

Preface
Given the recent explosion of genetic discoveries, 2007 is becoming known to human geneticists as the ‘year of genome-wide association studies’. In fact, more genetic risk factors for common diseases were identified in one year than had been collectively reported prior to 2007. In particular, this year witnessed the uncovering of many susceptibility genes that influence the susceptibility to individual immune-mediated diseases, and others that are shared across more than one disease. Although much work remains to be done, we discuss what effect these studies are having on our understanding of disease pathogenesis and their potential impact on future immunology studies.
Although the field of human genetics has been a promising approach to identify the key pathways that lead to immune-mediated diseases, most of the discoveries in this area have been limited until recently to disorders caused by deleterious mutations in single genes 1. In monogenic diseases, genomic regions that contain the causal gene can be identified using family-based linkage studies owing to the strong penetrance of these mutations. When similar study designs were used to identify genes involved in the predisposition to more common immune-mediated diseases such as Crohn's disease, multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus (SLE) and type one diabetes (T1D), success was limited. These difficulties were encountered based on the fact that such diseases are due to multiple different genetic risk factors, and that non-genetic risk factors can also play a role. Consequently, in common diseases, each individual risk factor confers a more modest risk that what is observed in monogenic disorders.
Powerful association-based approaches test more directly the causal genetic variation than linkage analyses2, and are consequently more useful for detecting the individual risk factors that confer only modest risk. Initially, association-based genetic studies were generally limited to the testing of modestly sized cohorts of patients with a small number of genetic variants in one gene or a small number of genes. Unfortunately, this primarily led to a very large number of false positive reports 3. This outcome was not that surprising given the low likelihood that an investigator could select the few causal genes from human genome, even armed with the best knowledge of disease pathogenesis.
The key to improving this approach was to probe the entire genome, as had been done in linkage analysis, and to combine the statistical power of an association study. In contrast to a typical genome-wide linkage scan that analyses a relatively small number (300-5000) of genetic markers, it was predicted that a GWAS would probably require typing ∼500,000 genetic markers known as single nucleotide polymorphisms (SNPs), owing to the low level of linkage disequilibrium expected for the human genome 4. However, at the time this prediction was made, the most comprehensive SNP map of the human genome only contained ∼2000 SNPs 5. Since then, substantial efforts have been made to catalogue and map the correlation patterns of SNPs (known as haplotypes) in order to facilitate genetic studies of human disease. Specifically, the International HapMap Project has catalogued the haplotype patterns across the human genome by analysing over 3 million SNPs, and has created a public resource in order that researchers can gain easy access to the data related to a gene of interest or for the entire genome6. In addition, while these efforts were ongoing, there was a commercial effort to develop platforms to allow the testing of hundreds of thousands SNPs in a single genotyping reaction. These technological platforms have progressed over time and now have the ability to type ∼1,000,000 SNPs in parallel.
These advances have enabled multiple genome-wide association studies (GWAS) of immune-mediated diseases in moderately sized cohorts. Specifically, in the last 12–18 months, numerous studies of 500–2000 cases and equivalent numbers of unrelated control subjects have been published for many of the diseases mentioned above. Have these GWAS led to new discoveries as hoped? The answer to that question is emerging as a resounding ‘yes’. The GWAS performed to date have revealed new pathways that contribute to disease development and, in addition, have identified common genes among immune-mediated diseases that had not previously been identified. This Review aims to provide information that will allow immunologists to understand the rapid conceptual and technological changes that have enabled GWAS, and to provide some of the tools necessary to overcome the challenges and to take advantage of the strengths of these types of studies. Furthermore, we attempt to provide some insight into how these discoveries will influence the path of both basic and clinical immunology research.
Understanding GWAS
In order to interpret the results of GWAS that investigate common diseases, it is important to have an understanding of the underlying genomic context. The human genome consists of ∼3 billion base pairs, and roughly 10 million positions in this genetic code are polymorphic in the human population at a significant frequency 7. It is these sequence differences that result in such phenotypic differences as height, skin colour, and differential resistance to infection or disease. Some of this variation will be neutral, some advantageous and some disadvantageous advantageous to an individual's health — and this can be context dependent. Such genetic variation can therefore be considered part of the normal variation that occurs within a species. The challenge is then to determine, in a time- and cost-efficient manner, which of the common variants contribute to disease susceptibility.
Although testing all known genetic variants in an association study would be the most direct approach, this currently is too costly and is not necessary given the structure of genetic variation in the human genome. Specifically, the information generated by the International HapMap Project details the correlation pattern between the SNPs in the human genome. In the cases where two or more SNPs are highly correlated with one another it is only necessary to test one of the SNPs as it will provide equivalent genetic information on the other SNPs that are part of the correlated group 6, 8. GWAS are therefore based on the concept that, by testing a large number of SNPs, it will be possible to detect disease-causing alleles, either by testing the actual causal variant or a SNP that is correlated with the causal variant.
Commercial genotyping chips have progressively increased the number of SNPs that can be typed in a single experiment (BOX 1). Moreover, these highly parallel genotyping platforms have also begun to incorporate structural variants known as copy number variants 9. Testing such a large number of variants can give rise to a large number of false positives, and thus it is crucial to conduct a sufficiently-powered study and to perform very rigorous quality control analyses on the genotype data prior to association testing (BOX 2) 10-12. In addition, it is important to perform replication studies in order to sort out the false positives from the true positives 13. The results of the replication study should be consistent (that is, with repeated associations to the same allele found) and reported for the same phenotypes in order to constitute true validation 13. One of the first successful GWAS, in which ∼100,000 SNPs in 96 cases and 50 control subjects were typed, identified a polymorphism in the gene encoding complement factor H which confers a risk for age-related macular degeneration 14. Since then, there have been numerous studies performed to identify risk factors for celiac disease, Crohn's disease, multiple sclerosis, rheumatoid arthritis, SLE, T1D and many other complex immune-mediated diseases (Table 1) 15-25.
BOX 1.
GWAS genotyping platforms
In recent years, two technologies have shared the market of genome-wide association studies (GWAS): Affymetrix GeneChips and Illumina BeadChips. Both platforms use the same basic principle of hybridization of genomic DNA fragments to a fixed probe. In the case of the Affymetrix Genechip, labelled DNA is hybridized to millions of probes synthesized on a 1.5 cm by 1.5 cm glass plate. For the Illumina Infinium Assay, Genomic DNA is hybridized to probes fixed on 3-micron silica beads that self assemble in microwells on planar silica slides. Allelic specificity is conferred by enzymatic-based extension and the labelling of the additional nucleotide. These microarray technologies require from 250ng to 750ng of genomic DNA per sample, and one sample is simultaneously tested for all markers. The Affymetrix standard workflow is five days, whereas the Illumina workflow is three days.
Since the beginning of the GWAS using single nucleotide polymorphisms (SNPs) in 2004, Affymetrix and later, Illumina, developed numerous chips that are increasingly powerful for capturing genomic variability (see Table for general specifications of chips). The latest chips contain probes for ∼1 million SNPs. SNP selection methods have also evolved in recent years. The earlier Affymetrix chips used unbiased SNP selection, targeting SNPs at regular intervals to cover the entire genome. Illumina. More recently, Affymetrix (Genechip 6.0) used the development of the HapMap database to select tag SNPs in order to maximize genomic coverage of the newest arrays. In addition, more recent chips have included other markers of genomic variation such as mitochondrial SNPs and copy number variant (CNV) probes.
Overview of genome-wide association microarray genotyping platforms.
| Affymetrix Genechip Human Mapping 100K | Affymetrix Genechip Human Mapping 500K | Affymetrix Genome-Wide Human SNP Array 6.0 | Illumina HumanHap300 | Illumina HumanHap550** | Illumina Human1M | |
|---|---|---|---|---|---|---|
| Autosomal SNPs | 116,204 | 500,568 | 906,600 | 318,237 | 561,466 | 1,072,820 |
| GC* CEU | n/a | 0.67 | 0.82 | 0.81 | 0.9 | 0.94 |
| GC* CHB+JPT | n/a | 0.64 | 0.82 | 0.68 | 0.86 | 0.92 |
| GC* YRI | n/a | 0.68 | 0.65 | 0.34 | 0.57 | 0.73 |
| Mitochondrial SNPs | 0 | 0 | 465 | 0 | 163 | 163 |
| CNVs (number of probes) | 0 | 0 | 946,000 | 54,480 | 98,656 | 206,665 |
| Number of CNV regions covered | 0 | 0 | 5,677 | 2,735 | 2,991 | 3,293 |
n/a= information not available
genomic coverage expressed in terms of fraction of SNPs (HapMap Phase 2) captured at a coefficient of determination (r2) >0.8
CEU= HapMap CEPH UTAH population
CHB+JPT= HapMap Chinese and Japanese populations
YRI= HapMap Yoruba population
An updated version of that chip called HumanHap650Y that contains additional SNPs selected to tag variation found in the YRI population
BOX 2.
Statistical Analyses of GWAS Data
Association studies are increasingly carried out in the context of unbiased genome-wide approaches. The technologies used, such as microarrays, allow for numerous assays to be carried out simultaneously. A fraction of the SNPs will perform suboptimally under these uniform conditions and can generate unreliable data. Therefore, steps of quality control and replication are used to ensure the validity of the results. The figure below illustrates the steps involved in a typical association study, using as an example the Wellcome Trust Case Control Consortium (WTCCC) genome-wide association studies (GWAS) of seven complex diseases and the following replication for the Crohn's disease-associated single nucleotide polymorphisms (SNPs) 16, 19.
The first step, the screen, is performed on a first cohort of cases and matched controls and/or family-based trios. This initial genotyping is followed by careful quality control steps to identify problematic SNPs (e.g. SNPs not in Hardy-Weinberg equilibrium) or DNAs (e.g. DNAs that appear to be contaminated by other samples). Low-quality markers and samples are rejected. The association tests are then performed on the clean dataset. In addition, poor matchin between cases and controls or the presence of genetically distinct subpopulations among the studied samples — a phenomenon called stratification — can give rise to false association signals. Stratification must be tested, and genomic control markers can then be used to calculate a correction factor that is applied to the association results.
Following the screen, a threshold of significance is chosen for the inclusion of markers in the replication step (Step 2). The replication markers are typed in a second, independent cohort. A quality control step is also applied to these data and appropriate correction factors are applied to the results. A true association signal is confirmed by the detection of a significant association in both screening and replication steps. Furthermore, this association has to be in the same direction, that is, the same allele has to be associated in both steps. Given the large number of hypotheses tested in GWAS (each SNP tests a different hypothesis), a stringent threshold is necessary to minimize the number of false positives reported in a GWAS, and the significance threshold of 5×10-7 is becoming broadly accepted. 16
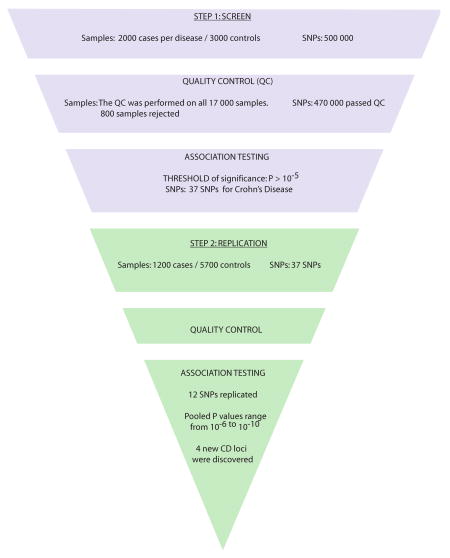
Table 1. Summary of recent GWAS and follow-up studies in immune-mediated diseases showing shared and disease-specific genetic risk factors.
| Gene/Region | Gene Name | Proposed Function | Disease | Approach | OR | CHR | Variation | RAF | Association with other Dx | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| NOD2 | Caspase-recruitment domain 15 | Sensing of cytosolic bacterial components | CD | AM/CG | 3.00 | 16q12 | R675W G881R 980fs | 0.11 0.02 0.01 | 28, 29 | |
| IBD5 | N/A | unknown | CD | AM | 1.30 | 5q31 | L503F | 0.46 | SLE | 88,30 |
| 10q21 locus | N/A | unknown | CD | GWAS | 0.60 | 10q21 | SNP | 0.13 | 20 | |
| PTPN2 | protein tyrosine phosphatase, non-receptor type 2 | TCR signalling | CD | GWAS | 1.30 | 18p11 | SNP | 0.19 | GD, T1D | 16, 89 |
| IL23R | Interleukin-23 receptor | generating & maintaining TH17 cells | CD | GWAS | 0.26 | 1p31 | R381Q | 0.07 | PSO | 18, 48 |
| ATG16L1 | autophagy 16-like 1 | Autophagy | CD | GWAS | 0.68 | 2q37 | T216A | 0.46 | 20, 36 | |
| NCF4 | neutrophil cytosolic factor 4 | Production of reactive oxygen species | CD | GWAS | 1.19 | 22q12 | SNP | 0.27 | 20 | |
| PTGER4 | prostaglandin E receptor 4 | Prostaglandin signalling | CD | GWAS | 1.33 | 5p13 | SNP | 0.66 | 15 | |
| IRGM | immunity-related GTPase family member M | Autophagy | CD | GWAS | 1.54 | 5q33 | SNP | 0.03 | 16, 19 | |
| 3p21 haplotype | N/A | See MST1 | CD | GWAS | 1.09 | 3p21 | multiple | 0.24 | 16 | |
| MST1 | Macrophage stimulating 1 | Regulates production of pro-inflammatory mediators | IBD | AM/CG | 1.30 | 3p21 | R689C | 0.23 | 46 | |
| IL2-IL21 locus | Interleukin-2 and -21 | Regulation of T-cell subsets | CeD | GWAS | 0.63 | 4q27 | SNP | 0.20 | MS, RA, T1D | 16,24,21 |
| IL2Ra | IL-2 receptor alpha chain | Regulatory T-cell function | MS | GWAS | 1.35 | 10p15 | SNP | 0.84 | GD, T1D | 16,21,90 |
| IL7Ra | IL-7 receptor alpha chain | Homoeostasis of memory T cells | MS | GWAS | 1.24 | 5p13 | T244I | 0.76 | 21 | |
| STAT4 | Signal transducer and activator of transcription 4 | Development of T-cell responses | RA | CG | 1.32 | 2q32 | SNP | 0.21 | SLE | 62, 66 |
| TRAF1-C5 locus | TNF receptor associated factor/complement component 5 | Mediates activation of NF-κB/mediates local inflammation | RA | GWAS | 1.32 | 9q33 | SNP | 0.48 | 22 | |
| TNFAIP3 | TNF alpha-induced protein 3 | TNF receptor signalling | RA | GWAS | 1.20 | 6q23 | SNP | 0.18 | 57 | |
| IRF5 | Interferon regulatory factor 5 | Transcriptional regulator of pro-inflammatory cytokines | SLE | CG | 1.60 | 7q32 | SNP | 0.48 | 91 | |
| TNFSF4 | TNF superfamily member 4 | Co-stimulation of activated CD4+ Tcells | SLE | CG | 1.28 | 1q25 | SNP | 0.20 | 67 | |
| BANK1 | B-cell scaffold protein with ankyrin repeats | BCR signalling | SLE | CG | 1.38 | 4q24 | R61H | 0.75 | 65 | |
| ITGAM | Integrin alpha-M | Clearance of immune complexes | SLE | GWAS | 1.70 | 16p11 | SNP | 0.11 | 66 | |
| FCGR2a | Low affinity receptor IIa for immunoglobulin Fc fragment | Clearance of immune complexes | SLE | GWAS | 0.74 | 1q23 | SNP | 0.43 | 66 | |
| BLK locus | B lymphocyte specific tyrosine kinase | B cell activation | SLE | GWAS | 1.39 | 8p23 | SNP | 0.25 | 25 | |
| PTPN22 | protein tyrosine phosphatase, non-receptor type 22 | TCR signalling | T1D | CG | 1.28 | 1p13 | R620W | 0.14 | GD, RA, SLE | 16, 66,85,92 |
| SH2B3 | SH2B adaptor protein 3 | TCR signalling | T1D | GWAS | 1.35 | 12q24 | R262W | 0.54 | 23 | |
| KIAA0350/CLEC16A | C-type lectin domain family 16, member A | unknown | T1D | GWAS | 1.19 | 16p13 | SNP | 0.71 | 23, 93 | |
| IFIH1 | Interferon induced with helicase C domain 1 | Receptor for viral dsRNA | T1D | GWAS | 0.86 | 2q24 | A946T | 0.37 | GD, RA | 16,94,95 |
This is a partial list of associations identified in recent GWAS which illustrates the common and disease-specific nature of genetic risk factors for immune-mediated diseases. CD, Crohn's Disease; CeD, Celiac Disease; GD, Grave's Disease; MS, Multiple Sclerosis; PSO, Psoriasis; RA, Rheumatoid Arthritis; SLE, Systemic Lupus Erythematosus; T1D, Type 1 Diabetes; IBD, Inflammatory Bowel Disease; AM, association mapping; CG, candidate gene association; GWAS, Genome-Wide Association Study; OR, Odds Ratio; RAF, Risk Allele Frequency in HapMap-European (CEU) population; TNF, tumour-necrosis factor; TCR, T-cell receptor.
The unbiased nature of GWAS is evident from some of the gene discoveries summarized in Table 1. For example, two genes that have recently been shown to have a role in autophagy were identified by GWAS to influence susceptibility to Crohn's disease 19, 20, although a role for autophagy in Crohn's disease had not been previously appreciated. In addition, it is exciting that comparing the results of one disease to those of another can reveal risk factors that influence susceptibility to more than one disease (Table 1). A striking example of this is the finding that a ∼500 kb genomic region on chromosome 4q27, that contains genes encoding interleukin-2 (IL-2) and IL-21, is associated with susceptibility to celiac disease, multiple sclerosis, rheumatoid arthritis, and T1D 24, 26.
It should be noted, however, that even when statistically significant associations are identified, that GWAS do not always identify the causal gene within an associated region. This might be due to the fact that the associated region contains more that one gene (as in the example above) or that it does not contain any known coding sequence (also known as a gene desert). An example of a gene desert is the association of Crohn's disease with a region of the chromosome 5p13 15 — although strongly associated with disease susceptibility this genomic region has no known genes. Further investigation led researchers to propose that the underlying mechanism of action of the associated region in Crohn's disease was through differential expression of the nearby prostaglandin E receptor 4 (PTGER4) gene. These results indicate that functional studies are often necessary to elucidate the biological mechanisms by which an associated region confers susceptibility15. It should also be noted, that, even when the association and linkage disequilibrium results point to a single causal gene, the casual sequence variant is not always easy to identify (TABLE 1).
In summary, not all identified and confirmed associations lead to the immediate identification of the causal gene and/or the causal variant. One feature that is true for all variants identified to date for common diseases is that the risk imparted by each of them is relatively modest — odds ratios usually range from 1.1 to 1.4 (Table 1). Analogous to the situation for non-genetic risk factors (e.g. the contribution of smoking to lung cancer risk), no single genetic risk factor can determine whether an individual will develop a disease or not; each variant can only modulate an individual's probability to developing disease. Furthermore, although the first GWAS studies have greatly increased the number of known genetic risk factors (for example, studies of Crohn's disease have increased the number of known risk factors from two genes in 2001 to eleven genes in 2008), it is important to recognize that many more remain unknown. Despite this incomplete picture, these discoveries have uncovered many new pathways that contribute to disease pathogenesis.
GWAS have revealed new pathways to disease
Chronic immune-mediated diseases affect approximately 3-5% of the population worldwide27. Most of the current therapies for such diseases rely on non-specific immunosuppressive approaches, in part due to our relative lack of knowledge of the specific mechanisms that cause disease onset and progression. Excitingly, the recent explosion in GWAS has had a dramatic effect on our ability to catalogue the true genetic risk factors for many of these diseases. Although we are still in the very early stages of GWAS, we highlight in the following sections some of the key findings that this approach has revealed in investigations of Crohn's disease, rheumatoid arthritis, SLE and multiple sclerosis genetics and discuss how these studies have shed light on new pathways that might be potential therapeutic targets.
Crohn's Disease
Crohn's disease and ulcerative colitis are debilitating, inflammatory diseases of the gastrointestinal tract, collectively known as the inflammatory bowel diseases (IBDs). Genetic studies have been particularly successful in the identification of genes for Crohn's disease. In 2001, one of the first successful discoveries of a causal gene for a complex trait was the identification of three genetic variants in the nucleotide-binding oligomerization domain protein 2 (NOD2) gene that were associated with Crohn's disease 28, 29. A second discovery came just a few months later with the identification of a haplotype, known as IBD5, of multiple genetic variants in a region containing five genes: interferon regulatory factor 1 (IRF1), solute carrier family members SLC22A5 and SLC22A4, PDZ- and LIM-containing protein 4 (PDLIM4) and prolyl-4 hydroxylase (P4HA2) 30. These discoveries were identified using the traditional approach of genome-wide linkage followed by association mapping of linked regions that had been so successful in the study of single gene disorders.
Although the difficulties encountered in identification of the causal gene for the IBD5 haplotype did foreshadow some of the challenges that lay ahead, it was widely felt that association studies that targeted linkage regions or candidate genes would continue to reveal the key pathways that could influence susceptibility to Crohn's disease. However, in the following five years, no further genes linked to Crohn's disease were identified. During that period, functional studies of NOD2 clearly indicated a central role for this protein in mediating interactions between microbes and the intestinal mucosa, and indicated that causal alleles might shift the balance such that minimally invasive bacteria could trigger the onset of Crohn's disease 31-33. These bacteria harbor various virulence factors involved in adhesion to and invasion of intestinal epithelial cultured cells.
The first four GWAS studies that investigated Crohn's disease using 300,000 or more SNPs were performed by the North American National Institute of Diabetes, Digestive and Kidney diseases (NIDDK) IBD Genetics Consortium, a Belgian–French group and the Wellcome Trust Case Control Consortium (WTCCC), and a total of eight novel genetic risk factors were identified (TABLE 1) 15, 16, 18, 20. This windfall in the discovery of multiple disease genes represented more than had previously been identified for any other immune-mediated diseases and had a dramatic effect on our understanding of the different pathways that are involved in IBD susceptibility and/or pathogenesis.
One of the most unexpected findings that have come from the Crohn's disease GWAS is that autophagy has an important role in disease pathogenesis. This is supported by the finding that two genes involved in autophagic processes — autophagy 16-like 1 (ATG16L1) and immunity-related GTPase family member M (IRGM) — were significantly associated with disease in GWAS studies 19, 20. Recent studies have demonstrated broad roles for autophagy in tumour suppression, organ development, neurodegeneration, innate immune responses, antigen presentation, T-cell homeostasis and in the clearance of intracellular microbes 34. In yeast, more that 30 genes required for autophagy have been identified, and the molecular cascade that regulates autophagy has recently been comprehensively reviewed 35.
The NIDDK study identified a non-synonymous amino acid change (Ala197Thr) in ATG16L1 that was associated with Crohn's disease, a finding that was reported in an independent association study of non-synonymous coding SNPs 20, 36. The NIDDK study suggested that the mechanism by which ATG16L1 genetic variants play a role in Crohn's disease pathogenesis might be through alterations in the sensing and clearance of gut microflora. Specifically, knock-down experiments in human cervical carcinoma cells (HeLa) indicated that ATG16L1 is essential for normal autophagosome formation following exposure to stimuli known to trigger autophagy, such as serum starvation, rapamycin or intracellular bacterial infection 20. Aside from this single study, very little is known about the role of ATG16L1 in autophagy and other cellular processes. ATG16L1 has been shown to form a trimolecular protein complex with ATG5 and ATG12 and, given that ATG5 is known to be required for lymphocyte development and signalling and especially for the maintenance of innate immune B1 B cells in peripheral tissues, it is probable that ATG16L1 has multiple functions in the immune system 6, 37. In addition to defense against pathogens, autophagic vacuoles might allow some bacterial and viral pathogens to establish a replicative niche and supply them with nutrients for growth38. As both excessive and defective autophagy might have deleterious effects, future studies should focus on the functional consequences of Crohn's disease-associated ATG16L1 variants in a cell- and tissue-specific context.
Currently even less is known about IRGM, another autophagy-related gene that was associated with Crohn's disease in the WTCCC study16. The authors were able to localize the association to the IRGM gene but, in the re-sequencing analyses, they were unable to identify the causal variant in patients 19. In humans, the IRG family consists of two members, IRGM and IRGC. In contrast to the structural diversity of IRG proteins in mice, the human protein lacks most of the N- and C-terminal domains that flank the GTP-binding domain, and the promoter lacks an interferon (IFN) response signature found in mouse IRG proteins39. Human IRGM has many splice variants, however the cellular distribution and biological function are unknown40, however, studies that demonstrate a role for human IRGM in the resistance to mycobacterial infection, indicate that the human protein probably also has an immune function. Although its exact role in Crohn's disease pathogenesis remains to be determined, it is likely that, together with ATG16L1, IRGM influences the host immune response to the gut microflora (FIG. 1a) 41.
Figure 1. Schematic representation of cell-specific signalling pathways mediated by Crohn's disease susceptibility genes.
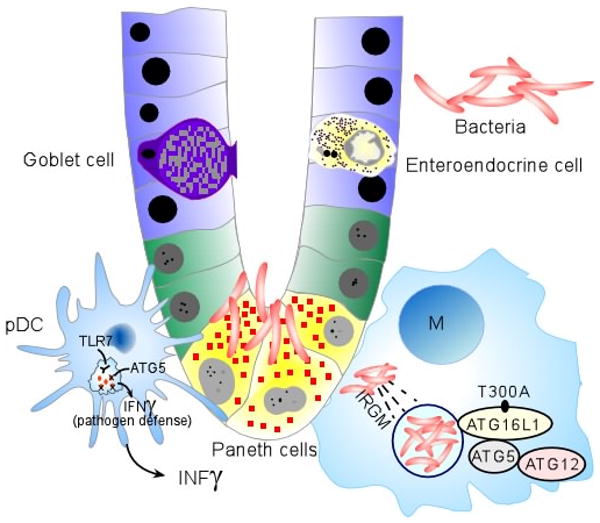
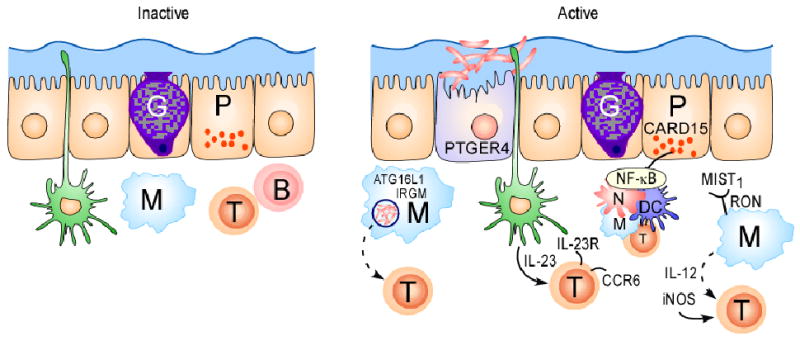
(a) Under homeostatic conditions, the mucus layer and tight junctions associated with intestinal epithelial cells maintain barrier integrity. A dynamic balance between host defence immune responses and luminal enteric bacteria at the mucosal frontier is central to the pathogenesis of Crohn's disease. Recent genetics studies have implicated autophagy, defects in innate immunity and imbalance between pathogenic and regulatory T cell populations as risk factors for Crohn's disease. Dendritic cells (DCs) extend dendrites between epithelial cells and can sense changes in luminal contents. Potential signalling pathways in inflammation, and the respective genes involved, are represented. Caspase-recruitment domain 15 (NOD2) has a role in the release of antimicrobial peptides and in sensing cytosolic microbial ligands; prostaglandin E receptor 4 (PTGER4) is involved in promoting epithelial restitution; the interleukin-23 receptor (IL23R) has a role in generating and maintaining of TH17 cells; autophagy 16-like 1 (ATG16L1) and immunity-related GTPase family member M (IRGM) are involved in and microbial defence and adaptive immunity; macrophage-stimulating factor 1 (MST1) has a role in down regulating inflammatory response.
In addition to the data suggesting that aberrant autophagic processes contribute to Crohn's disease through abnormal handling of gut microflora, multiple genes identified in GWAS of Crohn's disease have further suggested that dysregulation of the innate immune system could have a dominant role in determining disease susceptibility. For example, the significant association of the neutrophil cytosolic factor 4 (NCF4) gene found in the NIDDK study supports this theory. NCF4 encodes the p40PHOX protein that has previously been shown to have an important role in NADPH oxidase activity and the production of reactive oxygen species (ROS) following phagocytosis, both of which are important for mounting effective antimicrobial immune responses 20, 42, 43. Moreover, the WTCCC study identified a region of ∼1Mb on chromosome 3p that contains multiple genes. This region was contained within the IBD9 locus (which spans ∼25 Mb) that was identified in early linkage studies of IBD 44, 45. Although the WTCCC study did not identify the causal gene in this region, a more recent study found a coding variant (R689C) in the macrophage stimulating 1 (MST1) gene (which encodes the macrophage-stimulating protein (MSP)) that might explain the WTCCC results 45, 46. It has been suggested that the R689C variant might influence the production of pro-inflammatory mediators such as nitric oxide, prostaglandin E2 (PGE2) and interleukin-12B (IL-12p40) in response to lipopolysaccharide (LPS) or cytokine-mediated macrophage activation (FIG. 1b) 46. Interestingly, results of another recent independent study indicate that variation in the gene encoding the receptor for MSP might also influence susceptibility to Crohn's disease 47.
It is tempting to conclude from these studies that Crohn's disease is uniquely due to disturbances in key pathways of the innate immune system; however, the adaptive immune system clearly also has an important role in disease susceptibility and/or pathogenesis (. The most striking example is that of IL-17-producing CD4+ T helper cells (TH17 cells). The first genetic evidence suggesting that TH17 cells are involved in human inflammatory diseases came from the report of an association of IL23R in the NIDDK GWAS of Crohn's disease 18. It is thought that IL-23 is an important growth-factor for TH17 cells, and the NIDDK study demonstrated that the Glu allele at residue 381 in the IL-23R protein conferred significant protection against developing Crohn's disease. The more common allele (which occurs at a frequency of ∼95%) at residue 381 is an Arg, which is highly conserved across multiple species including the opposum 18. Although functional studies aimed at determining the mechanistic impact of the glutamine at residue 381 are ongoing, it is interesting to note that this genetic variant also showed a similar protective effects in a psoriasis cohort, consistent with the clinical observations that psoriasis and Crohn's disease are co-segregated with high frequency 48.
IL-23 is a heterodimeric cytokine comprised of linked p19 and p40 subunits and is produced by a broad array of cells including dendritic cells (DCs) and macrophages 49. Many microbial signals, such as Toll-like receptor (TLR) ligands, LPS, and the synthetic RNA mimic polyIC, enhance the expression of IL-23 subunits, resulting in bioactive production of this cytokine in the setting of mucosal disruption. In response to IL-6 and tumor growth factor beta (TGFβ), naïve CD4+ T cells upregulate IL-23R, providing a positive autocrine signal for the expansion of TH17 cells. However, little is known so far about the IL-23R signalling pathways that control human TH17-cell differentiation and function in models of autoimmunity 50. A recent report by Annunziato et al. demonstrated that two populations of TH17 cells are found in patients with Crohn's disease — one subset is characterized by the expression of chemokine receptor 6 (CCR6), IL-23R and retinoic-acid-receptor-related orphan receptor γ (RORγT), a transcription factor important for TH17-cell differentiation, and the second subset by the production of IFNγ 51.
It is noteworthy that CCR6-expressing TH17 cells are recruited to inflamed joints in rheumatoid arthritis through chemokine beta family ligand 20 (CCL20) production and contribute to disease pathogenesis, although there is currently no evidence for a genetic association of the genes involved in TH17-cell biological pathways with rheumatoid arthritis 52. Future studies should aim to elucidate the precise functional deficits associated with the heterogeneous populations of TH17 cells in the pathogenesis of Crohn's disease and other immune-mediated diseases, and to address the functions of these cell populations in patients carrying genetic variants associated with disease susceptibility.
Multiple sclerosis
Multiple sclerosis is an autoimmune disorder characterized by inflammation, demyelination and axonal degeneration in the central nervous system. Decades of studying the cellular and molecular immunology of patients with multiple sclerosis and of experimental disease models indicate that cytokines and adaptive immune responses are involved in disease pathogenesis. Despite this knowledge, the only known genetic risk factor for MS prior to GWAS has been genes within the Major Histocompatibility Complex (MHC) 53. A recent GWAS study of patients with multiple sclerosis indeed demonstrated the existence of causal genetic variation in genes encoding cytokine receptors and molecules associated with adaptive immunity. Specifically, the first GWAS of multiple sclerosis, which was performed by the International MS Genetics Consortium (IMSGC) 21, revealed that the genetic locus with the strongest genetic effect in this study was, not surprisingly, the MHC. In addition, this study revealed that disease was also associated with the IL-7 receptor alpha chain gene (IL7Ra) and IL-2 receptor alpha chain (IL2Ra) genes. The reported association between IL7Ra and multiple sclerosis was supported by two other independent studies 54, 55. IL-7 is important for homeostasis of the memory T-cell pool and might be important for promoting the expansion of autoreactive T cells in patients with multiple sclerosis 21, 54, 55.
In one of these studies, the authors offered mechanistic insight into the function of the non-synonymous polymorphism T244L in IL7RA. In vitro studies indicated that differential splicing of IL-7RA changed the expression from the soluble form of the protein to the membrane-bound form, which could lead to compromised immune responses to multiple sclerosis-related antigens 54. In contrast, the causal variation in IL2Ra leading to MS has not yet been identified. It is notable, however, that IL2Ra is also associated with T1D, not only providing support to the concept that genetic risk factors can either be disease-specific or common to multiple related diseases but providing a rationale for examining functional consequences of associated genetic variants in different phenotypic contexts 21, 56. Taken together, these data support the long-held view that the regulation of the expression of interleukins is of central importance in multiple sclerosis pathogenesis. Importantly, these studies were successful for pinpointing the specific factors involved and provide hints that enhanced knowledge of alternative splicing events could be crucial to understanding immune-disease susceptibility.
Rheumatoid Arthritis
Rheumatoid arthritis is a common autoimmune disease characterized by chronic, destructive and debilitating inflammation of the joints. There have been two recent GWAS that have revealed that genomic regions on chromosomes 6q23 and 9q33.2 are specifically associated with rheumatoid arthritis 22, 57. Examination of the 6q23 locus revealed that the tumour-necrosis factor (TNF) alpha-induced protein 3 gene (TNFAIP3) is located within this associated region. This gene encodes a protein that acts as a checkpoint in TNF-induced nuclear factor-κB (NF-κB) activation that has both ubiquitin-ligase activity and de-ubiquitylating activity specific for molecules in TNF receptor signalling pathways16, 57. Although a causal variant that could formally implicate this gene in rheumatoid arthritis pathogenesis has not yet been identified, deep re-sequencing and subsequent association analyses will hopefully identify a causative non synonymous SNP or a regulatory SNP that alter the expression of TNFAIP3.
The associated SNP in the 9q33 region was found to be in linkage disequilibrium with two genes also relevant to chronic inflammation: TRAF1 (which encodes TNR receptor–associated factor 1) and C5 (which encodes complement component 5). Although complement activation in a chronic inflammatory disease such as rheumatoid arthritis is likely, it is probable that the causal gene within the TRAF1–C5 locus is TRAF1 as not only is TRAF1 believed to be a negative regulator of TNFR and T-cell receptor (TCR) signalling, it is also known to bind TNFAIP3 58, 59. Taken together, the implication of TNFAIP3 and TRAF1 as the likely causative genes in the 6q23 and 9q33 chromosomal regions support a major role for TNF-mediated pathogenic mechanisms, a hypothesis that is entirely consistent with the fact that anti-TNF biologics are currently the most effective therapies for RA60, 61.
In addition to identification of the above risk factors by GWAS, a recent candidate gene association study identified that genetic variation surrounding the signal transducer and activator of transcription 4 (STAT4) gene is also associated with rheumatoid arthritis 62. It has been shown that STAT4-dependent signalling downstream of the IL-12R has a crucial role in the development of TH1-cell responses. Based on these results, it is tempting to speculate that STAT4 polymorphisms associated with rheumatoid arthritis and SLE might result in enhanced IL-12-driven signalling, increased IFNγ levels and persistent inflammation.
Another gene that has been identified as a common risk factor for to multiple immune-mediated diseases including rheumatoid arthritis is protein tyrosine phosphatase, non-receptor type 22 (PTPN22), which has a function in regulating the signalling from the TCR and therefore important in dampening the activation signals initiated by ligand binding to cell surface TCR. The involvement of PTPN22 in multiple autoimmune diseases places this gene in the company of general autoimmunity genes such as CTLA4 & MHC (TABLE 1, BOX 3) 63. Taken together, the genes first discovered by either targeted or genome-wide association approaches support that T-cell signalling pathways have a predominant role in determining rheumatoid arthritis susceptibility (FIG. 2).
BOX 3.
The use of human cells to validate functional implications of polymorphisms
One of the major challenges ahead is to identify the functional consequences of the associated polymorphisms that are identified by genome-wide association studies (GWAS). An elegant example of how this has been accomplished is that of the protein tyrosine phosphatase, non-receptor type 22 (PTPN22) gene that is as been associated with susceptibility to rheumatoid arthritis, systemic lupus erythematosus (SLE), Grave's disease and type 1 diabetes (T1D) 85. PTPN22 encodes a phosphatase that is involved in T-cell receptor (TCR) signalling. Specifically, it is known that activation of the TCR initiates a complex intracellular signalling programme that offers both great sensitivity to antigen and multiple levels of regulatory control over the response to antigen. Following engagement of the TCR complex, protein tyrosine kinases become activated which then phosphorylate tyrosine-containing immunoreceptor tyrosine based activation motifs (ITAMs) in the cytoplasmic tails of TCR–CD3 chains. Phosphatases in turn dampen this response, providing a means of turning signalling on and off. The genetic variant associated with autoimmunity is a nucleotide change that results in a single amino acid substitution from Arg to Trp at position 620 (R620W). To gain mechanistic insight into the function of PTPN22 in autoimmunity, Rieck and colleagues investigated the function of lymphocytes isolated from patients who are heterozygous or homozygous for the R620W variant 86. Patients homozygous for the R620W variant showed a profound deficit in T-cell responses to antigen stimulation, and heterozygosity was associated with reduced responsiveness of memory CD4+ T cells following TCR stimulation. These results seem paradoxical, as autoimmunity seems more likely to arise from hyper-responsive rather than hypo-responsive T cells. However, it is known that the strength of the TCR stimulus is one mechanism by which autoreactive immune cells are selected for deletion during thymic development. Reduced receptor-mediated signals could impair normal T-cell selection and result in the survival of a population of autoreactive lymphocytes, or loss of the regulatory mechanisms by which such cells are normally held in check. This study underscores the importance of studying genetic variants within an affected population to yield data relevant to gene function within the disease state.
Figure 2. Interactions between innate immune cells and adaptive immune cells are central SLE and rheumatoid arthritis pathogenesis.
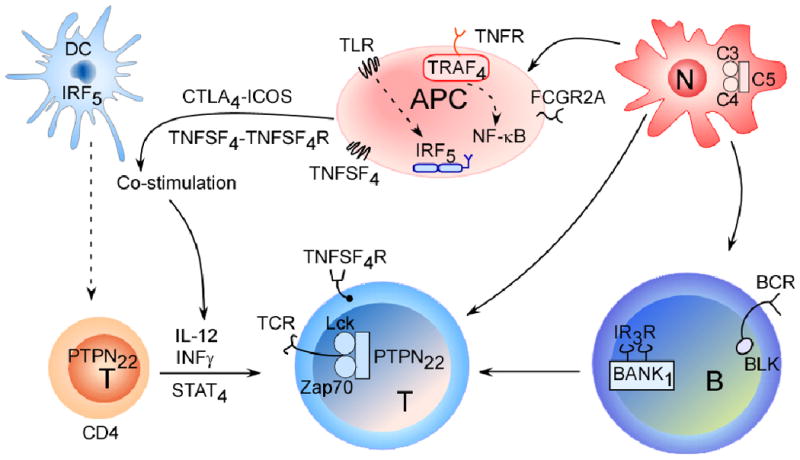
GWAS have highlighted both common and unique genetic variants that are associated with rheumatoid arthritis and systemic lupus erythematosus (SLE). The depicted cells and signalling pathways illustrate the multiple components of innate and adaptive immune responses that can contribute to autoimmune responses in these diseases. The proteins encoded by candidate genes associated with rheumatoid arthritis and SLE are depicted. Ultimately collaboration between different cell types initiates local and systemic inflammation. The complement cascade (involving complement component 5 (C5)), which influences both rheumatoid arthritis and SLE, reports danger signals to the surrounding mileu through neutrophil recruitment and activation. Antigen presenting cells are equipped to eliminate pathogens and provide cytokines and co-stimulatory cues (such as tumour-necrosis factor superfamily 4 (TNFSF4)) to T cells. Other adaptor proteins, such as tumour-necrosis factor receptor-associated factor 4 (TRAF4), and the activation of transcription factors such as signal transducer and activator of transcription 4 (STAT4) and interferon-regulatory factor 5 (IRF5), which is involved in type 1 interferon (IFN) production, direct the secretion of specific cytokines and chemokines. The involvement of autoantibodies (which are cell-derived) as well as type 1 IFNs in classic SLE demonstrate that disease pathology involves a dysfunction in both the innate and adaptive immune responses.
Systemic Lupus Erythematosus (SLE)
The first quarter of 2008 has witnessed an explosion in the number of genetic discoveries for SLE a result of the use of GWAS approaches. SLE is a prototypic, multisystem autoimmune disease primarily affecting women of child-bearing age, usually characterized by the loss of tolerance to nuclear antigens and the production of autoantibodies. The STAT4 and PTPN22 genes that had previously been identified as risk factors for SLE by targeted association studies were also identified in GWAS studies of patients with SLE (TABLE 1) 62, 64. The revelation of several other SLE-specific risk factors by recent GWAS and additional targeted studies provide compelling evidence that genetic variation in genes having key functions in the adaptive immune system are involved in the pathogenesis of the disease. 17, 25, 65-67.
Similar to some other autoimmune diseases, alleles of the exogenous peptide antigen presenting HLA class II proteins have long been associated with the risk of SLE 68. Gene expression studies and analysis of plasma cytokine profiles of patients with the disease have previously suggested a strong link between SLE pathogenesis and IFN signalling 69, 70. Therefore, the discovery of SNPs that affected alternative splicing and altered steady-state levels of IRF5, a component of the interferon-signalling pathway, in patients with SLE was a major step forward 71.
Although not all of the recent findings reported at the beginning of 2008 have been found to be functionally significant, the identification of ITGAM (integrin alpha-M, also known as complement receptor type 3 alpha subunit) might provide clues as to how impaired clearance of immune complexes affects the pathogenesis of SLE. Previous studies have demonstrated that ITGAM is important for the adherence of neutrophils and monocytes to inflamed endothelium, and also in the phagocytosis of complement coated particles72, 73. Therefore ITGAM might play a role in the recruitment of innate immune cells and in immune complex clearance, the latter being a process thought to be impaired in a subset of SLE patients.
Finally, the identification of the association with B cell scaffold protein with ankyrin repeats (BANK1), provides a new entry point for the study of role of B cells in SLE, a cell population long known to be involved in the pathogenesis of the disease. Earlier studies showed that BANK1 deficiency in mice resulted in a hyper-reactive B-cell phenotype through enhanced CD40-mediated AKT activation 74, which is potentially one of the key pathways for the production of autoantibodies. Despite the fact that the current picture of how these genes contribute to SLE is still incomplete (FIG. 2), it is becoming clear that an understanding of the common and disease-specific risk factors will be crucial for the appropriate design of targeted therapies. For example, one could envisage a SLE-specific therapeutic approach that aims to deplete hyperactive B cells by targeting BANK1-related signalling pathways, thereby influencing autoantibody production and the regulation of adaptive immune responses. Additional RA-associated genes that are common to SLE include: STAT4 and TRAF1-C5 22, 62. However our understanding of the functional effects and the mechanism of action of these genetic variants is substantially incomplete, the lack of a coding variant and or knockout function suggest that these variants may control level and protein function. STAT4 dependant signaling via the IL-12 receptor plays a critical role in the development of Th1 type T cell responses 75. Based on these results its is tantalizing to speculate that STAT4 polymorphisms associated with RA and Lupus may result in enhanced IL-12 driven signalling, increased IFN-γ levels and persistent inflammation.
Challenges Ahead
We hope that the information presented in the previous sections has convinced the reader that GWAS studies have truly revolutionized our ability to catalogue the genetic risk factors involved in many immune-mediated diseases in humans. Despite these successes, there is no doubt that these discoveries are only the tip of the iceberg — the genes identified to date can only account for a minor portion of the overall genetic component of disease risk. Moreover, given that these discoveries have predominantly emerged over the last 18 months, there is much still to learn regarding the function of the associated genetic risk variants, and the mechanistic framework presented in this Review is by necessity incomplete and can only be considered a starting point. Going forward, there are three main challenges:
Obtain a comprehensive picture of genetic risk
As summarized in Table 1, the risk conferred by any individual associated variant is very modest. Even taken together, the set of risk factors discovered to date can account for less than 10% of the overall genetic risk. Why have the GWAS performed to date not identified all of the factors that contribute to genetic risk? One reason is that these studies have been underpowered in terms of sample size — cohorts in the tens of thousands are needed to have sufficient power to detect the majority of real but modest genetic effects 10, 11. Although such numbers appear daunting, they are feasible when considering the sample sizes recruited for some large international multicentre clinical trials. Alternatively, combining individual association studies to obtain large sample sizes is greatly simplified by the fact that the current genotyping platforms have relatively fixed content and in fact recent meta-analyses of GWAS have proven very successful 76-78. Another reason is that, for the large number of genes that have been discovered, many of the causal variants have not yet been identified. As such, the associated variants identified in the GWAS might only be imperfectly correlated with the causal variants and, therefore, their genetic effect on disease susceptibility might be underestimated. The identification of causal variants will likely require re-sequencing efforts in large sample sizes, as even some genes for which we have already identified a causal variant are likely to contain additional as-yet unidentified causal variants. Finally, one other important reason why the data to date has not provided a comprehensive picture of genetic risk is that the entire genome has not yet been studied by these GWAS approaches. Other types of polymorphisms, which might include polymorphisms in copy number, mRNA precursors or repeat elements, could also contribute to genomic variation and human disease, an example of which has recently been demonstrated for psoriasis 79. Clearly, a combination of approaches will be necessary to complete this picture.
Develop an understanding of disease mechanisms
One of the major roles that the field of immunology will have in the coming years in determining the underlying mechanisms of disease susceptibility and progression is to place the genetic findings of association studies into a functional context. Success in this area will likely require a slight shift in the approaches that immunologists and geneticists currently use when working together. For example, given the significant success of human GWAS, it is likely that the study of gene function using animal models of disease (such as knock-in and knock-out models) will have a predominant role over the use of animals for disease gene identification. The major shift, however, will need to come from an integrated approach that allows the study of gene function as well as an emphasis on the study of the causal and protective variants identified in the human studies (an example is described in BOX 3). These approaches, in order to be successful, will depend on our ability to access live cells and tissue specimens from genetically defined individuals and to develop high-throughput functional assays to understand the biological pathways identified. In addition, it will be necessary to combine data from disparate sources in order to obtain a more global picture of the biological networks that are perturbed by the causal variants (FIG. 3). A number of such integrated pathway and network approaches are emerging that will enhance our understanding of the fundamentals of disease pathogenesis 80-84.
Figure 3. Schematic work-flow to place candidate genes in disease specific signalling pathways.
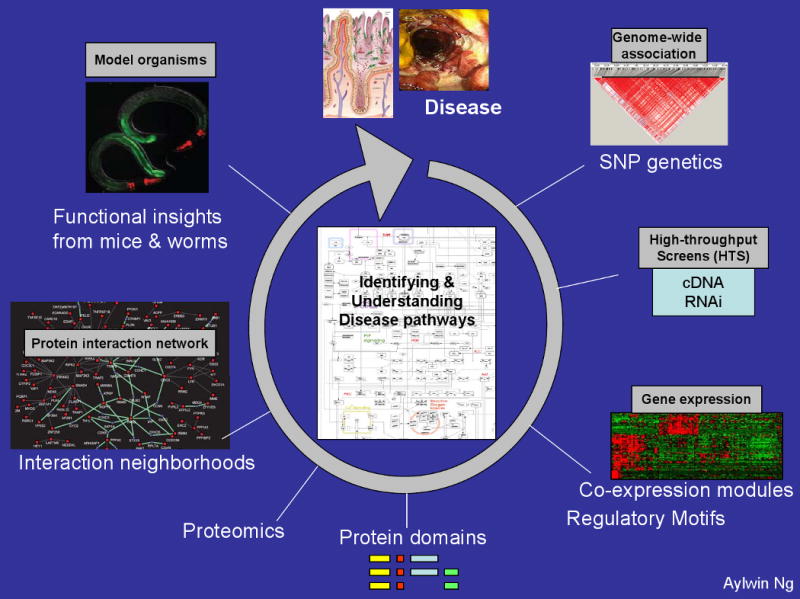
a) Fine mapping of causal variants associated with disease susceptibility. Genes identified as associated will be deeply re-sequenced in patients and controls to identify potentially functional polymorphisms explaining association as well as additional rare, penetrant mutations in the same gene. The goal is to sequence cases and controls for the exons and nearby evolutionarily conserved sequence of each gene to identify risk alleles when compared to control patients. b) targeted RNA interference screens in epithelial and immune cells to place candidate genes in known pathways 81 c) microarray analysis of peripheral blood and intestinal biopsy samples from Crohn's disease patients to examine gene expression changes associated with identified causal variants d) functional analysis of domains surrounding causal variants; for example, nucleotide-binding oligomerization domain protein 2 (NOD2) and leucine-rich repeat (LRR) domains 87 e) bioinformatics approaches to identify interactions between pathways f) use of model organisms, such as zebra fish and worms, to elucidate microbe–host cell interactions.
Translate findings into medical practice
In order for these discoveries to have a practical impact on society, they must eventually be applied to clinical practice. A GWAS-derived information-driven approach has the potential to shift the emphasis away from symptom control and alleviation towards targeting of the proximal causal factors in disease, which would represent a major victory for medicine. Immune-mediated diseases have the greatest potential for success given our ability to readily access, assess and modulate components of the immune system.
It is important to consider that some associated variants predispose to disease whereas others are protective. The existence of both predisposing and protective alleles in a single molecule (for example the Crohn's disease-associated variants in IL23R) illustrates that a meticulous and comprehensive understanding of the mechanisms involved in disease pathogenesis is required in order to develop safe and effective therapeutics. Finally, although no single causal variant is likely to have any diagnostic value due to the modest risk conferred by each individual allele, a more immediate payoff could be the development of comprehensive risk models which would incorporate an individual's current immune status and genetic and non-genetic risk factors in order to provide the necessary tools (such as biomarkers and predictive algorithms) to aid in clinical management.
In conclusion, although the initial GWAS have only identified a small portion of the entire set of genetic risk factors for common immune-mediated diseases, they have already had a significant impact on our understanding of several of the key biological pathways involved in disease pathogenesis. The path forward for the further identification of additional genetic risk factors relies largely on our ability to study much larger cohorts of well-phenotyped patients. The greatest challenges ahead will be the integration of genetic knowledge and immunological studies, and the eventual application of this new knowledge to improving current clinical treatment options and the management of patients with these debilitating immune-mediated disorders.
Acknowledgments
The authors would like to thank C. Lefebvre, A. Huett, A. Ng and C. Labbé for their help in the preparation of this manuscript. RJX is supported by the following grants AI062773, DK 43351 and CCIB development funds. JDR is funded by grants from the National Institutes of Allergy and Infectious Diseases (AI065687; AI067152) from the National Institute of Diabetes and Digestive and Kidney Diseases (DK064869; DK062432) and the Crohn's and Colitis Foundation of America (SRA512).
Glossary
- Allele
In association studies, the term allele is most commonly used to refer to markers. An allele can also be described as one specific version of a marker inherited from a parent (such as A or T, for example, in the case of an A/T single nucleotide polymorphism). Allele is also used in a broader context when referring to a specific version of an entire gene
- Association testing/association-based approaches
A genetic variant is genotyped in a population for which phenotypic information is available (such as disease occurrence or a range of different trait values). If a correlation is observed between genotype and phenotype, there is said to be an association between the variant and the disease or trait
- Autophagy
a cellular process in which cytoplasmic organelles and macromolecular complexes are entrapped in double membrane bound vesicles for delivery to lysosomes and subsequent degradation. This process is involved in constitutive turnover of proteins and organelles and is central to cellular activities that maintain a balance between synthesis and breakdown of various proteins
- B1 B cells
B1 cells are B cells that express the cell-surface molecule CD5, which can bind to another B cell surface protein, CD72. CD5-CD72 is thought to mediate B cell–B cell interaction. B-1 cells are found primarily in the peritoneum and have a role in T-cell-independent antibody production
- Causal gene
The specific gene that is responsible for disease risk conferred by a genomic region that has been identified by an association
- Causal variant
The specific DNA sequence that functionally gives rise to the increased risk conferred by the causal gene or genomic region identified by an association
- Copy number variant (CNV)
A genomic variant characterized by differences in the number of copies of specific repeated DNA fragments that range from 1 kb to several Mb long. CNVs can contain entire genes, and can be used as markers in association studies
- Gene desert
Large genomic segments devoid of genes. These segments tend to harbour limited sequence conservation. The rare conserved region can contain transcriptional regulatory elements
- Genetic markers
Genomic variants used as positional tools to find association between specific DNA fragments and certain a phenotype or disease
- Genome-wide linkage scan
A linkage study performed with markers located across the entire genome. Traditionally performed with ∼300 markers of simple sequence length repeats (SSLPs) and more recently with ∼5,000 single nucleotide polymorphisms
- Genotyping
A test designed to identify the genetic constitution of an individual, that is, the alleles present at one or more specific loci
- Haplotype
a combination or pattern of alleles at multiple linked loci that are inherited together
- HAPMAP
Genetic resource created by the International HapMap Project (www.hapmap.org). The HapMap (short for haplotype map) is a catalogue of common genetic variants that occur in human beings. It describes what these variants are, where they occur in the genome, and how they are distributed among people within populations and among populations in different parts of the world
- Hardy-Weinberg equilibrium
A fundamental principle in genetics stating that the genotypic frequencies of a large, randomly mating population remain constant in the absence of mutation, migration, natural selection, or random drift
- Immunoreceptor tyrosine-based activation motif (ITAM)
ITAMs are protein motifs that exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. This motif may be dually phosphorylated on tyrosines that link antigen receptors to downstream signalling molecules
- Knock-down/knock-in experiments
Molecular biology experiments aimed to decrease the expression of endogenous genes (knock-down) or to introduce a specific allele of a given gene (knock-in). Most commonly, the knock-down approach is used in vitro whereas the knock-in approach in vivo
- Linkage study
A study aimed at establishing linkage between a genetic marker and a disease locus. Linkage is based on the tendency for genes and genetic markers to be inherited together because of their location near one another on the same chromosome
- Linkage disequilibrium (LD)
A situation in which alleles in a chromosomal region occur together more often than can be accounted for by chance, indicating that the alleles are in close proximity on the DNA strand and are most likely to be passed on together within a population
- Locus
The specific position of a gene or a marker on a chromosome
- Odds ratio
A measure of relative risk, usually estimated from studies that compare patients with a disease with control subjects. In a genetic disease context, it is defined as the ratio of the odds of having an allele or a genotype while being affected by the disease to the odds of as healthy control being affected
- Penetrance
The likelihood, under given environmental conditions, with which a specific phenotype is expressed by individuals with a specific genotype. For example, if 50% of the people with a gene “X” that is known to cause the disease in fact develop the disease, then the penetrance of gene X is 0.5
- Replication study
A study designed to test (or replicate) a prior and explicit genetic hypothesis. For example, replication studies are performed to validate the findings of a GWAS; thus discriminating false positives from true positives identified in the GWAS.
- Single nucleotide polymorphism (SNP)
A genomic variant in which a single base in the DNA differs from the usual base at that position. SNPs are the most common type of variation in the human genome
- Simple sequence length polymorphism (SSLP)
also known as a microsatellite marker; a DNA variant consisting of short repeat of up to four nucleotides. The units are repeated from ten to hundreds of time.
- Tag SNP
A representative SNP in a region of the genome with high linkage disequilibrium (LD) Tag SNPs allow a reduction in the number of SNPs that must be genotyped while permitting the generation of the same amount of information. The HapMap Project catalogued tag SNPs in the entire genome
- TH17 cells
Th17 cells are a recently-identified subset of CD4+ T helper cells that are protective against extracellular microbes, but are responsible for autoimmune disorders in mice and men
Biographies
Ramnik Xavier is Associate Professor of Medicine at Harvard Medical School and investigator at the Center for Computational and Integrative Biology at Massachusetts General Hospital. In addition Dr Xavier serves as core member of the NIH funded Center for the study of inflammatory bowel disease at Massachusetts General Hospital. His laboratory uses genomics, computation and genetic approaches in mice to identify genes and signalling pathways in immunity and cancer. Recent studies have also focussed on functional validation of genetic variants associated with Crohn's disease.
John D. Rioux is an Associate Professor of Medicine at the Université de Montréal and the Montreal Heart Institute. After obtaining a PhD in Immunology, Dr. Rioux spent 10 years at the Broad Institute of MIT and Harvard developing and applying approaches to study the genetics of complex immune diseases. Since 2005, his laboratory in Montreal has continued to identify genetic risk factors for inflammatory disorders, their biological consequences and their potential use in clinical practice.
Footnotes
- A description of how genome-wide association studies (GWAS) have dramatically influenced our ability to identify genetic risk factors for complex immune-mediated diseases.
- Provides the information necessary for immunologists to better understand the rapid advances in our knowledge regarding genetic variation and in the technologies to probe this variation that have enabled GWAS, and to provide some understanding of both the analytical challenges and the strengths of this type of study.
- A summary of the recent GWAS studies that have investigated immune-mediated diseases including Crohn's disease, multiple sclerosis, rheumatoid arthritis and systemic lupus erythematosus.
- A discussion of how the knowledge emanating from these GWAS are revealing how previously unknown biological pathways can contribute to disease pathogenesis, highlighting both disease-specific pathways and pathways common to more than one immune-mediated disease.
- discussion of the challenges that lie ahead: how can we complete the mechanistic picture of how genetic variation in multiple different genes leads to disease development? How these data will impact the design of future immunology studies, and how can we apply this knowledge to clinical practice in the future?
References
- 1.Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584–9. doi: 10.1038/nature03723. [DOI] [PubMed] [Google Scholar]
- 2.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 3.Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999;22:139–44. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]
- 5.Wang DG, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–82. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- 6.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 8.Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–32. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- 9.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 11.Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet. 2005;6:109–18. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- 12.Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7:781–91. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- 13.Chanock SJ, et al. Replicating genotype-phenotype associations. Nature. 2007;447:655–60. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 14.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Libioulle C, et al. Novel Crohn Disease Locus Identified by Genome-Wide Association Maps to a Gene Desert on 5p13.1 and Modulates Expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nath SK, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152–4. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 18.Duerr RH, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parkes M, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rioux JD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hafler DA, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–62. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 22.Plenge RM, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study. N Engl J Med. 2007;357:1199–209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todd JA, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–64. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Heel DA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007;39:827–9. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hom G, et al. Association of Systemic Lupus Erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358:900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 26.Zhernakova A, et al. Novel association in chromosome 4q27 region with rheumatoid arthritis and confirmation of type 1 diabetes point to a general risk locus for autoimmune diseases. Am J Hum Genet. 2007;81:1284–8. doi: 10.1086/522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maas K, et al. Cutting edge: molecular portrait of human autoimmune disease. J Immunol. 2002;169:5–9. doi: 10.4049/jimmunol.169.1.5. [DOI] [PubMed] [Google Scholar]
- 28.Hugot JP, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 29.Ogura Y, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 30.Rioux JD, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29:223–8. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- 31.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–59. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Kim YG, et al. The Cytosolic Sensors Nod1 and Nod2 Are Critical for Bacterial Recognition and Host Defense after Exposure to Toll-like Receptor Ligands. Immunity. 2008;28:246–57. doi: 10.1016/j.immuni.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Wehkamp J, Schmid M, Stange EF. Defensins and other antimicrobial peptides in inflammatory bowel disease. Curr Opin Gastroenterol. 2007;23:370–8. doi: 10.1097/MOG.0b013e328136c580. [DOI] [PubMed] [Google Scholar]
- 34.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–77. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hampe J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 37.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–83. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 39.Taylor GA. IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007;9:1099–107. doi: 10.1111/j.1462-5822.2007.00916.x. [DOI] [PubMed] [Google Scholar]
- 40.Bekpen C, et al. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005;6:R92. doi: 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–41. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 42.Ellson CD, et al. Neutrophils from p40phox-/- mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med. 2006;203:1927–37. doi: 10.1084/jem.20052069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suh CI, et al. The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcgammaIIA receptor-induced phagocytosis. J Exp Med. 2006;203:1915–25. doi: 10.1084/jem.20052085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rioux JD, et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66:1863–70. doi: 10.1086/302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satsangi J, et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 46.Goyette P, et al. Gene-centric Association Mapping of Chromosome 3p implicates MST1 in IBD pathogenesis. Mucosal Immunology. 2008;1:131–138. doi: 10.1038/mi.2007.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beckly JB, et al. Two-stage candidate gene study of chromosome 3p demonstrates an association between nonsynonymous variants in the MST1R gene and Crohn's disease. Inflamm Bowel Dis. 2008;14:500–7. doi: 10.1002/ibd.20365. [DOI] [PubMed] [Google Scholar]
- 48.Cargill M, et al. A Large-Scale Genetic Association Study Confirms IL12B and Leads to the Identification of IL23R as Psoriasis-Risk Genes. Am J Hum Genet. 2007;80:273–390. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kastelein RA, Hunter CA, Cua DJ. Discovery and Biology of IL-23 and IL-27: Related but Functionally Distinct Regulators of Inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 50.Acosta-Rodriguez EV, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 51.Annunziato F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–61. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirota K, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204:2803–12. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yeo TW, et al. A second major histocompatibility complex susceptibility locus for multiple sclerosis. Ann Neurol. 2007;61:228–36. doi: 10.1002/ana.21063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gregory SG, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–91. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 55.Lundmark F, et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet. 2007;39:1108–13. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- 56.Vella A, et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet. 2005;76:773–9. doi: 10.1086/429843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plenge RM, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–82. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsitsikov EN, et al. TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–57. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- 59.Chung JY, Park YC, Ye H, Wu H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. 2002;115:679–88. doi: 10.1242/jcs.115.4.679. [DOI] [PubMed] [Google Scholar]
- 60.Elliott MJ, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 61.Weinblatt ME, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253–9. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]
- 62.Remmers EF, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–86. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vang T, et al. Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol. 2008;26:29–55. doi: 10.1146/annurev.immunol.26.021607.090418. [DOI] [PubMed] [Google Scholar]
- 64.Criswell LA, et al. Analysis of Families in the Multiple Autoimmune Disease Genetics Consortium (MADGC) Collection: the PTPN22 620W Allele Associates with Multiple Autoimmune Phenotypes. Am J Hum Genet. 2005;76:561–71. doi: 10.1086/429096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kozyrev SV, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:211–6. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- 66.Harley JB, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Graham DS, et al. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat Genet. 2008;40:83–9. doi: 10.1038/ng.2007.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fernando MM, et al. Identification of Two Independent Risk Factors for Lupus within the MHC in United Kingdom Families. PLoS Genet. 2007;3:e192. doi: 10.1371/journal.pgen.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baechler EC, et al. Gene expression profiling in human autoimmunity. Immunol Rev. 2006;210:120–37. doi: 10.1111/j.0105-2896.2006.00367.x. [DOI] [PubMed] [Google Scholar]
- 70.Bauer JW, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3:e491. doi: 10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Graham RR, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104:6758–63. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lim J, et al. An essential role for talin during alpha(M)beta(2)-mediated phagocytosis. Mol Biol Cell. 2007;18:976–85. doi: 10.1091/mbc.E06-09-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schymeinsky J, Mocsai A, Walzog B. Neutrophil activation via beta2 integrins (CD11/CD18): molecular mechanisms and clinical implications. Thromb Haemost. 2007;98:262–73. [PubMed] [Google Scholar]
- 74.Yokoyama K, et al. BANK regulates BCR-induced calcium mobilization by promoting tyrosine phosphorylation of IP(3) receptor. Embo J. 2002;21:83–92. doi: 10.1093/emboj/21.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaplan MH. STAT4: a critical regulator of inflammation in vivo. Immunol Res. 2005;31:231–42. doi: 10.1385/IR:31:3:231. [DOI] [PubMed] [Google Scholar]
- 76.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lettre G, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet. 2008;40:584–91. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–45. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hollox EJ, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40:23–5. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pujana MA, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39:1338–49. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 81.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 82.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–35. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emilsson V, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–8. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 84.Keller MP, et al. A Gene Expression Network Model of Type 2 Diabetes Links Cell Cycle Regulation in Islets with Diabetes Susceptibility. Genome Res. 2008 doi: 10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol. 2006;18:207–13. doi: 10.1016/j.smim.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 86.Rieck M, et al. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. 2007;179:4704–10. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]
- 87.Palsson-McDermott EM, O'Neill LA. Building an immune system from nine domains. Biochem Soc Trans. 2007;35:1437–44. doi: 10.1042/BST0351437. [DOI] [PubMed] [Google Scholar]
- 88.De Jager PL, et al. The role of inflammatory bowel disease susceptibility loci in multiple sclerosis and systemic lupus erythematosus. Genes Immun. 2006;7:327–34. doi: 10.1038/sj.gene.6364303. [DOI] [PubMed] [Google Scholar]
- 89.Ueda H, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–11. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 90.Brand OJ, et al. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves' disease using a multilocus test and tag SNPs. Clin Endocrinol (Oxf) 2007;66:508–12. doi: 10.1111/j.1365-2265.2007.02762.x. [DOI] [PubMed] [Google Scholar]
- 91.Graham RR, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–5. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 92.Velaga MR, et al. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves' disease. J Clin Endocrinol Metab. 2004;89:5862–5. doi: 10.1210/jc.2004-1108. [DOI] [PubMed] [Google Scholar]
- 93.Hakonarson H, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–4. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 94.Smyth DJ, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–9. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 95.Sutherland A, et al. Genomic polymorphism at the interferon-induced helicase (IFIH1) locus contributes to Graves' disease susceptibility. J Clin Endocrinol Metab. 2007;92:3338–41. doi: 10.1210/jc.2007-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]