfide. J Biol Chem. 2007;282:31302–31307. doi: 10.1074/jbc.M705236200. [DOI] [PubMed] [Google Scholar]
- Molinari M, Helenius A. Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature. 1999;402:90–93. doi: 10.1038/47062. [DOI] [PubMed] [Google Scholar]
- Oliver DB, Cabelli RJ, Dolan KM, Jarosik GP. Azide-resistant mutants of Escherichia coli alter the SecA protein, an azide-sensitive component of the protein export machinery. Proc Natl Acad Sci USA. 1990;87:8227–8231. doi: 10.1073/pnas.87.21.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne AR, Rapoport TA. Protein translocation is mediated by oligomers of the SecY complex with one SecY copy forming the channel. Cell. 2007;129:97–110. doi: 10.1016/j.cell.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Powers T, Walter P. Co-translational protein targeting catalyzed by the Escherichia coli signal recognition particle and its receptor. EMBO J. 1997;16:4880–4886. doi: 10.1093/emboj/16.16.4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley AP. The complete general secretory pathway in gram-negative bacteria. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowska A, Mayer MP, Hoffmann A, Merz F, Zachmann-Brand B, Schaffitzel C, Ban N, Deuerling E, Bukau B. Dynamics of trigger factor interaction with translating ribosomes. J Biol Chem. 2008;283:4124–4132. doi: 10.1074/jbc.M708294200. [DOI] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA. Conservation and diversity of the cellular disulfide bond formation pathways. Antioxid Redox Signal. 2006;8:797–811. doi: 10.1089/ars.2006.8.797. [DOI] [PubMed] [Google Scholar]
- Sone M, Akiyama Y, Ito K. Differential in vivo roles played by DsbA and DsbC in the formation of protein disulfide bonds. J Biol Chem. 1997a;272:103490–10352. doi: 10.1074/jbc.272.16.10349. [DOI] [PubMed] [Google Scholar]
- Sone M, Kishigami S, Yoshihisa T, Ito K. Roles of disulfide bonds in bacterial alkaline phosphatase. J Biol Chem. 1997b;272:6174–6178. doi: 10.1074/jbc.272.10.6174. [DOI] [PubMed] [Google Scholar]
- Tian P, Bernstein HD. Identification of a post-targeting step required for efficient cotranslational translocation of proteins across the Escherichia coli inner membrane. J Biol Chem. 2009;284:11396–11404. doi: 10.1074/jbc.M900375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ureta AR, Endres RG, Wingreen NS, Silhavy TJ. Kinetic analysis of the assembly of the outer membrane protein LamB in Escherichia coli mutants each lacking a secretion or targeting factor in a different cellular compartment. J Bacteriol. 2007;189:446–454. doi: 10.1128/JB.01103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W, Schekman R. Protein translocation across biological membranes. Science. 2005;310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.