

Abstract
Background/Aims
Wilson disease (WD) is a disorder of copper transport caused by mutations within the ATP7B gene. WD is phenotypically variable and can present with predominantly hepatic or neurologic manifestations. The mechanisms responsible for this variability are unknown. GP73, a Golgi membrane protein, is expressed in hepatocytes in response to acute and chronic liver disease.
Methods
Hepatocyte GP73 expression was examined in the livers of WD patients by semiquantitative immunohistochemistry. GP73 mRNA levels were measured in mice with a deletion of the WD gene (Atp7b−/−) by real-time PCR, and these values were compared to the concomitant histological abnormalities and previously reported copper levels.
Results
Hepatocyte GP73 expression was more frequently observed in patients with hepatic versus neurologic presentation (79% vs. 30%, p<0.05). Furthermore, GP73 expression was significantly higher (44.7 ± 14.0 vs. 2.0 ± 0.81, p<0.05) in patients with hepatic phenotype. In Atp7b−/− mice, GP73 mRNA was significantly elevated at 20–46 weeks of age, coincident with extensive hepatic inflammation and fibrosis, but not at six weeks, when hepatic histology was normal despite significant copper overload. GP73 mRNA levels normalized concomitantly with the resolution of hepatic injury at 60-weeks, however in tumor-like nodules GP73 was strikingly elevated.
Conclusion
Increased hepatocyte GP73 expression is more commonly a feature of hepatic than neurologic WD, and is triggered in response to inflammation, fibrosis, and dysplasia, rather than copper overload.
Keywords: Atp7b, Copper, Golgi, Knockout mouse, Liver
1. Introduction
GP73 (Golm1, Golph2) is an integral Golgi membrane protein of unknown function that is present at high levels in hepatocytes of patients with acute and chronic hepatitis and hepatocellular cancer (HCC; [1–3]). In normal livers, GP73 is constitutively expressed in biliary epithelial cells, whereas hepatocyte expression is minimal and typically limited to zone 1.
The time course and extent of GP73 upregulation in liver disease has been previously studied in detail. In acute hepatitis, GP73 is expressed in the majority of hepatocytes, and cellular levels of the protein are increased up to 100-fold. In chronic hepatitis C- or alcohol-induced liver disease, GP73 expression increases gradually, and in parallel with the fibrosis stage. Maximal GP73 expression is present in fully-established cirrhosis, with uniform, high-level expression comparable to acute liver disease [1, 2]. GP73 is also highly expressed in hepatocytes of patients with HCC. Additionally, GP73 is secreted into the serum of HCC patients following furin-mediated proteolytic cleavage of its N-terminus [4], a feature that can be exploited for the diagnosis of HCC [5]. These studies suggest that GP73 expression is a marker of hepatocellular injury, fibrosis, and dysplasia.
Wilson disease (WD) is an inherited disorder caused by mutations in the Cu2+ transporting ATPase, ATP7B. ATP7B is localized to the trans-Golgi network and is normally expressed at high levels in hepatocytes and in the central nervous system (CNS). The loss of ATP7B activity results in a marked accumulation of copper in the liver and brain of affected patients. WD is characterized by marked phenotypic variability with two distinct presentations: predominant hepatic manifestations (fulminant hepatitis, chronic hepatitis or cirrhosis, but normal neuropsychiatric examination), and CNS symptoms (psychosis, dementia) in the absence of apparent hepatitis. Hepatic copper is elevated in both cases and the biological basis of this variability is unknown [6].
Hepatic GP73 expression is increased in various forms of acute and chronic liver disease [1, 3]. Based on its association with the hepatic injury response, we reasoned that hepatic GP73 expression might be a marker for the hepatic phenotype of WD. We tested this hypothesis in a cohort of well-defined WD patients who had undergone liver biopsy or resection. In order to further examine the relationship between GP73 expression, hepatocellular copper overload, and hepatic injury in WD, we studied GP73 expression in mice with a null mutation of Atp7b, the murine homolog of the WD gene. Our studies suggest that hepatocellular inflammation, fibrosis, and dysplasia are the driving forces for the increased GP73 expression in WD, whereas hepatocellular copper overload alone is not sufficient.
2. Patients and Methods
2.1. Patients
The study involved 31 subjects from a previously described WD registry [6]. The demographic and clinical data of the study subjects are provided in Table 1. Patients were classified as having primarily hepatic or neurologic disease using published criteria [6, 7]. The “hepatic” classification required the exclusion of neurologic symptoms by a detailed clinical neurological examination at the time of diagnosis, and the “neurologic” presentation was defined by the presence of neurological and/or psychiatric symptoms at the time of diagnosis. Nineteen subjects presented with symptoms of WD-related liver disease, 10 patients presented with neurologic manifestations of WD without clinically overt liver disease, and 2 subjects were asymptomatic siblings of WD study patients. The presence/absence of Kayser-Fleischer rings (KFR) was determined on opthamologic examination by slit-lamp analysis. Clinical assays and ATP7B mutation analyses were performed as previously described [8]. Serum ceruloplasmin (Cpl) was measured by radial immunodiffusion (NOR-Partigen Coeruloplasmin, Behring, Marburg, Germany; normal range: 20–60 mg/dL).
Table 1.
Characteristics of Patients with Hepatic or Neurologic WD
| # | GP73 IH score |
Age at Dx |
Age at Onset |
Sex | Cpl (mg/L) |
KFR | Neu* | Liver− Histology † |
Rhodanine | Hepatic Copper (mg/g) |
WD Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hepatic WD | |||||||||||
| 1 | 0 | 11 | 11 | f | 17.1 | − | − | Fibrosis | − | 847 | H1069Q/? |
| 2 | 5 | n/a | n/a | f | >9 | − | − | Cirrhosis | + | 1115 | H1069Q/? |
| 3 | 0 | 24 | 24 | m | 21.0 | − | − | Cirrhosis | − | 79 | ?/? |
| 4 | 5 | 15 | 15 | m | 14.6 | − | − | Cirrhosis | − | 1288 | H1069Q/H1069Q |
| 5 | 5 | 15 | 14 | f | n/a | − | − | Cirrhosis | + | 758 | H1069Q/? |
| 6 | 10 | 19 | 19 | f | n/a | − | − | Cirrhosis | + | 590 | I1148T/? |
| 7 | 60 | 33 | m | low | − | − | Cirrhosis | + | 264 | H1069Q/? | |
| 8 | 10 | 12 | 12 | f | <9 | − | − | Cirrhosis | − | 164 | H1069Q/? |
| 9 | 120 | 10 | 10 | f | 18.4 | − | − | Fibrosis | + | 1278 | H1069Q/R816S |
| 10 | 120 | 10 | 10 | m | 7.0 | + | − | Cirrhosis | − | 311 | V883A−fs/V883A−fs |
| 11 | 0 | 31 | 31 | f | 20.4 | + | − | Fibrosis | + | 115 | 2299insC/G1030S |
| 12 | 120 | 31 | 18 | m | 21.6 | _ | − | Cirrhosis | + | 2060 | G710A/G710A |
| 13 | 5 | 17 | 16 | f | 6.8 | + | − | CH | − | 525 | 3400delC/? |
| 14 | 5 | 10 | 10 | m | 8.0 | + | − | Fibrosis | − | 31 | V1217 1218Ldel/? |
| 15 | 0 | 14 | 14 | f | 21.0 | − | − | Fibrosis | − | 1000 | H1069Q/R969Q |
| 16 | 200 | 40 | 8 | m | 6.0 | − | − | Cirrhosis | n/a | 645 | R1319X/? |
| 17 | 60 | 27 | 27 | f | 14.0 | + | − | Cirrhosis | + | 963 | H1069Q/H1069Q |
| 18 | 5 | 16 | 16 | m | 8.0 | + | − | Cirrhosis | + | 950 | H1069Q/L1305P |
| 19 | 120 | 23 | 20 | f | 8.1 | + | + | Cirrhosis | n/a | 266 | H1069Q/H1069Q |
| Neurologic WD | |||||||||||
| 20 | 5 | 27 | 27 | m | 7.0 | + | + | Fibrosis | − | 571 | H1069Q/H1069Q |
| 21 | 5 | 20 | 20 | f | 7.0 | + | + | Cirrhosis | + | 320 | H1069Q/H1069Q |
| 22 | 5 | 20 | 20 | f | 3.9 | + | + | CH | − | 1700 | H1069Q/H1069Q |
| 23 | 0 | 39 | 24 | m | 20.0 | + | + | Cirrhosis | + | 50 | H1069Q/H1069Q |
| 24 | 0 | 45 | 41 | f | 6.2 | + | + | CH | − | 475 | K35X/? |
| 25 | 0 | 17 | 15 | m | 6.2 | + | + | Cirrhosis | + | 192 | G710S/L1305P |
| 26 | 0 | 14 | 14 | m | 9.8 | + | + | URH | − | 1448 | H1069Q/V1217− |
| 27 | 0 | 16 | 16 | f | 7.0 | + | + | CH | + | 269 | H1069Q/W779X |
| 28 | 0 | 32 | 18 | m | 11.4 | + | + | Fibrosis | − | 386 | H1069Q/H1069Q |
| 29 | 5 | 47 | 44 | f | 9.1 | + | + | URH | n/a | 164 | G710S/G710S |
| Asymptomatic siblings | |||||||||||
| 30 | 5 | 32 | 32 | f | 17.4 | + | − | Cirrhosis | + | 533 | H1069Q/H1069Q |
| 31 | 0 | 25 | 25 | f | low | − | − | CH | − | 844 | H1069Q/H1069Q |
Neurological symptoms at presentation “−“ = absent, “+” = present
liver histology on biopsy or explant: CH, chronic hepatitis; URH, unspecific reactive hepatitis.
2.2. Histological Evaluation of Liver Biopsies
Three- to 5-mm segments of the core of fresh liver biopsy specimens obtained by the Menghini technique were air-dried in an Eppendorf tube. Copper content was determined by flame atomic absorption spectroscopy according to Kingston and Jassie [9] using a commercial copper standard (Titrisol/Merck, Darmstadt, Germany). The inter- and intra-assay variations of hepatic copper content were 1.8% and 3.0%, respectively. The remaining liver biopsy specimens were fixed in 7.5% buffered formalin and embedded in paraffin. Serial 3μm sections were stained with hematoxylin & eosin (H&E), chromaniline blue, and Prussian blue according to standard methods. All slides were re-examined by a single pathologist unaware of the clinical data, and the activity of the lesions was scored according to the recommendations of Batts and Ludwig [10]. Intracytoplasmic copper accumulation was determined specifically by the p-dimethylaminobenzylidene-m-rhodanine method [8]. To exclude fading of staining as reason for rhodanine negativity, new sections were made in all negative samples and were freshly stained and reread. The amount of stainable copper was graded as present or absent.
2.3. GP73 Immunohistochemistry
GP73 immunoreactivity was detected in 5μm liver sections with a two-stage, indirect immunoenzymatic-immunohistochemical assay (DAKO EnVision, Dako Corporation, Carpinteria, CA). Endogenous peroxidase activity was blocked by a 5-min preincubation in 0.03% hydrogen peroxide. Sections were incubated for 30 min with rabbit polyclonal GP73-specific antisera (1:1000 v/v) as previously described [3]. After washes, sections were incubated with a peroxidase-labeled polymer conjugated to goat anti-rabbit immunoglobulin for 30 min. GP73 immunoreactivity was visualized using the chromogen 3-amino-9-ethylcarbazole (AEC, DAKO Corporation). Samples were counterstained with Mayer’s Hematoxylin. Images were captured using a Nikon DXM1200 digital still camera with ACT-1 software (Nikon, Tokyo, Japan).
2.4. Semiquantitative GP73 Immunohistochemical Scoring System
GP73 staining was assessed using a semiquantitative scoring system. The percentage of GP73-positive hepatocytes was determined for each sample in 20 adjacent, non-overlapping high-power fields. The staining intensity was graded as absent or barely detectable (0), present (1), or strong (2), using previously validated control samples of normal and cirrhotic livers as standards [1]. The percentage of GP73-expressing hepatocytes was calculated for each field, and multiplied with the corresponding staining intensity score to yield a mean GP73 immunohistochemistry (IH) score for each sample. The GP73 quantitation was performed by a single observer (CJF) who was unaware of the clinical diagnoses of the samples.
2.5. Mice
The generation of mice carrying a null mutation of the Atp7b gene has been previously described [11]. Mice were propagated on a C57BL x 129S6/SvEv background. Animals were housed at the Oregon Health and Science University Department of Comparative Medicine according to National Institutes of Health guidelines outlined in the “Guide for the Care and Use of Laboratory Animals” as previously described [12]. Mice were euthanized at 6-, 20, 32, 46, or 60-weeks of age. Their livers were removed, and portions were snap frozen in liquid nitrogen, and stored at −80°C until analysis. Copper measurements were performed by atomic absorption spectrometry as previously described [12].
2.6. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from livers of wild-type and Atp7b−/− mice at 6–60 weeks (n=6 per group) and isolated using TRIZOL reagent (Invitrogen, Carlsbad, CA) according to manufacturer’s guidelines. The RNA was further purified using an (RNAeasy clean-up kit (Qiagen, Hilden, Germany). One-step RT–PCR was performed with a LightCycler instrument (Roche, Pleasanton, CA) in a volume of 20 μl containing 50ng of total RNA, 0.5μM of each primer, LightCycler SYBR Green RT–PCR Master Mix and Quanti Tect RT Mix (Qiagen, Valencia, CA). Reverse transcription was performed at 50°C for 20min. RT-PCR amplification of GP73 and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) internal control were performed as described [12] using QuantiTect® Primers (Mm_Golm1_va.1_SG and Mm_Gapd_2_SG, respectively; Qiagen). The product sizes were confirmed by separation on agarose gel and SYBR-Green staining. The abundance of GP73 mRNA in each sample was normalized to GAPDH. GP73 mRNA levels in Atp7b−/− and wild-type mice were compared using the 2ΔΔct method and the software REST gene and the Taylor algorithm [13, 14].
2.7. Histology of mouse livers
Livers were fixed in 10% buffered formalin at pH 7.4 for 48–72 hours, transferred to 80% ethanol, and embedded in paraffin. Sections (5 μm) were H&E stained and analyzed as previously described [12] on a Zeiss Axioscope 50 microscope and photographed with a Nikon Coolpix 990 digital camera.
2.8. Statistical Analysis
The proportions of GP73-expressing and non-expressing hepatocytes in hepatic vs. neurologic WD patients were compared by Chi-Square analysis. GP73 immunohistochemical scores were compared by rank sum testing. Additional statistical comparisons were performed by t-testing for samples with unequal variance, and by Chi-Square analysis. A p-value of <0.05 was considered significant.
3. Results
3.1. Severity of Liver Diseases in Hepatic vs. Neurologic Presentation
Nineteen subjects were categorized with “hepatic” WD (male/female: 8/11, age: 10–40, fulminant WD: 5, haemolytic anemia: 2, decompensated cirrhosis: 3, chronic liver disease: 9). Ten subjects were assessed with “neurologic” WD (male/female: 5/5, age: 14–44, including 2 patients with history of liver disease), Table 1. Histologic analysis of liver samples (Fig. 1) in patients with hepatic WD showed cirrhosis (n=13; 68%), hepatitis with advanced fibrosis (n=5; 26%), and chronic hepatitis (n=1; 5.3%). In comparison, histologic findings in patients with neurologic presentation included inactive cirrhosis (n=3; 30%), inactive hepatitis with fibrosis (n=2; 20%), chronic hepatitis (n=3; 30%), and non-specific changes (n=2; 20%).
Fig. 1.

Distribution of histological liver disease in patients with hepatic vs. neurologic presentations of WD. Percent of WD patients with predominantly hepatic (open bars) or neurologic (hatched bars) involvement with the diagnosis shown.
3.2. Biochemical and Clinical Comparison Between WD Groups
Patients with neurologic and hepatic presentation were comparable with respect to the age of onset and diagnosis, hepatic copper content, gender distribution, and Rhodanine staining (Table 2). KFR were more prevalent in patients with neurologic presentation. Serum Cpl levels were significantly higher in patients with hepatic presentation compared to neurologic patients.
Table 2.
Inter-group Comparison Between Patients with Hepatic or Neurologic Presentation
| Hepatic | Neurologic | Statistical significance | |
|---|---|---|---|
| Age at diagnosis (Years) | 19 ± 9 | 28 ± 12 | n.s. |
| Age at onset (Years) | 17 ± 7 | 24 ± 11 | n.s. |
| Serum Cpl (mg/L) | 13 ± 6 | 9 ± 4 | P=0.048 |
| Hepatic copper (mg/g) | 697 ± 596 | 557 ± 559 | n.s. |
| Gender | 8m 11f | 5m 5f | n.s. |
| KFR | 12(−) 7(+) | 0(−) 10(+) | P=0.0103 |
| Rhodanine | 9(+) 8(−) | 4(+) 5(−) | n.s. |
Groups were compared using two-sample, two-tailed t tests assuming unequal variances, or by Chi-Square analysis.
3.3. GP73Expression
GP73 expression in hepatocytes was detected in 15 of 19 patients (79%) with hepatic WD (weak: 8, strong: 7), but in only 3 of 10 patients (30%, χ2 13.5, p<0.01) with neurologic WD (Fig. 2). GP73 IH staining of livers from patients with a hepatic WD presentation was significantly more intense compared to liver sections of patients with a neurologic presentation (Fig. 2C).
Fig. 2.

Hepatocyte expression of GP73 in WD with predominant hepatologic or neurologic manifestation. GP73 IH staining in livers from patients with primary hepatologic (A) or neurologic (B) presentations of WD. In (A), robust immunoreactivity is present in hepatocytes, with a pericanalicular pattern. Little or no immunoreactivity is present in fibrous septa and in non-hepatocyte cell types within the lobules. In (B) minimal or no immunoreactivity is present in hepatocytes. Individual perisinusoidal cells show GP73 immunoreactivity. Original Magnification 20×. (C) GP73 IH staining intensity scores (mean ± standard error) of hepatocyte GP73 expression in WD patients with primary liver and neurological involvement. Significance levels; *, p<0.05.
3.4. GP73 mRNA Levels in Livers of Wild-Type and Atp7b−/− Mice
GP73 mRNA was present in low levels in the livers of wild-type control mice of all ages and was normalized to a value of 1.0. At 6-weeks GP73 mRNA levels in Atp7b−/− mice were similar to control (Fig. 3). In contrast, GP73 mRNA levels were significantly upregulated in Atp7b−/− mice at 20- and 32-weeks, and to a lesser extent at 46-weeks. GP73 mRNA levels decreased over time and in regenerating tissue at 60-weeks were comparable to control. In contrast, in “tumors” that had been dissected from surrounding regenerating liver tissues at 60-weeks, GP73 mRNA was dramatically elevated. Tumor-like regions in 60 week-old mice were multifocal with features of both HCC and cholangiocarcinoma and have been described in detail previously [12].
Fig. 3.

Upregulation of GP73 mRNA in Atp7b−/− mice. Whole-liver GP73 mRNA levels (mean ± SD) were quantified in wild-type and Atp7b−/− mice by RT-PCR relative to the internal standard (GAPDH). GP73 mRNA values from Atp7b−/− mice are expressed relative to normalized values from wild-type mice at each time point. GP73 mRNA levels were measured separately in micro-dissected tumor tissues from 60-weeks old Atp7b−/− mice (open bar). Statistical significances were calculated using REST gene software with a Taylor algorithm. Significance levels; *, p < 0.05.
3.5. Hepatic Copper Levels and Histologic Abnormalities in Atp7b−/− Mice
Hepatic copper levels were significantly elevated in Atp7b−/− mice compared to controls at all examined time points [12]. Liver histology at 6-weeks appeared normal, despite markedly elevated serum transaminases indicative of acute hepatocellular damage (Fig. 4). Marked hepatocellular injury, inflammation, and focal fibrosis were evident at the 20- and particularly the 32 and 46-week time points [12]. These histological changes correlated with significant upregulation of GP73 mRNA. Interestingly, the inflammatory changes had largely resolved at 60-weeks, in parallel with a decline in GP73 mRNA levels in regenerating liver tissue.
Fig. 4.
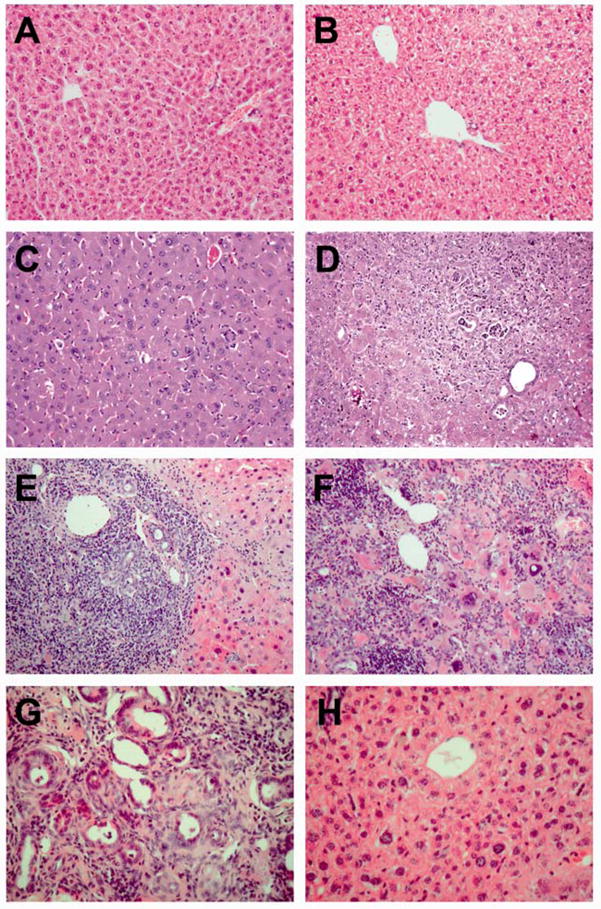
Histology of Atp7b−/− mouse livers. H&E stained liver sections from a 6 week-old wild-type female (A), a 32 week old wild-type female (B), a 6 week-old Atp7b−/− female showing normal histology (C), a 20 week-old Atp7b−/− female showing hepatitis and a bile duct lesion (D), a 32 week-old Atp7b−/− female showing severe hepatitis and a microabcess (E), a 46 week-old Atp7b−/− female showing severe necrosis and hepatitis (F), a tumor from a 60 week-old Atp7b−/− female showing neoplastic bile duct proliferation (G), and regenerating liver tissue with normal histology (H) from the same animal as (G). Original magnification 100× (A, B), 200× (C–G).
4. Discussion
Increased hepatocyte expression of GP73 has been observed in a wide range of acute and chronic liver diseases and hepatocellular cancer [1, 2]. While the function of GP73 remains unknown, the available data indicate that its expression is induced in response to acute inflammation, progressive fibrosis, and hepatocellular cancer development.
This is the first study to examine GP73 expression in WD, and to compare its expression pattern in patients with predominantly hepatic or neurologic involvement. The majority of our patients with a hepatic presentation of WD had advanced liver disease and cirrhosis. In agreement with previous studies, high GP73 expression was observed in most cases with advanced liver disease, with a pattern of GP73 immunoreactivity similar to that seen in cirrhosis from other causes. In comparison, the overall severity of liver disease was less pronounced in patients presenting with neurologic symptoms. In our group of patients with neurologic presentation, the overall hepatocyte GP73 expression was lower compared to hepatic patients. This difference held up even in the subgroups of patients with fully-established cirrhosis: the average GP73 IH score of cirrhotic patients with hepatic presentation was 55.4 (n=13) as compared to 1.67 (n=3) in patients with neurologic presentation, although the sample size was insufficient for statistical analysis. These data indicate that hepatocyte GP73 expression in WD patients may be differentially regulated in a primarily hepatic vs. neurologic disease course.
The underlying mechanisms for the phenotypic differences between primary hepatic vs. neurologic WD are unknown, and neither the extent of copper storage, nor the type of mutation explain differences in phenotypic presentations. In agreement with earlier reports, we noted a trend towards a later disease onset in patients with neurologic involvement, although this difference did not reach statistical significance [15, 16]. Assuming a continued increase of GP73 expression over time, a later disease onset in patients with neurologic disease might contribute to the observed lower hepatic GP73 levels in this group.
Similar to previous studies [17], we noted higher serum Cpl concentrations in patients with hepatic disease presentation. In general, serum Cpl levels are depressed in human WD, due to the reduction in enzymatically active, copper-containing Cpl. However inflammation can be associated with increased Cpl levels [18, 19]. Increases in systemic or hepatic inflammation might be involved in the upregulation of GP73 expression, similar to previous studies in patients with chronic hepatitis C infection or alcoholic liver disease [1].
In order to better understand the factors contributing to upregulation of hepatocyte GP73 in WD, we studied Atp7b−/− mice [11, 12]. Atp7b−/− mice do not show neurologic symptoms, but display striking sequential changes in liver histology, coupled with hepatic copper accumulation and alterations in enzymatic and biochemical profiles. By early adulthood (six weeks post-partum), their hepatic copper contents are increased 20–40-fold, reaching levels that are comparable to those observed in human WD [12]. At this time, maximal transaminase release is observed in the absence of inflammation, fibrosis, or other histological damage [12] and GP73 is expressed at low levels comparable to wild-type mice, similar to our previous findings [20]. Elevated ALT levels at this stage may be a result of early damage on a subcellular level-for example mitochondrial and nuclear damage not readily visible by light microscopy [12]. This suggests that GP73 expression is induced by specific signals rather than in response to general liver damage.
Copper accumulation results in severe hepatocellular injury, inflammation, bile duct proliferation, fibrosis, and steatosis within 2–3 months after birth. Extensive atypical bile duct proliferation and portal fibrosis occurs in these mice beginning by 20 weeks, and particularly after 36 weeks of age. The biliary epithelial cells of the abnormal bile ducts display high nuclear-cytoplasmic ratio, hyperchromasia, and dispolarity, consistent with dysplasia and possible neoplastic transformation, although the malignant nature of these changes needs further investigation. Areas with highly abnormal bile duct structures are macroscopically visible, and can be distinguished from the surrounding, more normal-appearing, regenerative areas [12].
In parallel to these changes, marked GP73 upregulation was observed, suggesting that cytokine release, neoplastic changes or other liver transcriptional events [12] can contribute to this regulation. Other factors potentially involved include copper-mediated oxidative stress, cholestasis, or sex hormone-related mechanisms [8, 21–23]. Strikingly, hepatic regeneration and the restoration of parenchymal structure at 46–60 weeks are associated with a decline in GP73 mRNA levels. This is in contrast to a marked upregulation of GP73 mRNA in micro-dissected biliary tumors at the 60 week time point. The dramatic upregulation of GP73 in dysplastic biliary tumors is intriguing in view of the known elevation of GP73 in hepatocellular and prostate cancer cells [24, 25], and should be further investigated. This finding raises the issue of WD-related hepatic carcinogenesis. Despite initial reports of reduced rates of HCC in WD, a growing number of case reports and small series indicate that HCC does occur as a complication of WD-induced liver disease [26, 27], suggesting that the phenotype in the Atp7b−/− mouse may be pertinent to the human disease process and hepatic carcinogenesis in general.
It is interesting to consider the increase of GP73 mRNA in the context of the overall transcriptome response in the WD liver. In a recent gene array analysis of 6-week old Atp7b−/− mice, Huster and colleagues observed a significant upregulation of transcriptional pathways involved in the M-phase of the mitotic cell cycle, cell division, chromosome structure and maintenance, electron transport, and DNA damage response. These changes may be related to the subsequent hepatic regeneration and bile-duct tumor formation [28], and therefore alone, they are insufficient to trigger the GP73 expression.
The normal ATP7B protein is localized to the trans-Golgi complex under steady-state conditions. GP73, despite being a resident cis-Golgi protein, transiently cycles through the trans-Golgi complex to the cell membrane and back to the Golgi via a unique endosomal retrieval pathway [29]. The presence of coil-coiled domains in the predicted GP73 structure is strongly suggestive of protein-protein interactions. Because GP73 transiently shares the same subcellular compartment with ATP7B, there is the potential for a functional or physical interaction. The known ATP7B interactome as determined by yeast two-hybrid studies includes ATOX1, glutaredoxin, COMMD1, PLZF, and the dynactin subunit p62 [15]. The dynactin interaction might be involved in the vesicular trafficking of ATP7B [30]. However, no physical interaction has been demonstrated between GP73 and ATP7B, and the GP73 interactome does not overlap with that of ATP7B (Molecular INTeraction Database “MINT”; http://mint.bio.uniroma2.it/mint/search/search.do; genename = “Golm1,” “ATP7B;” data not shown).
In summary, our study demonstrates that the GP73 expression in WD is related to the degree and severity of the hepatocellular injury and fibrosis, whereas hepatic copper overload alone does not appear to drive the GP73 expression. The striking parallel between GP73 expression and liver damage in WD needs further investigation, and suggests the utility of GP73 as a prognostic indicator for hepatic injury in patients with a primary hepatic presentation WD.
Acknowledgments
We gratefully acknowledge Doerthe Kuester, M.D. at Otto-von-Guericke-University, Magdeburg, Germany for her help with microscopic images. We also thank Mrs. Ines Sommerer, University of Leipzig for invaluable technical help.
This study was supported by a VA Merit Award to C.J.F., a German Research Foundation Grant HU932/3-2 to D.H., and an NIH grant (R21 DK075659) to S.L.
Abbreviations
- WD
Wilson Disease
- ATP7B
ATPase copper transporting, beta polypeptide
- GP73
Golgi protein 73 kDa
- HCC
hepatocellular carcinoma
- CNS
central nervous system
- KFR
Kayser-Fleischer rings
- Cpl
ceruloplasmin, H&E, hematoxylin & eosin
- IH
immunohistochemistry
- RT-PCR
Real-Time Polymerase Chain Reaction
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- Dx
diagnosis
- ATOX1
metallochaperone antioxidant-1
- COMMD1
copper metabolism MURR1 domain
- PLZF
promyelocytic leukemia zinc finger protein
- MINT
Molecular INTeraction database
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iftikhar R, Kladney RD, Havlioglu N, Schmitt-Graff A, Gusmirovic I, Solomon H, et al. Disease- and cell-specific expression of GP73 in human liver disease. Am J Gastroenterol. 2004;99:1087. doi: 10.1111/j.1572-0241.2004.30572.x. [DOI] [PubMed] [Google Scholar]
- 2.Kladney RD, Bulla GA, Guo L, Mason AL, Tollefson AE, Simon DJ, et al. GP73, a novel Golgi-localized protein upregulated by viral infection. Gene. 2000;249:53. doi: 10.1016/S0378-1119(00)00136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kladney RD, Cui X, Bulla GA, Brunt EM, Fimmel CJ. Expression of GP73, a resident Golgi membrane protein, in viral and nonviral liver disease. Hepatology. 2002;35:1431. doi: 10.1053/jhep.2002.32525. [DOI] [PubMed] [Google Scholar]
- 4.Bachert C, Fimmel C, Linstedt AD. Endosomal trafficking and proprotein convertase cleavage of cis Golgi protein GP73 produces marker for hepatocellular carcinoma. Traffic. 2007;8:1415. doi: 10.1111/j.1600-0854.2007.00621.x. [DOI] [PubMed] [Google Scholar]
- 5.Marrero JA, Romano PR, Nikolaeva O, Steel L, Mehta A, Fimmel CJ, et al. GP73, a resident Golgi glycoprotein, is a novel serum marker for hepatocellular carcinoma. J Hepatol. 2005;43:1007. doi: 10.1016/j.jhep.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 6.Ferenci P. Wilson’s Disease. Clin Gastroenterol Hepatol. 2005;3:726. doi: 10.1016/s1542-3565(05)00484-2. [DOI] [PubMed] [Google Scholar]
- 7.Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 8.Ferenci P, Steindl-Munda P, Vogel W, Jessner W, Gschwantler M, Stauber R, et al. Diagnostic value of quantitative hepatic copper determination in patients with Wilson’s Disease. Clin Gastroenterol Hepatol. 2005;3:811. doi: 10.1016/s1542-3565(05)00181-3. [DOI] [PubMed] [Google Scholar]
- 9.Kingston HM, Jassie LB. Microwave energy for acid decomposition at elevated temperatures and pressures using biological and botanical samples. Anal Chem. 1986;58:2534. doi: 10.1021/ac00125a038. [DOI] [PubMed] [Google Scholar]
- 10.Batts KP, Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am J Surg Pathol. 1995;19:1409. doi: 10.1097/00000478-199512000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Buiakova OI, Xu J, Lutsenko S, Zeitlin S, Das K, Das S, et al. Null mutation of the murine ATP7B (Wilson disease) gene results in intracellular copper accumulation and late-onset hepatic nodular transformation. Hum Mol Genet. 1999;8:1665. doi: 10.1093/hmg/8.9.1665. [DOI] [PubMed] [Google Scholar]
- 12.Huster D, Finegold MJ, Morgan CT, Burkhead JL, Nixon R, Vanderwerf SM, et al. Consequences of copper accumulation in the livers of the Atp7b−/− (Wilson disease gene) knockout mice. Am J Pathol. 2006;168:423. doi: 10.2353/ajpath.2006.050312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Bie P, Muller P, Wijmenga C, Klomp LW. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673. doi: 10.1136/jmg.2007.052746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panagiotakaki E, Tzetis M, Manolaki N, Loudianos G, Papatheodorou A, Manesis E, et al. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B) Am J Med Genet A. 2004;131:168. doi: 10.1002/ajmg.a.30345. [DOI] [PubMed] [Google Scholar]
- 17.Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, Madl C, et al. Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology. 1997;113:212. doi: 10.1016/s0016-5085(97)70097-0. [DOI] [PubMed] [Google Scholar]
- 18.Fox PL, Mukhopadhyay C, Ehrenwald E. Structure, oxidant activity, and cardiovascular mechanisms of human ceruloplasmin. Life Sci. 1995;56:1749. doi: 10.1016/0024-3205(95)00146-w. [DOI] [PubMed] [Google Scholar]
- 19.Ritland S, Skrede S, Johansen O. A long-term follow-up study of the hepatic copper and serum ceruloplasmin concentrations in patients with chronic liver disease. Scand J Gastroenterol. 1982;17:545. [PubMed] [Google Scholar]
- 20.Wright LM, Yong S, Picken MM, Rockey D, Fimmel CJ. Decreased survival and hepato-renal pathology in mice with C-terminally truncated GP73 (GOLPH2) Int J Clin Exp Pathol. 2009;2:34. [PMC free article] [PubMed] [Google Scholar]
- 21.Ferenci P. Pathophysiology and clinical features of Wilson disease. Metab Brain Dis. 2004;19:229. doi: 10.1023/b:mebr.0000043973.10494.85. [DOI] [PubMed] [Google Scholar]
- 22.Nagasaka H, Inoue I, Inui A, Komatsu H, Sogo T, Murayama K, et al. Relationship between oxidative stress and antioxidant systems in the liver of patients with Wilson disease: hepatic manifestation in Wilson disease as a consequence of augmented oxidative stress. Pediatr Res. 2006;60:472. doi: 10.1203/01.pdr.0000238341.12229.d3. [DOI] [PubMed] [Google Scholar]
- 23.Samuele A, Mangiagalli A, Armentero MT, Fancellu R, Bazzini E, Vairetti M, et al. Oxidative stress and pro-apoptotic conditions in a rodent model of Wilson’s disease. Biochim Biophys Acta. 2005;1741:325. doi: 10.1016/j.bbadis.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Lu Y, Yi Y, Liu P, Wen W, James M, Wang D, et al. Common human cancer genes discovered by integrated gene-expression analysis. PLoS ONE. 2007;2:e1149. doi: 10.1371/journal.pone.0001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varambally S, Laxman B, Mehra R, Cao Q, Dhanasekaran SM, Tomlins SA, et al. Golgi protein GOLM1 is a tissue and urine biomarker of prostate cancer. Neoplasia. 2008;10:1285. doi: 10.1593/neo.08922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwadate H, Ohira H, Suzuki T, Abe K, Yokokawa J, Takiguchi J, et al. Hepatocellular carcinoma associated with Wilson’s disease. Intern Med. 2004;43:1042. doi: 10.2169/internalmedicine.43.1042. [DOI] [PubMed] [Google Scholar]
- 27.Xu R, Bu-Ghanim M, Fiel MI, Schiano T, Cohen E, Thung SN. Hepatocellular carcinoma associated with an atypical presentation of Wilson’s disease. Semin Liver Dis. 2007;27:122. doi: 10.1055/s-2007-967203. [DOI] [PubMed] [Google Scholar]
- 28.Huster D, Purnat TD, Burkhead JL, Ralle M, Fiehn O, Stuckert F, et al. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J Biol Chem. 2007;282:8343. doi: 10.1074/jbc.M607496200. [DOI] [PubMed] [Google Scholar]
- 29.Puri S, Bachert C, Fimmel CJ, Linstedt AD. Cycling of early Golgi proteins via the cell surface and endosomes upon lumenal pH disruption. Traffic. 2002;3:641. doi: 10.1034/j.1600-0854.2002.30906.x. [DOI] [PubMed] [Google Scholar]
- 30.Lim CM, Cater MA, Mercer JF, La Fontaine S. Copper-dependent interaction of dynactin subunit p62 with the N terminus of ATP7B but not ATP7A. J Biol Chem. 2006;281:14006. doi: 10.1074/jbc.M512745200. [DOI] [PubMed] [Google Scholar]