

Abstract
Protein kinase Cι (PKCι) drives transformed growth of non-small cell lung cancer (NSCLC) cells through the Rho family GTPase Rac1. We show here that PKCι activates Rac1 in NSCLC cells by formation of a PKCι–Par6α complex that drives anchorage-independent growth and invasion through activation of matrix metalloproteinase-10 (MMP-10) expression. RNAi-mediated knockdown of PKCι, Par6α or Rac1 expression inhibits NSCLC transformation and MMP-10 expression in vitro. Expression of wild-type Par6α in Par6α-deficient cells restores transformation and MMP-10 expression, whereas expression of Par6α mutants that either cannot bind PKCι (Par6α-K19A) or couple to Rac1 (Par6α-ΔCRIB) do not. Knockdown of MMP-10 expression blocks anchorage-independent growth and invasion of NSCLC cells and addition of catalytically active MMP-10 to PKCι- or Par6α-deficient cells restores anchorage-independent growth and invasion. Dominant-negative PKCι inhibits tumorigenicity and MMP-10 expression in subcutaneous NSCLC tumors. MMP-10 and PKCι are coordinately overexpressed in primary NSCLC tumors, and tumor MMP-10 expression predicts poor survival in NSCLC patients. Our data define a PKCι–Par6α–Rac1 signaling axis that drives anchorage-independent growth and invasion of NSCLC cells through induction of MMP-10 expression.
Keywords: non-small cell lung cancer, Rac1, PB1 domain, cellular invasion, anchorage-independent growth
Introduction
We recently demonstrated that atypical protein kinase Cι (PKCι) is an oncogene in non-small cell lung cancer (NSCLC; Regala et al., 2005b). PKCι promotes anchorage-independent growth and tumorigenicity of NSCLC cells through the Rho family GTPase, Rac1 (Regala et al., 2005a). Expression of a dominant-negative, kinase-deficient PKCι (kdPKCι) in NSCLC cells inhibits Rac1 activity, and blocks anchorage-independent growth in culture and tumorigenicity in vivo (Regala et al., 2005a). Conversely, expression of a constitutively active Rac1 allele (RacV12) can restore anchorage-independent growth and tumorigenicity of NSCLC cells expressing kdPKCι (Regala et al., 2005a). Transgenic overexpression of the PB1 domain of PKCι (PKCι amino acids 1–113) in NSCLC cells inhibits Rac1 activity and transformation, consistent with a role for the PB1 domain of PKCι in the regulation of Rac1 (Regala et al., 2005a). The gold salt aurothiomalate (ATM) selectively inhibits the PB1–PB1 domain interaction between PKCι and Par6 in vitro by binding to cysteine 69 within the PB1 domain of PKCι (Erdogan et al., 2006; Stallings-Mann et al., 2006). ATM blocks Rac1 activity and inhibits anchorage-independent growth of NSCLC cells, suggesting a role for PB1–PB1 domain interactions between PKCι and Par6 in Rac1 activation and transformation (Erdogan et al., 2006; Stallings-Mann et al., 2006).
Here, we demonstrate the critical involvement of Par6α in NSCLC transformation. We find that Par6α is a key component of a PKCι–Par6α–Rac1 signaling axis that drives anchorage-independent growth and invasion of NSCLC cells. Reconstitution studies unambiguously demonstrate that PB1–PB1 domain interaction between PKCι and Par6α is necessary for Rac1 activity, and for anchorage-independent growth and invasion of NSCLC cells. In addition, we identify the matrix metalloproteinase-10 (MMP-10) as a critical gene target of the PKCι–Par6α–Rac1 signaling axis that is required for anchorage-independent growth and invasion of NSCLC cells. Finally, we show that PKCι and MMP-10 are coordinately overexpressed in primary human NSCLC tumors, and that MMP-10 expression is predictive of poor survival of NSCLC patients. These findings demonstrate a requisite role for Par6α in PKCι-dependent transformation and identify MMP-10 as a critical effector of the PKCι–Par6α complex in NSCLC cells.
Results
PB1–PB1 domain interactions between PKCι and Par6α are required for anchorage-independent growth and invasion of NSCLC cells
We first assessed whether PB1–PB1 domain interaction between PKCι and Par6 is required for NSCLC cell transformation using interfering RNA (RNAi) technology. Human H1703 NSCLC cells were chosen for analysis as they harbor PRKCI gene amplification, a commonly observed genetic alteration that drives PKCι expression in primary NSCLC tumors (Regala et al., 2005b). The target sequences of all RNAi reagents used in this study are given in Supplementary Figure 1. Cell populations stably expressing two independent lentiviral RNAi constructs targeting PKCι exhibited a significant reduction in PKCι mRNA abundance and protein expression (Supplementary Figure 2A). RNAi-mediated knockdown of PKCι correlated well with inhibition of anchorage-independent growth (Supplementary Figure 2B), consistent with our previous finding that expression of kdPKCι inhibits anchorage-independent growth of NSCLC cells (Regala et al., 2005a).
The atypical PKC isozymes PKCι and PKCζ share ~72% sequence homology at the amino-acid level, raising the possibility that RNAi to PKCι could affect PKCζ expression. However, quantitative real-time PCR (qPCR) demonstrated that PKCι-RNAi had no effect on PKCζ mRNA or protein expression, and that PKCζ-RNAi also had no effect on PKCι mRNA or protein expression (Supplementary Figures 2C and D). Furthermore, the inhibition of anchorage-independent growth observed in PKCι-RNAi cells was not seen in PKCζ-RNAi cells (Supplementary Figure 2E). These data demonstrate that the PKCι-RNAi and PKCζ-RNAi constructs are specific and that PKCι, but not PKCζ, plays a critical role in anchorage-independent growth of NSCLC cells.
We used a similar approach to evaluate the role of Par6 in NSCLC cell transformation. All three human Par6 isoforms, Par6α, Par6β and Par6γ, are expressed in H1703 cells (Supplementary Figure 2F). Given its abundance, we targeted Par6α for RNAi-mediated knockdown and functional analysis. Three independent lentiviral Par6α-RNAi constructs induced efficient knockdown of Par6α mRNA expression (Supplementary Figure 2G) and reduced anchorage-independent growth consistent with the level of Par6α mRNA knockdown (Supplementary Figure 2H). Par6α-RNAi caused specific knockdown of Par6α expression with no appreciable effect on Par6β or Par6γ mRNA abundance (Supplementary Figure 2I). Furthermore, Par6α-RNAi had no effect on PKCι mRNA; and PKCι-RNAi had no effect on Par6α mRNA demonstrating the specificity of these RNAi constructs (Supplementary Figure 2J). Thus, both PKCι and Par6α play a critical role in NSCLC cell anchorage-independent growth.
Expression of the PB1 domain of PKCι in NSCLC cells inhibits anchorage-independent growth (Regala et al., 2005a). Likewise ATM, which specifically binds the PB1 domain of PKCι, inhibits anchorage-independent growth of NSCLC cells (Erdogan et al., 2006; Stallings-Mann et al., 2006). Therefore, we assessed the ability of wild-type PKCι and two PB1 domain mutants of PKCι (PKCι-K20A and PKCι-D63A) to support anchorage-independent growth in PKCι-RNAi cells (Figure 1a). Our most efficient PKCι-RNAi construct targeted the 3′-UTR of the PKCι mRNA, making it possible to achieve stable PKCι transgene expression in PKCι-RNAi cells to levels similar to that of endogenous PKCι found in NT cells (Figure 1a, inset). Expression of either wild-type PKCι or PKCι-K20A restores anchorage-independent growth to levels similar to control NT cells whereas expression of PKCι-D63A does not (Figure 1a). The ability of PKCι mutants to support anchorage-independent growth correlated directly with their ability to bind Par6α (Supplementary Figure 3B). These data strongly indicate that binding of PKCι to Par6α is important for transformation.
Figure 1.
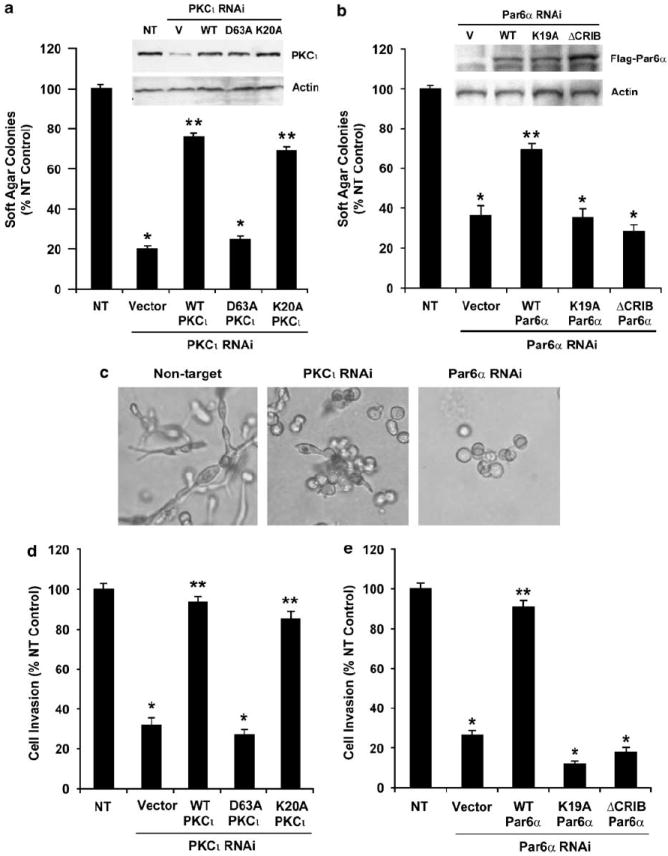
PB1–PB1 domain interaction between protein kinase Cι (PKCι) and Par6α is required for anchorage-independent growth and invasion of non-small cell lung cancer (NSCLC) cells. (a) A PKCι mutant that cannot bind Par6α (PKCι-D63A) does not support anchorage-independent growth. The indicated PKCι proteins were stably transfected into PKCι-RNAi cells and the cells were assessed for anchorage-independent growth. Results are expressed as % NT control. Values represent the mean ± s.e.m., n = 5. Asterisk (*) denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to PKCι-RNAi cells expressing empty pBabe control vector (Vector). Inset: Immunoblot analysis using PKCι antibody demonstrating similar expression of each PKCι mutant. Actin served as a control for loading. (b) Par6α mutants that cannot bind PKCι (Par6α-K19A) or couple to Rac1 (Par6-ΔCRIB) do not support anchorage-independent growth. Anchorage-independent growth in soft agar for NT and Par6α RNAi cells expressing the indicated Par6α mutant. Inset: Immunoblot analysis using anti-FLAG antibody demonstrating expression of each of the Par6α mutant proteins. Results represent mean ± s.e.m., n = 5. Asterisk (*) denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to Par6α-RNAi cells expressing empty control vector. (c) Representative photomicrographs of NT, PKCι-RNAi and Par6α-RNAi cells grown in three-dimensional Matrigel cultures. (d) Cellular invasion requires PKCι that can bind Par6α. NT and PKCι-RNAi cells expressing the indicated PKCι mutant were assessed for cellular invasion through Matrigel-coated chambers as described in Materials and methods. Results are expressed as % NT control. Values represent the mean ± s.e.m., n = 5. Asterisk (*) denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to PKCι-RNAi cells expressing empty pBabe control vector (Vector). (e) Cellular invasion requires Par6α that can bind PKCι and couple to Rac1. NT and Par6α-RNAi cells expressing the indicated Par6α mutant were assessed for cellular invasion through Matrigel-coated chambers as described in Materials and methods. Results are expressed as in (d) * denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to Par6α-RNAi cells expressing empty pBabe control vector (Vector).
Par6α binds PKCι through its PB1 domain and couples to Rac1 through a distinct Cdc42, Rac1 interaction binding (CRIB) domain (Joberty et al., 2000; Noda et al., 2001). To assess the importance of the PB1 and CRIB domains of Par6α in NSCLC transformation, we generated Par6α alleles mutated in these protein interaction domains (Supplementary Figure 3A). Wild-type Par6α binds PKCι whereas the PB1 domain mutant Par6α-K19A does not (Supplementary Figure 3C). The Par6α CRIB domain mutant (Par6α-ΔCRIB) impairs binding of Par6α to Rac1 (Qiu et al., 2000) without affecting PKCι binding (Supplementary Figure 3C). To assess the ability of these Par6α proteins to support anchorage-independent growth, we expressed FLAG-tagged Par6α mutants in Par6α-RNAi cells (Figure 1b). As our most efficient Par6α-RNAi construct targeted the coding region of the Par6α mRNA, we introduced two silent mutations within the target region of the human Par6α cDNA to generate RNAi-resistant mutants as described in Supplementary Materials and methods. Immunoblot analysis demonstrated similar levels of wild-type Par6α, Par6α-K19A and Par6α-ΔCRIB mutants in these cells (Figure 1b, inset). Expression of wild-type Par6α significantly restored anchorage-independent growth in Par6α-RNAi cells whereas neither the PKCι-binding mutant Par6α-K19A nor the Rac1 uncoupled mutant Par6α-ΔCRIB did (Figure 1b).
H1703/NT cells grown in three-dimensional Matrigel cultures exhibited elongated cell bodies and prominent cellular protrusions invading into the surrounding matrix (Figure 1c, left panel), morphology consistent with a highly invasive phenotype (Kleinman and Martin, 2005). In contrast, both PKCι-RNAi and Par6α-RNAi cells exhibited rounded morphology with few cellular projections, suggesting a less invasive phenotype (Figure 1c, middle panel and right panel). Invasion assays confirmed that NT cells are highly invasive whereas PKCι-RNAi and Par6-RNAi cells exhibit a significantly reduced invasive potential (Figures 1d and e). Invasion of PKCι-RNAi cells was significantly restored by re-expression of either wild-type PKCι or PKCι-K20A but not by PKCι-D63A (Figure 1d). Likewise, cellular invasion was restored in Par6α-RNAi cells by re-expression of wild-type Par6α but not Par6α-K19A or Par6α-ΔCRIB (Figure 1e). Taken together, these data demonstrate that the PB1–PB1 domain interaction between PKCι and Par6α is required for both anchorage-independent growth and invasion of NSCLC cells.
Rac1 is a critical effector of PKCι and Par6α
Our published data demonstrate that Rac1 is a critical downstream effector of PKCι in NSCLC cells (Regala et al., 2005a; Stallings-Mann et al., 2006). Figure 1 demonstrates that Par6α plays a requisite role in transformation that involves both the PB1 domain and the CRIB domain of Par6α. These data predict that Rac1 activity in NSCLC is regulated by the PKCι–Par6α complex. Consistent with this hypothesis, both PKCι-RNAi and Par6α-RNAi cells exhibited decreased Rac1 activity compared to NT cells (Figure 2a). Furthermore, expression of a constitutively active Rac1 mutant, RacV12, restored anchorage-independent growth in both PKCι-RNAi and Par6α-RNAi cells although having no significant effect on NT cells (Figure 2b). Expression of RacV12 in PKCι-RNAi and Par6α-RNAi cells also restored the invasive morphology in three-dimensional Matrigel culture (Figure 2c) and invasion through Matrigel-coated chambers (Figure 2d). Thus, Rac1 is a critical effector of PKCι-Par6α in NSCLC transformation. To directly assess the importance of Rac1 in NSCLC cell transformation, we generated Rac1-RNAi cells that exhibit efficient knockdown of Rac1 mRNA and protein expression (Supplementary Figure 4A). Rac1-RNAi cells exhibited decreased anchorage-independent growth similar to that observed in PKCι-RNAi and Par6α-RNAi cells (Supplementary Figure 4B). Furthermore, Rac1-RNAi cells exhibit rounded morphology in three-dimensional Matrigel culture and significantly reduced cellular invasion (Supplementary Figures 4C and D). Expression of constitutively active PKCι in Rac1 RNAi cells was unable to restore cellular invasion, confirming that Rac1 is required downstream of PKCι in transformation (Supplementary Figure 4E). Thus, Rac1 activity is regulated in NSCLC cells by the PKCι–Par6α complex and Rac1 is required downstream of this complex for NSCLC cell transformation.
Figure 2.
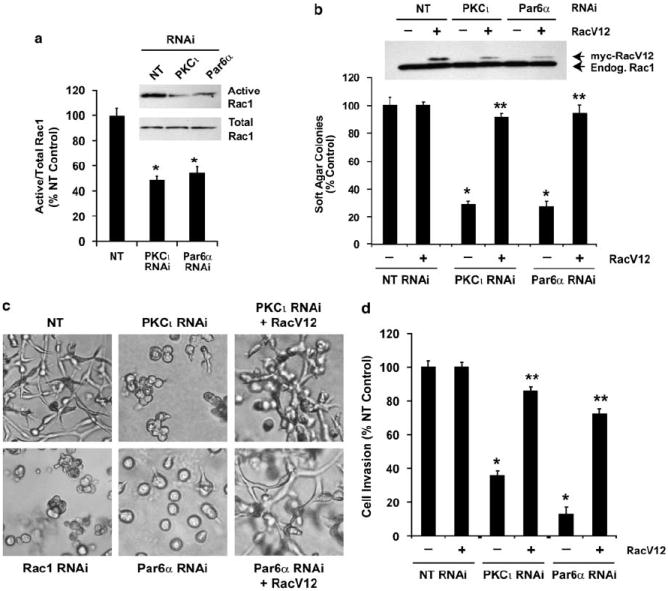
Rac1 is a critical effector of protein kinase Cι (PKCι)-, Par6α-dependent transformation. (a) RNAi-mediated knockdown of PKCι and Par6α inhibits cellular Rac1 activity. NT, PKCι-RNAi and Par6α-RNAi cells were assayed for Rac1 activity as described in Materials and methods. Representative immunoblot results are shown for active (GTP-bound) and total Rac1. Quantitative analysis of Rac1 activity from NT, PKCι-RNAi and Par6α-RNAi cells is also shown. Results represent the mean of the ratio of Active to Total Rac1 ± s.e.m. and are presented as % NT control, n = 3. Results are representative of three independent experiments. (b) RacV12 reconstitutes anchorage-independent growth in PKCι and Par6α-deficient cells. NT, PKCι-RNAi and Par6α-RNAi cells were transfected with LZRS retrovirus expressing RacV12 (+) or an empty control (−) LZRS virus and assessed for anchorage-independent growth in soft agar. Results are presented as % NT control and values represent the mean ± s.e.m., n = 5. Asterisk (*) denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to corresponding RNAi cells expressing empty control LZRS virus (−). (c) Cellular morphology of NT, PKCι-RNAi, Par6α-RNAi, Rac1-RNAi, PKCi-RNAi + RacV12 and Par6α-RNAi + RacV12 cells grown in three-dimensional Matrigel culture. (d) RacV12 reconstitutes invasion in PKCι and Par6α-deficient cells. NT, PKCι-RNAi and Par6α-RNAi cells were transfected with LZRS retrovirus expressing RacV12 (+) or an empty control (−) LZRS virus and assessed for invasion through Matrigel-coated chambers. Results are presented as % NT control and values represent the mean ± s.e.m., n = 5. Asterisk (*) denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to corresponding RNAi cells expressing empty control LZRS virus (−).
Identification of MMP-10 as a target of oncogenic PKCι in NSCLC
We next determined downstream targets of PKCι involved in transformation by conducting gene expression analysis of H1703 NT and PKCι-RNAi cells (see Figure 1a). This analysis identified 10 candidate PKCι target genes based on fold-change (> twofold) and P-value (< 0.05) listed in Table 1. PKCι was among the 10 regulated genes, confirming efficient knockdown of PKCι mRNA in the cells used for the expression analysis.
Table 1.
Protein kinase Cι gene targets in primary lung cancers
| H1703 | Tumor samples |
Gene name | Target RefSeqs | ||
|---|---|---|---|---|---|
| PKCι RNAi/NT | P-value | PKCι Lo/Hi | P-value | ||
| 0.139 | 0.001 | 2.062 | 0.943 | kynureninase (l-kynurenine hydrolase) | NM_003937 |
| 0.167 | 0.001 | 0.595 | 0.063 | stanniocalcin 1 | NM_003155 |
| 0.199 | 0.001 | 0.173 | 0.001 | matrix metallopeptidase 10 (stromelysin 2) | NM_002425 |
| 0.222 | 0.001 | 0.205 | 0.001 | protein kinase C iota | NM_002740 |
| 0.254 | 0.006 | 0.104 | 0.001 | keratin-associated protein 2-1|keratin-associated protein 2-4 | NM_203405 |
| 3.915 | 0.001 | 0.864 | 0.075 | pleckstrin homology-like domain, family B, member 2 | NM_145753 |
| 3.502 | 0.027 | 1.189 | 0.541 | SMC1 structural maintenance of chromosomes 1-like 2 (yeast) | NM_148674 |
| 3.500 | 0.046 | 1.671 | 0.821 | olfactory receptor, family 2, subfamily H, member 1 | NM_030883 |
| 3.412 | 0.001 | 0.462 | 0.009 | KDEL (Lys-Asp-Glu-Leu) containing 2 | NM_153705 |
| 7.392 | 0.008 | 0.462 | 0.109 | aldehyde dehydrogenase 1 family, member A1 | NM_000689 |
Bold face indicates PKCι gene targets that correlate with PKCι expression in primary lung cancers.
To identify PKCι target genes most likely to be relevant to the human disease, we interrogated a public domain database containing gene expression data from 35 primary human NSCLC tumors (Garber et al., 2001) for correlations between the expression of each of the nine identified genes and PKCι (Table 1). Kendall’s τ rank correlation analysis revealed that only two of the nine candidate genes, Keratin associated protein 26–1 (KRTAP 26-1) and MMP-10, showed a significant correlation with PKCι expression in primary lung tumors consistent with that observed in H1703 cells. A third gene, KDEL, showed an association with PKCι in primary tumors that was opposite to that predicted from the expression analysis of H1703 cells. MMP-10, also known as stromelysin 2, was judged to be the most promising candidate as a target for PKCι-mediated transformation and was subjected to further analysis.
MMP-10 expression is regulated through the PKCι–Par6α–Rac1 signaling axis
We next assessed whether MMP-10 expression is regulated through the PKCι–Par6α–Rac1 signaling axis. qPCR analysis revealed that MMP-10 expression is significantly inhibited in PKCι-RNAi, Par6α-RNAi and Rac-RNAi NSCLC cells when compared to NT control cells (Figure 3a). Furthermore, expression of wild-type Par6α in Par6α-RNAi cells restored MMP-10 expression whereas neither Par6α-K19A nor Par6α-ΔCRIB did so (Figure 3b). Finally, expression of RacV12 in PKCι-RNAi and Par6α-RNAi cells restored MMP-10 expression to levels comparable to NT cells while having little effect on MMP-10 expression in NT cells (Figure 3c). Thus, MMP-10 expression is regulated through the PKCι–Par6α–Rac1 signaling axis.
Figure 3.
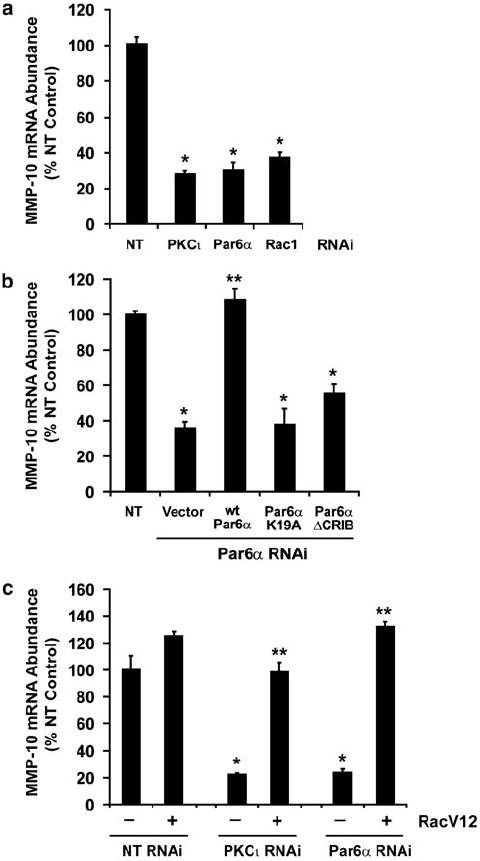
Matrix metalloproteinase-10 (MMP-10) expression in non-small cell lung cancer (NSCLC) cells is regulated through the protein kinase Cι (PKCι)–Par6α–Rac1 signaling axis. (a) MMP-10 expression is regulated by PKCι, Par6α and Rac1. MMP-10 mRNA abundance was determined in NT, PKCι-RNAi, Par6α-RNAi and Rac1-RNAi cells by quantitative real-time PCR (qPCR) as described in Materials and methods. Results are expressed as % NT control. Values represent the mean ± s.d.; n = 3. Asterisk (*) denotes P < 0.05 compared to NT control. (b) MMP-10 expression is regulated through the PKCι–Par6α–Rac1 signaling axis. qPCR was used to assess MMP-10 mRNA abundance in NT cells and in Par6α-RNAi cells expressing either pCMV vector control, wild-type Par6α, Par6α-K19A or Par6α-ΔCRIB. Results are expressed as % NT control. Values represent the mean ± s.d.; n = 3. Asterisk (*) denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to Par6α-RNAi/vector cells. (c) MMP-10 expression is restored by expression of RacV12 in PKCι-RNAi and Par6α-RNAi cells. NT, PKCι-RNAi and Par6α-RNAi cells were stably transfected with LZRS virus expressing RacV12 or empty control LZRS virus. MMP-10 mRNA abundance was determined by qPCR as described above. Results are expressed as % NT control. Values represent mean ± s.d.; n = 3. Asterisk (*) denotes P < 0.05 compared to NT control. ** denotes P < 0.05 compared to the indicated RNAi cells expressing control LZRS vector.
MMP-10 is required for anchorage-independent growth and invasion of NSCLC cells
We next assessed whether MMP-10 plays an important role in NSCLC transformation. Two independent RNAi constructs targeting MMP-10 significantly knocked down MMP-10 RNA abundance (Figure 4a) and caused a commensurate inhibition of anchorage-independent growth (Figure 4b). A similar inhibition of anchorage-independent growth was observed in cells treated with the general MMP inhibitor GM6001 (Figure 4b). MMP-10-RNAi inhibited MMP-10 mRNA but had no effect on PKCι, Par6α or Rac1 mRNA abundance (Figure 4c) indicating that the cellular effects of MMP-10 knockdown are not caused by regulating expression these genes. In addition, both MMP-10-RNAi cells and cells treated with GM6001, exhibited rounded morphology in three-dimensional Matrigel cultures (Figure 4d) and reduced invasion through Matrigel-coated chambers (Figure 4e). Thus, MMP-10 plays a critical role in NSCLC anchorage-independent growth and invasion of NSCLC cells.
Figure 4.
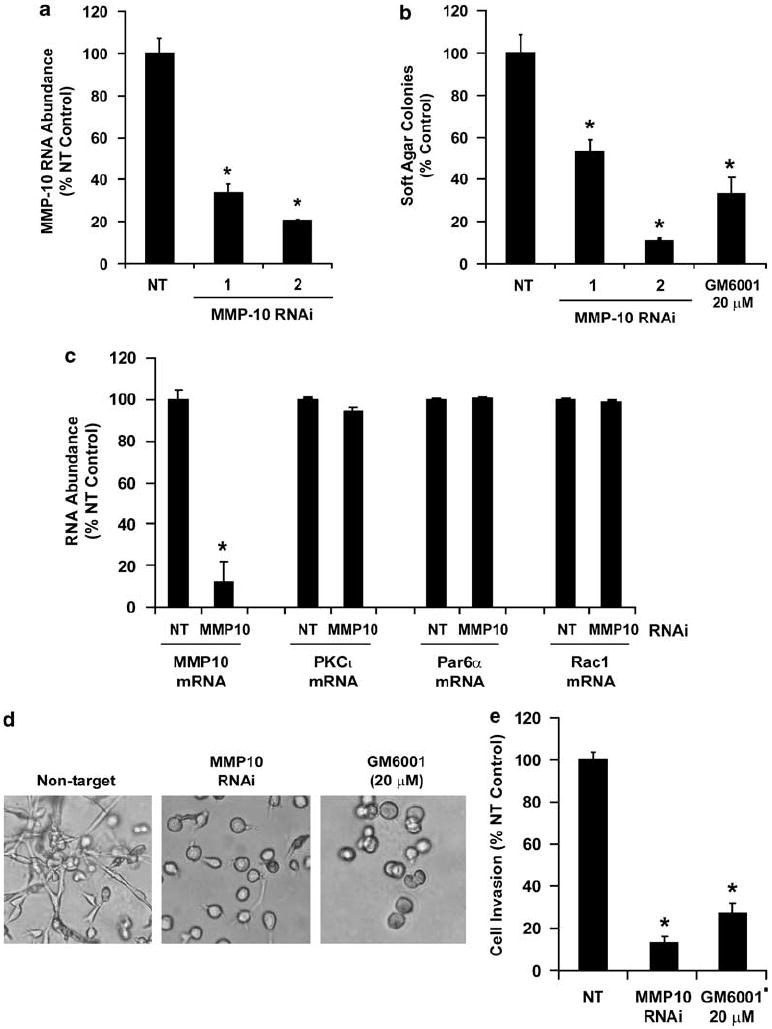
Matrix metalloproteinase-10 (MMP-10) plays a critical role in anchorage-independent growth and invasion of non-small cell lung cancer (NSCLC) cells. (a) RNAi-mediated knockdown of MMP-10 expression using three independent lentiviral MMP-10-RNAi constructs. Quantitative real-time PCR (qPCR) results are expressed as % NT control. Values represent the mean ± s.d., n = 3. Asterisk (*) denotes P < 0.05 relative to NT control. (b) MMP-10 RNAi inhibits anchorage-independent growth. Effect of MMP-10-RNAi constructs and treatment with the MMP inhibitor GM6001 (20 μm) on anchorage-independent growth in soft agar. Results represent the mean ± s.e.m., n = 5 and are expressed as % NT control cells; * denotes P < 0.005 relative to NT control. (c) MMP-10 RNAi selectively inhibits MMP-10. qPCR was used to assess expression of MMP-10, protein kinase Cι (PKCι), Par6α and Rac1 in NT and MMP-10 RNAi cells as described in Materials and methods. Results are expressed as % NT control and values represent the mean ± s.d.; n = 3. Asterisk (*) denotes P < 0.005 relative to NT control. (d) Representative photomicrographs of NT, MMP-10-RNAi, and NT cells treated with the MMP inhibitor GM6001 (20 μm) grown in three-dimensional Matrigel culture. (e) Effect of MMP-10 RNAi and GM6001 treatment on cellular invasion through Matrigel-coated chambers. Results represent the mean ± s.e.m., n = 5. Results are expressed as % NT control; * denotes P < 0.005 relative to NT control.
MMP-10 is a critical effector of PKCι-mediated transformation
We next assessed whether MMP-10 is a critical effector of the oncogenic PKCι–Par6α–Rac1 signaling axis. For this purpose, we assessed the ability of catalytically active recombinant MMP-10 enzyme to reconstitute anchorage-independent growth (Figure 5a) and cellular invasion (Figure 5b) in PKCι- and Par6α-RNAi cells. Addition of recombinant MMP-10 significantly restored anchorage-independent growth and invasion to both PKCι- or Par6α-deficient cells. Interestingly, addition of an equivalent amount of catalytically active MMP-3 (stromelysin 1), the MMP species most closely related to MMP-10, did not restore anchorage-independent growth or invasion, indicating that MMP-10 plays a selective role in cellular transformation downstream of PKCι and Par6α.
Figure 5.
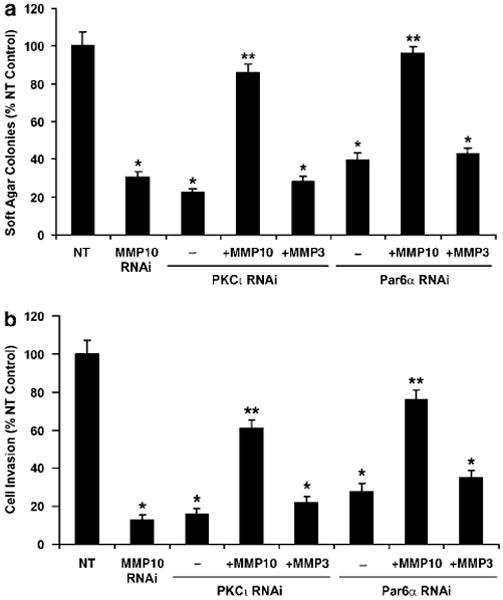
Matrix metalloproteinase-10 (MMP-10) is a critical effector of protein kinase Cι (PKCι) and Par6α-mediated transformation of non-small cell lung cancer (NSCLC) cells. (a) Active MMP-10 enzyme restores anchorage-independent growth to PKCι- and Par6α-deficient cells. NT, MMP-10-RNAi, PKCι-RNAi and Par6α-RNAi cells were plated in soft agar and assessed for anchorage-independent growth as described in Materials and methods. PKCι-RNAi and Par6α-RNAi cells were plated in the presence of 10 U/ml of recombinant, catalytically active human MMP-10 (+ MMP-10), MMP-3 (+ MMP-3) or diluent (−) as indicated. Results are presented as % NT control. Values represent the mean ± s.e.m., n = 5. Asterisk (*) denotes P < 0.05 compared to NT control; ** denotes P < 0.05 compared to indicated RNAi cells in the absence of MMP-10. (b) MMP-10 restores cellular invasion in PKCι- and Par6α-deficient cells. Cell invasion through Matrigel-coated chambers was performed as described in Materials and methods. MMP treatments and data analyses are as described in (a).
PKCι regulates MMP-10 expression in NSCLC tumors in nude mice
We next assessed whether PKCι regulates MMP-10 expression in NSCLC tumors in vivo. We previously showed that expression of a dominant-negative, kinase-deficient allele of PKCι (kdPKCι) in A549 NSCLC cells inhibits tumorigenicity in nude mice (Regala et al., 2005a). A549 NSCLC cell tumors expressing kdPKCι were significantly smaller than A549 cell tumors expressing the control expression plasmid pBabe (Figure 6a). qPCR analysis revealed a significant reduction in MMP-10 mRNA in A549/kdPKCι tumors, whereas expression of the most well-characterized MMP species expressed in these cells, MMP-2 and MMP-9, was unaffected by kdPKCι expression (Figure 6b). Immunohistochemical analysis of A549/kdPKCι and A549/pBabe tumors confirmed reduced expression of MMP-10 protein in kdPKCι-expressing tumors (Figure 6c). Thus, PKCι selectively regulates MMP-10 expression in NSCLC cell tumors in vivo.
Figure 6.
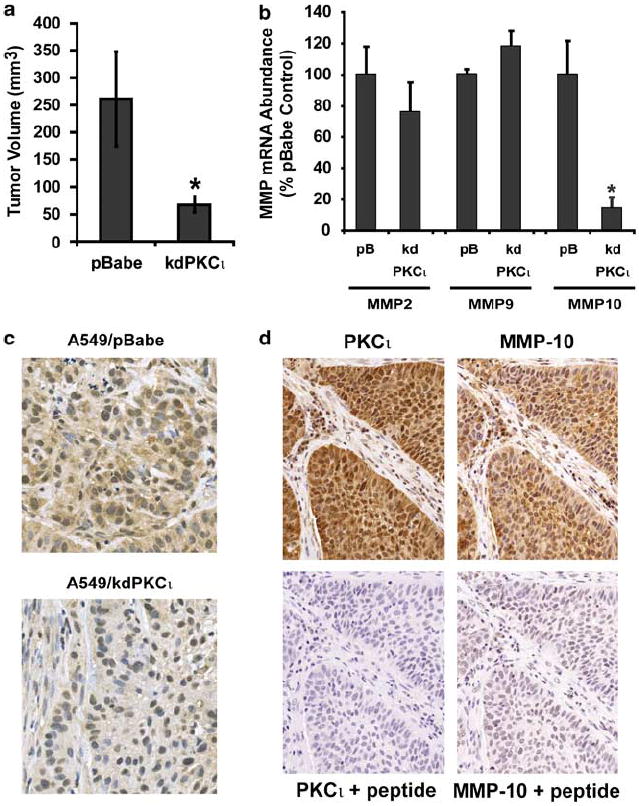
Expression of dominant-negative, kinase-deficient protein kinase Cι (PKCι) inhibits matrix metalloproteinase-10 (MMP-10) expression in non-small cell lung cancer (NSCLC) tumors in vivo. A549 NSCLC lung cancer cells were stably transfected with a dominant-negative, kinase-deficient PKCι allele (kdPKCι) or a control vector, pBabe. Tumor cells were injected subcutaneously into nude mice as described in Materials and methods. (a) Expression of kdPKCι inhibits tumor growth. A549/pBabe and A549/kdPKCι tumors were measured 15 days after subcutaneous inoculation as described in Materials and methods. Results are expressed as tumor volume (mm3). Values represent the mean ± s.e.m., n = 6. Asterisk (*) denotes P < 0.05 compared to pBabe control. (b) RNA from A549/pBabe and A549/kdPKCι tumors was isolated and assessed for MMP-2, MMP-9 and MMP-10 mRNA abundance by quantitative real-time PCR (qPCR) as described in Materials and methods. Data are expressed as % pBabe control. Values represent the mean ± s.e.m.; n = 6. Asterisk (*) denotes P < 0.05 compared to pBabe control. (c) MMP-10 protein expression is inhibited in A549/kdPKCι tumors. Immunohistochemistry (IHC) for MMP-10 was performed on A549/pBabe and A549/kdPKCι tumors as described in Materials and methods. Results are representative of the six tumors in each genotype. (d) PKCι and MMP-10 are coexpressed in primary NSCLC. IHC for PKCι and MMP-10 from a representative primary NSCLC tumor is shown. IHC was performed as described in Materials and methods. Specificity of the staining for PKCι and MMP-10 was confirmed by inclusion of specific antigen peptide in the primary antibody preparation (+ peptide). Higher magnification images can be seen in Supplementary Figure 5.
Unlike most other MMP species, MMP-10 is expressed primarily in NSCLC tumor cells not the surrounding stromal elements (Cho et al., 2004; Gill et al., 2004), a pattern of expression similar to that of PKCι in NSCLC tumors (Regala et al., 2005b). Immunohistochemical analysis demonstrated coexpression of high levels of PKCι and MMP-10 in primary NSCLC tumor cells (Figure 6d). Specificity of immunohistochemical staining for PKCι and MMP-10 was confirmed using an excess of antigenic peptide.
MMP-10 expression in human NSCLC tumors predicts poor survival
We next assessed whether there was a direct correlation between PKCι and MMP-10 expression in NSCLC tumors. The open source genomic profiles of 35 primary NSCLC tumor samples were organized into tertiles based on PKCι expression as described in Supplementary Materials and methods. The median value for PKCι expression for the high tertile was 1762 (n = 11, range 947–4502), whereas the median value for the middle tertile was 746 (n = 12, range 545–947, P > 0.0007 relative to the high group) and the median for the low tertile was 357 (n = 12, range 148–527, P > 0.0007 relative to the high group). The analysis revealed a statistically significant correlation between PKCι and MMP-10 expression across these groups (Figure 7a).
Figure 7.
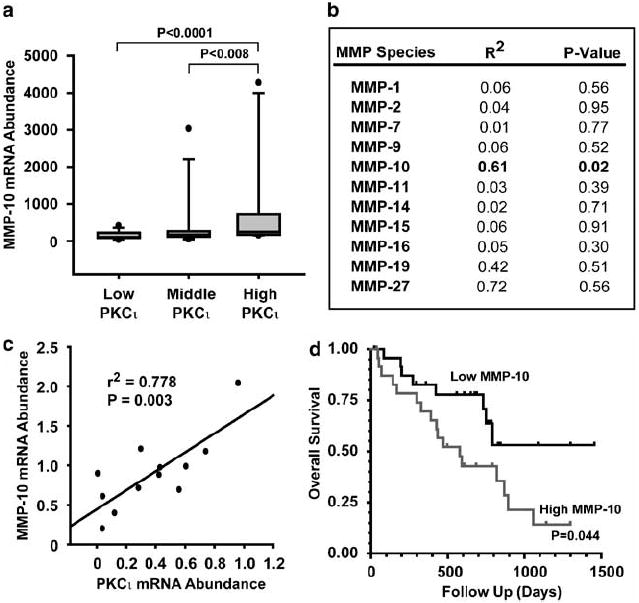
Matrix metalloproteinase-10 (MMP-10) is overexpressed in primary non-small cell lung cancer (NSCLC) tumors. (a) MMP-10 expression correlates with protein kinase Cι (PKCι) expression in primary NSCLC tumors. Microarray data for 35 primary NSCLC were analysed for PKCι and MMP-10 expression as described in Materials and methods. Cases were force ranked based on PKCι expression, binned into tertiles and assessed for correlation between PKCι and MMP-10 expression across the groups. P-values are shown for the comparisons across the groups. (b) The correlation between PKCι and MMP-10 expression is specific. Linear regression analysis of microarray expression data for a correlation between PKCι and MMP species. A correlation coefficient and P-value for each potential correlation is presented. (c) MMP-10 and PKCι expression correlate in stage I NSCLC tumors. Linear regression analysis demonstrates a positive correlation between PKCι and MMP-10 mRNA abundance in stage I NSCLC cases. Twelve stage I primary NSCLC tumors were analysed for PKCι and MMP-10 expression by quantitative real-time PCR (qPCR). (d) MMP-10 expression predicts poor survival in NSCLC patients. Sixty primary NSCLC tumors were analysed for MMP-10 expression by qPCR and the cases stratified into quartiles based on MMP-10 expression. Kaplan–Meier survival analysis was performed and survival curves for the first (high MMP-10) and fourth (low MMP-10) quartiles are shown. P = 0.044 between the low and high MMP-10 groups.
MMP-10 is a member of a large family of structurally related matrix metalloproteases, many members of which have been implicated in various aspects of tumor biology (Egeblad and Werb, 2002). Therefore, linear regression analysis was used to determine whether PKCι correlated with expression of other MMP species present in the dataset. Of the 11 MMP species present, only MMP-10 expression showed a statistically significant correlation with PKCι expression (R2 = 0.61; P = 0.02) demonstrating that the correlation between PKCι and MMP-10 is highly specific (Figure 7b).
Both MMP-10 and PKCι expression is elevated in NSCLC independent of tumor stage, suggesting that elevated expression of these genes is an early event in lung carcinogenesis (Gill et al., 2004; Regala et al., 2005b). qPCR analysis of 12 stage I NSCLC cases obtained from the Mayo Clinic lung tumor bank demonstrated a strong positive correlation between PKCι mRNA and MMP-10 mRNA abundance (r2 = 0.778, P = 0.003; n = 12; Figure 7c). Furthermore, Kaplan–Meier analysis of 60 NSCLC cases for which survival data were available demonstrated that NSCLC patients whose tumors express high MMP-10 levels (top quartile) exhibited significantly worse survival than those whose tumors express low MMP-10 (bottom quartile; Figure 7d). Thus, MMP10 expression profiling may be of prognostic significance.
Discussion
Atypical PKCι is an oncogene that drives anchorage-independent growth through the Rho family GTPase Rac1 (Regala et al., 2005b). Here we provide direct genetic evidence that the polarity protein Par6α plays a requisite role in NSCLC transformation by binding PKCι and coupling PKCι to Rac1. PKCι or Par6α mutants that are incapable of binding to each other do not support transformation, nor does a Par6α mutant that is uncoupled from Rac1. Taken together, these data define a PKCι–Par6α–Rac1 signaling axis that is required for anchorage-independent growth and invasion. Our present results are consistent with our earlier work suggesting the involvement of the PB1 domain of PKCι in anchorage-independent growth of NSCLC cells (Regala et al., 2005a; Erdogan et al., 2006; Stallings-Mann et al., 2006) and provide conclusive genetic and biochemical evidence that Rac1 is a critical downstream effector of PKCι and Par6α in NSCLC transformation. However, we cannot formally rule out the possibility of an additional role for Rac1 as an upstream modulator of the PKCι–Par6 complex. Accumulating evidence demonstrates that the two atypical PKC isozymes, PKCι and PKCζ, are not functionally redundant but rather serve distinct, often divergent, roles in many cell types (reviewed in Fields and Regala, 2007). Our data demonstrate that PKCι and PKCζ play distinct roles in lung tumorigenesis; whereas PKCι is required for NSCLC transformation, PKCζ is dispensible. Our results are consistent with the fact that PKCι, but not PKCζ, is overexpressed in primary NSCLC tumors and cell lines, and that PRKCI is uniquely targeted for genetic alteration in NSCLC tumors (Regala et al., 2005a, b).
A major goal of this study was to identify critical target(s) of PKCι that mediate cellular transformation of NSCLC cells. Our identification of MMP-10 as a critical effector of oncogenic PKCι is both surprising and novel. The MMPs are a large family of structurally related zinc-dependent endoproteases that play central roles in tumor invasion, metastasis, angiogenesis and tumor cell proliferation (Egeblad and Werb, 2002). However, relatively little is known about MMP-10 function in human cancer. The stromelysin subfamily of MMPs (stromelysin 1 (MMP-3), stromelysin 2 (MMP-10) and stromelysin 3 (MMP-11)) is overexpressed in NSCLC (Delebecq et al., 2000; Bodey et al., 2001; Gill et al., 2004; Kren et al., 2006). Interestingly, MMP-3 and MMP-11 are predominantly expressed in stromal elements surrounding lung tumors whereas MMP-10 is highly overexpressed in NSCLC tumor cells but not stroma (Gill et al., 2004). MMP-10 expression is elevated in NSCLC tumors independent of tumor grade, stage, type or lymph node status, suggesting that MMP-10 is associated with early tumor growth (Gill et al., 2004). Interestingly, higher levels of MMP-10 are observed in primary stage IB NSCLC tumors that recur following surgical resection (Cho et al., 2004), suggesting that MMP-10 expression could be useful in identifying NSCLC patients at high risk of recurrence. Our data also indicate that MMP-10 expression profiling may be of prognostic significance. Interestingly, MMP-10 overexpression has also been observed in squamous cell carcinomas of the head and neck (Muller et al., 1991; O-Charoenrat et al., 2001), oral cavity (Impola et al., 2004) and esophagus (Mathew et al., 2002). These tumor types harbor frequent chromosome 3q26 and PRKCI amplification (Pimkhaokham et al., 2000; Imoto et al., 2001; Snaddon et al., 2001; Osada and Takahashi, 2002). It is tempting to speculate that these tumors overexpress MMP-10 as a result of PRKCI amplification and resultant PKCι overexpression.
Despite a considerable literature demonstrating that MMP-10 is overexpressed in human tumors, only one study has assessed the potential functional role of MMP-10 in tumorigenesis. Overexpression of exogenous MMP-10 in mouse T-cell lymphoma cells leads to more rapidly growing tumors in nude mice than control cells, suggesting a role for MMP-10 in lymphoma cell growth (Van Themsche et al., 2004). However, the question of whether endogenous MMP-10 was required for tumor growth was not assessed in this study. Our current study provides direct evidence that MMP-10 is a critical effector of the PKCι–Par6α–Rac1 signaling axis that is required for anchorage-independent growth and invasion of NSCLC cells. The fact that recombinant active MMP-10, but not MMP-3, can support NSCLC transformation suggests that these two highly related MMP species serve distinct, nonoverlapping functions in NSCLC cell biology. Future investigation will be required to elucidate the molecular mechanism(s) by which MMP-10 exerts its oncogenic effects. In summary, our present study provides important new insights into the role of PKCι signaling in NSCLC transformation. We demonstrate that Par6α plays a critical role in transformation as a component of an oncogenic PKCι–Par6α–Rac1 signaling axis that drives anchorage-independent growth and invasion through specific induction of MMP-10 expression. Our data also demonstrate that MMP-10 may be a useful prognostic marker for NSCLC patients, and an attractive target for development of targeted therapies for treatment of NSCLC.
Materials and methods
Cell lines, antibodies and enzymes
Human H1703 and A549 NSCLC cell lines were obtained from American Type Culture Collection (Manassas, VA, USA) and maintained as suggested by the supplier. Antibodies used were as follows: PKCι (BD Transduction Laboratories, Franklin Lakes, NJ, USA) cat no. 610176 for immunoblot analysis; Santa Cruz Biotechnology (sc-727) for immunohistochemistry), PKCζ (Cell Signaling, Beverly, MA, USA, 9372), actin (Cell Signaling 14967), FLAG (Sigma A-8592), Rac1 (BD Transduction Laboratories 610651), GFP (Molecular Probes/Invitrogen, Carlsbad, CA, USA, A11120) and MMP-10 (Abcam 38930). The MMP inhibitor GM6001 was from Calbiochem, San Diego, CA, USA (364206). Recombinant human MMP-10 (30.7 U/μg; 1 U = 100 pmol/min at 37 °C) was obtained from Biomol (SE-329). Recombinant human PKCι and PKCζ proteins were from Upstate Biotechnology (Lake Placid, NY, USA). Recombinant human MMP-3 (81 U/μg) was a generous gift from Dr E Radisky (Mayo Clinic, Jacksonville, FL, USA).
Lentiviral RNAi-mediated gene knockdown and qPCR
Lentiviral vectors containing short hairpin RNAi against human PKCι, PKCζ, Par6α, Rac1 and MMP-10 were obtained from Sigma-Aldrich Mission shRNA library (St Louis, MO, USA). A nontarget control lentiviral vector containing a short hairpin that does not recognize any human or mouse genes (NT-RNAi) was used as a negative control in all RNAi experiments. For RNAi transfection, cells were seeded in 100-mm plates and grown to 70–80% confluency. The supernatant was removed from the cells and 3 ml of complete culture media containing polybrene (6 μg/ml) was added. After 10 min at room temperature, 400 μl of viral supernatant (multiplicity of infection ~ 3) was added. Following 24 h incubation at 37 °C, cells were washed and grown for 24 h in 10ml of fresh culture media containing 10% fetal bovine serum. Populations of stably transfected cells were selected in 5 μg/ml puromycin. RNAi constructs were analysed for efficiency of target gene knockdown using qPCR assays using TaqMan technology from Applied Biosystems (Foster City, CA, USA). All RNAi target sequences used in this study are provided in supplemental materials (Supplementary Figure 1). qPCR assays and reagents for human PKCι, PKCζ, Par6α, Par6β, Par6γ, Rac1, MMP-2, MMP-9 and MMP-10 were obtained from Applied Biosystems and gene expression was analysed on an Applied Biosystems 7900HT sequence analyser.
Soft agar growth assays
Anchorage-independent growth was assayed by the ability of cells to grow as colonies in soft agar as described previously (Regala et al., 2005a). In some cases, the selective MMP inhibitor GM6001 (20 μm) was added to the agar. Cell colonies were visualized and quantified under a dissecting microscope (Olympus, Melville, NY, USA) after 4 weeks in culture.
Three-dimensional cultures
H1703 cell transfectants were seeded into 96-well plates (1000 cells per well) onto a layer of 35 μl of Matrigel Growth Factor Reduced Basement Membrane Matrix (BD Biosciences, San Jose, CA, USA). Once attached, 150 μl of 10% Matrigel Basement Membrane Matrix containing 1% serum diluted in culture medium was placed on top of the cells. This top layer was removed and replenished every other day. Cells were visualized microscopically (Olympus) 7 days after plating and cell were photographed to observe cellular morphology. Images were captured using ImagePro software.
Cellular invasion assay
Cellular invasion was measured in 24-well plate transwell chambers containing inserts coated with Matrigel basement membrane (Corning Costar, Cambridge, MA, USA) as described previously (Zhang et al., 2004). In some experiments, the selective MMP inhibitor GM6001 (20 μm) was added to medium in the upper and lower chambers. Cell attachment was verified using control inserts coated with collagen (Becton Dickinson, Franklin Lakes, NJ, USA) and was unaffected by any of the genetic disruptions.
Immunohistochemistry
Immunohistochemistry was performed on 5-μm paraffin-embedded sections as described previously (Regala et al., 2005b). PKCι was detected using a PKCι antibody (1:400 dilution in phosphate buffered saline (PBS)/Tween 20; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and MMP-10 was detected using an MMP10 antibody (1:100 dilution in PBS/Tween 20; Abcam, Cambridge, MA, USA). Specificity of immunostaining was confirmed by antigen peptide competition in which a 200-fold molar excess of PKCι or MMP-10 peptide (provided by antibody supplier) was preincubated overnight at 4 °C with aliquots of the appropriately diluted antibody prior to use. Images were captured using the ScanScope scanner (Aperio Technologies, Vista, CA, USA) and analysed using Aperio ImageScope software.
Analysis of NSCLC tumors grown in nude mice for MMP expression
A549 human NSCLC cells stably infected with recombinant retrovirus (pBabe) containing a dominant-negative, kinase-deficient human PKCι mutant allele (kdPKCι) or empty control pBabe virus (Regala et al., 2005a) were injected subcutaneously into athymic nude mice (Harlan-Sprague-Dawley, Indianapolis, IN, USA) and allowed to establish ectopic tumors as described previously (Regala et al., 2005a). Tumors were excised 30 days after inoculation and RNA extracted for qPCR analysis of mRNA abundance as described previously (Regala et al., 2005a). qPCR assay reagents for human MMP-2, MMP-9 and MMP-10 were obtained from Applied Biosystems. The use of nude mice, and all animal procedures, was authorized under an approved IACUC protocol.
Catalytic MMP enzyme assay
MMP activity was measured using a colorimetric assay and the thiopeptolide substrate acetyl-Pro-Leu-Gly-S-Leu-Leu-Gly-OEt (Biomol International, Plymouth Meeting, PA, USA). Enzymatic hydrolysis of the thioester bond produces a sulfhydryl group, which reacts with DTNB [5,5′-dithiobis(2-nitrobenzoic acid), Ellman’s reagent] to form 2-nitro-5-thiobenzoic acid, which is detected by an increase in absorbance at 412 nm (ε = 13 600 /m/cm at pH ≥ 6.0). Substrate hydrolysis was monitored on a Varian Cary 100 UV/Vis spectrophotometer equipped with a Peltier-thermostatted multicell changer at 37 °C, and initial rates were determined from the linear phase of reaction. MMP-3 enzyme concentration (0.97 μg/μl) was determined by UV absorbance at 280 nm, using a calculated molar extinction coefficient of 28 420/m/cm. MMP-10 enzyme concentration was 0.45 μg/μl as indicated by the manufacturer. Equal amounts of MMP-3 or MMP-10 activity (10 U/ml) was added to H1703 cell cultures and assessed for invasion and soft agar growth described above.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr Evette Radisky for recombinant human MMP-3 enzyme and expertise in protease activity assays; Dr Eric Edell, Aaron Bungum, Capella Weems and Ying Zhang for assistance in acquisition, processing and analysis of human lung cancer tissue samples; Jennifer Havens and the Mayo Clinic RNA Interference Technology Resource for RNAi reagents and Pam Kreinest and Brandy Edenfield for immunohistochemistry analysis. This work was supported in part through grants from the National Institutes of Health (CA081436) and a Team Science Project grant from the James and Esther King Biomedical Research Program to APF.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Bodey B, Bodey B, Jr, Groger AM, Siegel SE, Kaiser HE. Invasion and metastasis: the expression and significance of matrix metalloproteinases in carcinomas of the lung. In Vivo. 2001;15:175–180. [PubMed] [Google Scholar]
- Cho NH, Hong KP, Hong SH, Kang S, Chung KY, Cho SH. MMP expression profiling in recurred stage IB lung cancer. Oncogene. 2004;23:845–851. doi: 10.1038/sj.onc.1207140. [DOI] [PubMed] [Google Scholar]
- Delebecq TJ, Porte H, Zerimech F, Copin MC, Gouyer V, Dacquembronne E, et al. Overexpression level of stromelysin 3 is related to the lymph node involvement in non-small cell lung cancer. Clin Cancer Res. 2000;6:1086–1092. [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Erdogan E, Lamark T, Stallings-Mann M, Lee J, Pellecchia M, Thompson EA, et al. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281:28450–28459. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- Fields AP, Regala RP. Protein kinase C iota: human oncogene, prognostic marker and therapeutic target. Pharmacol Res. 2007;55:487–497. doi: 10.1016/j.phrs.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill JH, Kirwan IG, Seargent JM, Martin SW, Tijani S, Anikin VA, et al. MMP-10 is overexpressed, proteolytically active, and a potential target for therapeutic intervention in human lung carcinomas. Neoplasia. 2004;6:777–785. doi: 10.1593/neo.04283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto I, Pimkhaokham A, Fukuda Y, Yang ZQ, Shimada Y, Nomura N, et al. SNO is a probable target for gene amplification at 3q26 in squamous-cell carcinomas of the esophagus. Biochem Biophys Res Commun. 2001;286:559–565. doi: 10.1006/bbrc.2001.5428. [DOI] [PubMed] [Google Scholar]
- Impola U, Uitto VJ, Hietanen J, Hakkinen L, Zhang L, Larjava H, et al. Differential expression of matrilysin-1 (MMP-7), 92 kD gelatinase (MMP-9), and metalloelastase (MMP-12) in oral verrucous and squamous cell cancer. J Pathol. 2004;202:14–22. doi: 10.1002/path.1479. [DOI] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15:378–386. doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Kren L, Goncharuk VN, Krenova Z, Stratil D, Hermanova M, Skrickova J, et al. Expression of matrix metalloproteinases 3, 10 and 11 (stromelysins 1, 2 and 3) and matrix metalloproteinase 7 (matrilysin) by cancer cells in non-small cell lung neoplasms. Clinicopathologic studies. Cesk Patol. 2006;42:16–19. [PubMed] [Google Scholar]
- Mathew R, Khanna R, Kumar R, Mathur M, Shukla NK, Ralhan R. Stromelysin-2 overexpression in human esophageal squamous cell carcinoma: potential clinical implications. Cancer Detect Prev. 2002;26:222–228. doi: 10.1016/s0361-090x(02)00035-1. [DOI] [PubMed] [Google Scholar]
- Muller D, Breathnach R, Engelmann A, Millon R, Bronner G, Flesch H, et al. Expression of collagenase-related metalloproteinase genes in human lung or head and neck tumours. Int J Cancer. 1991;48:550–556. doi: 10.1002/ijc.2910480412. [DOI] [PubMed] [Google Scholar]
- Noda Y, Takeya R, Ohno S, Naito S, Ito T, Sumimoto H. Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells. 2001;6:107–119. doi: 10.1046/j.1365-2443.2001.00404.x. [DOI] [PubMed] [Google Scholar]
- O-Charoenrat P, Rhys-Evans PH, Eccles SA. Expression of matrix metalloproteinases and their inhibitors correlates with invasion and metastasis in squamous cell carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg. 2001;127:813–820. [PubMed] [Google Scholar]
- Osada H, Takahashi T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene. 2002;21:7421–7434. doi: 10.1038/sj.onc.1205802. [DOI] [PubMed] [Google Scholar]
- Pimkhaokham A, Shimada Y, Fukuda Y, Kurihara N, Imoto I, Yang ZQ, et al. Nonrandom chromosomal imbalances in esophageal squamous cell carcinoma cell lines: possible involvement of the ATF3 and CENPF genes in the 1q32 amplicon. Jpn J Cancer Res. 2000;91:1126–1133. doi: 10.1111/j.1349-7006.2000.tb00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu RG, Abo A, Steven Martin G. A human homolog of the C. elegans polarity determinant Par-6 links Rac and Cdc42 to PKCzeta signaling and cell transformation. Curr Biol. 2000;10:697–707. doi: 10.1016/s0960-9822(00)00535-2. [DOI] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005a;280:31109–31115. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, et al. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005b;65:8905–8911. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- Snaddon J, Parkinson EK, Craft JA, Bartholomew C, Fulton R. Detection of functional PTEN lipid phosphatase protein and enzyme activity in squamous cell carcinomas of the head and neck, despite loss of heterozygosity at this locus. Br J Cancer. 2001;84:1630–1634. doi: 10.1054/bjoc.2001.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006;66:1767–1774. doi: 10.1158/0008-5472.CAN-05-3405. [DOI] [PubMed] [Google Scholar]
- Van Themsche C, Alain T, Kossakowska AE, Urbanski S, Potworowski EF, St-Pierre Y. Stromelysin-2 (matrix metalloproteinase 10) is inducible in lymphoma cells and accelerates the growth of lymphoid tumors in vivo. J Immunol. 2004;173:3605–3611. doi: 10.4049/jimmunol.173.6.3605. [DOI] [PubMed] [Google Scholar]
- Zhang J, Anastasiadis PZ, Liu Y, Thompson EA, Fields AP. Protein kinase C βII induces cell invasion through a Ras/MEK–, PKCiota/RAC 1-dependent signaling pathway. J Biol Chem. 2004;279:22118–22123. doi: 10.1074/jbc.M400774200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.