

Abstract
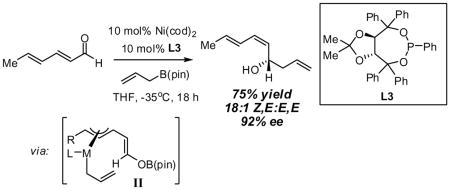
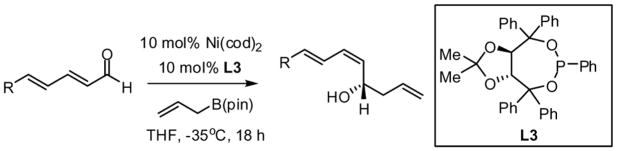
The nickel-catalyzed enantioselective addition of allylboronic acid pinacol ester - allylB(pin) – to α,β,γ,δ-unsaturated aldehydes is described. This reaction results in a remarkable inversion in substrate olefin geometry providing the Z,E configured reaction product in good enantioselectivity and olefin stereoselectivity. The reaction appears to proceed by conversion of the dienal to an unsaturated π-allyl complex, followed by reductive elimination via transition state (II). Enantioselectivities range from 73–94% ee for a range of δ-substituted dienals when chiral ligand L3 is employed.
3,3′-Reductive elimination of bis(allyl)metal species results in C-C bond-forming allyl coupling at a site remote from the metal center. This mechanistic postulate was recently described computationally by Echavarren1 and can be used to understand an array of reactions from Yamamoto’s allylative dearomatization reaction2 to the Tsuji allylation.3 Recently, we reported that Ni and Pd complexes can catalyze the enantioselective conjugate addition of allylboronic acid pinacol ester (allylB(pin), 2, Scheme 1) to dialkylidene ketones (i.e. 1, Scheme 1).4 Mechanistic studies suggest that this conjugate addition also proceeds by 3,3′-reductive elimination, albeit in this case from unsaturated π-allyl complex I. Structure I is obtained by boron Lewis acid-promoted electron transfer from either Ni(0) or Pd(0) to the enone, followed by transmetalation of the allyl group from boron to the transition metal.5 Notably, simple enones and their derivatives are inert under the reaction conditions, a feature which is attributed to the reaction mechanism.
Scheme 1.
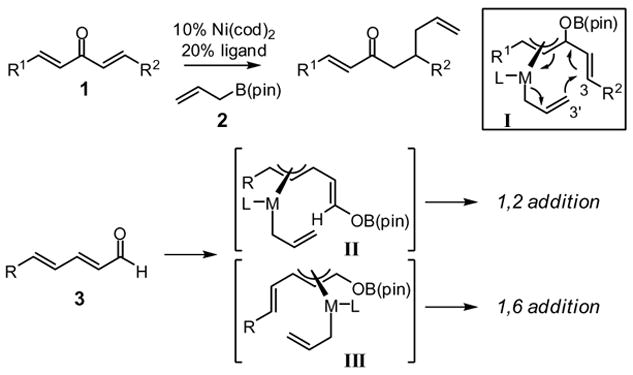
Considering the utility of the allyl unit in organic synthesis, we have begun to investigate whether 3,3′-reductive elimination might enable new types of allylation reactions.6 In light of the structural requirements of transition state I in Scheme 1, we considered it plausible that α,β,γ,δ-unsaturated aldehydes (3) might access intermediates such as II or III, thereby gaining access to a path for low-barrier 3,3′-reductive elimination. In this report, we describe the first examples of this allylation, a process that delivers the 1,2 addition product and results in a remarkable inversion of substrate olefin geometry. Significantly, with appropriate chiral ligands, reactions of substrate class 3 can occur with high enantioselection and provide products which are not readily accessed by other catalytic allylation reactions.
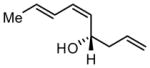
To investigate the rapidity with which catalysis of the dienal allylation might proceed, the reaction between sorbic aldehyde (4) and allylB(pin) was examined (Scheme 2). These reactants undergo non-catalyzed reaction at room temperature in THF solvent achieving >95% conversion to (E,E)-5 after 15 hours (initial [substrates] = 0.5 M). Remarkably, in the presence of Ni(cod)2 and PCy3, the reaction is complete in 40 minutes and, rather than delivering 1,2 addition product (E,E)-5, (E,Z)-5 is the predominant reaction product. Consistent with the discussion above, the rate acceleration appears restricted to dienals (crotonaldehyde reacts with comparable rates both in the presence and absence of catalyst) and like reactions through I which deliver the E-enolate,4 reaction through II furnishes the Z alkene.
Scheme 2.

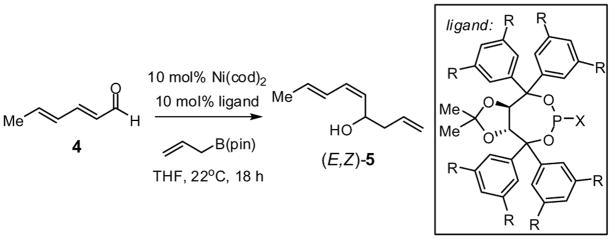
Previous investigations of the catalytic enantioselective conjugate allylation reaction employed taddol-derived phosphonite L1 for optimal selectivity.7 As depicted in Table 1, this ligand structure is effective for the asymmetric 1,2-allylation of aldehyde 4, however, enantioselectivity is minimal (entry 1). Analysis of related ligand structures revealed that phosphonites are generally superior to phosphoramidites and that phosphonite L3 is optimal; in THF at room temperature 65% yield of alcohol (E,Z)-5 was obtained with an enantiomeric purity of 72% ee. Notably, the reaction with ligand L3 occurs rapidly at room temperature with complete conversion achieved in 25 minutes! The high rate of this reaction suggested that it might occur efficiently even at lower temperature. Indeed at −35 °C, the catalytic reaction still occurs and, while 18 hours are required to achieve complete conversion, the enantioselectivity and olefin stereoselectivity are enhanced significantly (Table 2).
Table 1.
Ni-Catalyzed Enantioselective Dienal Allylation.
 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand | R | X | %yielda | [E,Z]:[E,E]b | %ee[E,Z]c |
| 1 | L1 | tBu | Ph | 62 | 20:1 | 19 |
| 2 | L2 | Me | Ph | 65 | >20:1 | 48 |
| 3 | L3 | H | Ph | 71 | 17:1 | 75 |
| 4 | L4 | H |  |
60 | >20:1 | 52 |
| 5 | L5 | Me | 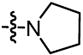 |
59 | >20:1 | 25 |
Isolated yield of purified material.
Determined by NMR analysis of unpurified product.
Determined by chiral GC analysis.
Table 2.
Scope of Ni-Catalyzed Enantioselective Dienal Allylationa
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | yield (%)b | (E,Z):(E:E) | %ee |
| 1 | 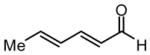 |
 |
84 | >20:1 | 88 |
| 2 | 75 | 18:1 | 92 | ||
| 3 | 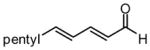 |
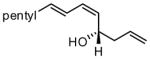 |
84 | >20:1 | 87 |
| 4 | 76 | >20:1 | 90 | ||
| 5 | 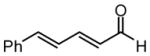 |
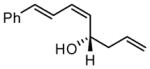 |
68 | >20:1 | 91 |
| 6 | 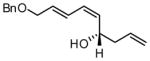 |
86 | 15:1 | 73 | |
| 7 | 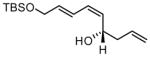 |
81 | 7:1 | 85 | |
| 8 | 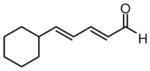 |
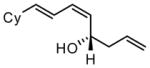 |
92 | >20:1 | 93 |
| 9 | 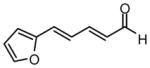 |
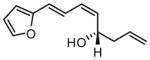 |
73 | 15:1 | 94 |
| 10 | 83 | 16:1 | 90 | ||
For entries 1,3, and 5–10: reaction carried out in a glovebox freezer. For entries 2 and 4, reaction carried out outside glovebox and quenched by addition of 30 equiv. of acetaldehyde, followed by warming to ambient. See the SI for details.
Isolated yield of purified material.
Under optimized conditions, many δ-substituted dienals also participate in the Ni-catalyzed enantioselective allylboration reaction. As shown in Table 2, the stereoselectivity is dependent upon the diene substituents even when these groups are five atoms away from the newly formed stereocenter. This observation is completely consistent with the proposed reaction mechanism. Of note, the reaction delivers the E,Z stereoisomer as the predominant product. The E,E olefin isomer, when observed, is racemic and assumed to arise from non-catalyzed reaction that occurs during room temperature work-up. Indeed, addition of 30 equivalents of acetaldehyde prior to work-up serves to eliminate much of the E,E product. Regarding substrate scope, aromatic and aliphatic substituents are tolerated at the δ carbon and provide products with high enantiomeric purity. Considering the lability of allylic oxygenated substituents in the presence of late transition metal catalysts, it is notable that the substrates in entries 6 and 7 provide good product yields and high levels of stereocontrol.
The functional group pattern present in the allylation products is useful for further manipulation. In particular, transformations that make use of directing effects8 and A(1,3) strain9 as stereocontrol elements can render substrate functionalization selective. For example, as shown in Scheme 3 these effects lead to selective epoxidation such that (E,Z)-5 is efficiently converted into epoxide 6 with a high level of stereocontrol.10 Richly functionalized epoxide 6 itself leads to a variety of building blocks that are not readily accessible by alterative strategies. For instance, ring-closing metathesis of 6 with the second generation Hoveyda-Grubbs catalyst furnishes novel epoxycyclohexadienol 7.11 Alternatively, Pd-catalyzed substitution of 6 with oxygen and carbon nucleophiles provides 8 and 9 as single isomers.10c,12
Scheme 3.

In conclusion, we have described a unique catalytic enantioselective allylation of unsaturated carbonyls that appears to occur by 3,3′-reductive elimination. The functional group pattern that is packaged in the reaction products is relatively unique and should be amenable to rapid complexity generation. Future studies in regards to the scope of this transformation and its use in asymmetric synthesis are in progress.
Supplementary Material
Acknowledgments
Frontier Scientific is appreciated for a donation of allylB(pin) as are the NIGMS (R01 GM-64451) and the NSF (DBI-0619576; BC Mass Spectrometry Center).
Footnotes
Supporting Information: Characterization and procedures. This material is free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Chem Eur J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Cárdenas DJ, Echavarren AM. New J Chem. 2004;28:338. [Google Scholar]
- 2.(a) Bao M, Nakamura H, Yamamoto Y. J Am Chem Soc. 2001;123:759. doi: 10.1021/ja003718n. [DOI] [PubMed] [Google Scholar]; (b) Lu S, Xu Z, Bao M, Yamamoto Y. Angew Chem Int Ed. 2008;47:4366. doi: 10.1002/anie.200800529. [DOI] [PubMed] [Google Scholar]; (c) Ariafard A, Lin Z. J Am Chem Soc. 2006;128:13010. doi: 10.1021/ja063944i. [DOI] [PubMed] [Google Scholar]
- 3.Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J Am Chem Soc. 2007;129:11876. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sieber JD, Liu S, Morken JP. J Am Chem Soc. 2007;129:2214. doi: 10.1021/ja067878w. [DOI] [PubMed] [Google Scholar]; (b) Sieber JD, Morken JP. J Am Chem Soc. 2008;130:4978. doi: 10.1021/ja710922h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For related oxidative additions, see: Brookhart M, Young GJ. J Chem Soc, Chem Comm. 1974:205.Johnson JR, Tully PS, Mackenzie PB, Sabat M. J Am Chem Soc. 1991;113:6172.Grisso BA, Johnson JR, Mackenzie PB. J Am Chem Soc. 1992;14:5160.Grennberg H, Gogoll A, Baeckvall JE. Organometallics. 1993;12:1790.Ogoshi S, Yoshida T, Nishida T, Morita M, Kurosawa H. J Am Chem Soc. 2001;123:1944. doi: 10.1021/ja0036099.Morita M, Inoue K, Ogoshi S, Kurosawa H. Organometallics. 2003;22:5468.Ogoshi S, Morita M, Kurosawa H. J Am Chem Soc. 2003;125:9020. doi: 10.1021/ja0361042.Morita M, Inoue K, Yoshida T, Ogoshi S, Kurosawa H. J Organomet Chem. 2004;689:894.
- 6.For recent reviews of catalytic enantioselective carbonyl allylation, see: Hall DG. Synlett. 2007:1644.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Denmark SE, Fu J. Chem Rev. 2003;103:2761. doi: 10.1021/cr020050h.For original investigations in this area, see: Furuta K, Mouri M, Yamamoto H. Synlett. 1991:561.Aoki S, Mikami K, Terada M, Nakai T. Tetrahedron. 1993;49:1783.Costa AL, Piazza MG, Tagliavini E, Trombini C, Umani-Ronchi A. J Am Chem Soc. 1993;115:7001.Keck GE, Geraci LS. Tetrahedron Lett. 1993;34:7827.
- 7.Review of taddol-derived phosphorous ligands: Feringa BL. Acc Chem Res. 2000;33:346. doi: 10.1021/ar990084k.Select examples, hydrogenation: ven den Berg M, Minnard AJ, Schudde EP, van Esch J, de Vries AHM, de Vries JG, Feringa BL. J Am Chem Soc. 2000;122:11539.Hydrosilation: Jensen JF, Svendsen BY, la Cour TV, Pedersen HL, Johannsen M. J Am Chem Soc. 2002;124:4558. doi: 10.1021/ja025617q.Hydroboration: Ma MFP, Li K, Zhou Z, Tang C, Chan ASC. Tetrahedron: Asymm. 1999;10:3259.For TADDOL-derived phosphonite, see: Seebach D, Hayakawa M, Sakaki J, Schweizer WB. Tetrahedron. 1993;49:1711.
- 8.Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307. [Google Scholar]
- 9.Hoffmann RW. Chem Rev. 1989;89:1941. [Google Scholar]
- 10.(a) Tanaka S, Yamamoto H, Nozaki H, Sharpless KB, Michaelson RC, Cutting JD. J Am Chem Soc. 1974;96:5254. doi: 10.1021/ja00823a042. [DOI] [PubMed] [Google Scholar]; (b) Rossiter BE, Verhoeven TR, Sharpless KB. Tetrahedron Lett. 1979:4733. [Google Scholar]; (c) Yoshida S, Asano M, Kobayashi Y. Tetrahedron Lett. 2005;46:7243. [Google Scholar]
- 11.Garber SB, Kingbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168. [Google Scholar]
- 12.(a) Trost BM, Molander GA. J Am Chem Soc. 1981;103:5969. [Google Scholar]; (b) Tsuji J, Kataoka H, Kobayashi Y. Tetrahedron Lett. 1981;22:2575. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.