

Abstract
Status epilepticus (SE) typically progresses into temporal lobe epilepsy (TLE) typified by complex partial seizures. Because sizable fraction of patients with TLE exhibit chronic seizures that are resistant to antiepileptic drugs, alternative therapies that are efficient for diminishing SE-induced chronic epilepsy have great significance. We hypothesize that bilateral grafting of appropriately treated striatal precursor cells into hippocampi shortly after SE is efficacious for diminishing SE-induced chronic epilepsy through long-term survival and differentiation into GABA-ergic neurons. We induced SE in adult rats via graded intraperitoneal injections of kainic acid, bilaterally placed grafts of striatal precursors (pre-treated with fibroblast growth factor-2 and caspase inhibitor) into hippocampi at 4 days post-SE, and examined long-term effects of grafting on spontaneous recurrent motor seizures (SRMS). Analyses at 9–12 months post-grafting revealed that, the overall frequency of SRMS was 67–89% less than that observed in SE-rats that underwent sham-grafting surgery and epilepsy-only controls. Graft cell survival was ~33% of injected cells and ~69% of surviving cells differentiated into GABA-ergic neurons, which comprised subclasses expressing calbindin, parvalbumin, calretinin and neuropeptide Y. Grafting considerably preserved hippocampal calbindin but had no effects on aberrant mossy fiber sprouting. The results provide novel evidence that bilateral grafting of appropriately treated striatal precursor cells into hippocampi shortly after SE is proficient for greatly reducing the frequency of SRMS on a long-term basis in the chronic epilepsy period. Presence of a large number of GABA-ergic neurons in grafts further suggests that strengthening of the inhibitory control in host hippocampi likely underlies the beneficial effects mediated by grafts.
Keywords: Caspase inhibitors, Cell transplantation, Dentate gyrus, Epilepsy, Fibroblast growth factor-2, GABA-ergic interneurons, Graft cell survival, Graft integration, Kainic acid, Neural grafting, Spontaneous seizures, Striatal precursor cells
Epilepsy, epitomized by recurrent spontaneous seizures due to hyperactivity and synchronization of activity within populations of neurons, affects over 50 million people (Strine et al., 2005). Temporal lobe epilepsy (TLE), characterized by progressive development of complex partial seizures and hippocampal neurodegeneration, is seen in ~30% of epileptic patients (Manford et al., 1992). Although the etiology of TLE is unknown in most cases (McNamara, 1999), it is typically seen after an initial precipitating injury (IPI) such as status epilepticus (SE), brain injury, tumors, meningitis, and encephalitis in other cases (French et al., 1993; Mathern et al., 1995; Mathern et al., 1996; Lewis, 2005). Seizures in TLE originate from the temporal lobe foci, which are associated with learning and memory impairments, reduced dentate neurogenesis and depression (Devinsky, 2004; Hattiangady et al., 2004; Detour et al., 2005; Pirttila et al., 2005). Because significant number of people (~25%) afflicted with epilepsy have seizures that cannot be controlled by antiepileptic drugs and surgical removal of the epileptic focus can lead to cognitive impairments (Litt et al., 2001; McKeown and McNamara, 2001), there is pressing need to develop alternative therapeutic approaches that considerably restrain chronic epilepsy after SE or head trauma.
Interventions that are efficacious for facilitating repair of the disrupted hippocampal circuitry may also restrain TLE after an IPI. Indeed, replacement of degenerated neurons in the injured hippocampus via grafting of fetal hippocampal cells has shown promise for restraining epileptogenic changes and controlling seizures (Shetty et al., 2005; Rao et al., 2007). Furthermore, it is believed that an increased excitatory neurotransmission found in the epileptic hippocampus is partly due to a reduced number of GABA-ergic interneurons (Shetty and Turner, 2000, 2001), loss of functional inhibition (Lloyd et al., 1986; Cornish and Wheal, 1989; During et al., 1995) and diminished numbers of GABA-ergic terminals (Ribak et al., 1986; Esclapez and Trottier, 1989). From this perspective, the idea of restraining spontaneous recurrent motor seizures (SRMS) in the epileptic hippocampus via grafting of cells that just release the inhibitory neurotransmitter GABA at the seizure focus has received considerable attention (Löscher et al., 2008). For instance, grafting of GABA-soaked beads, immortalized GABA-ergic cells, cells that are engineered to produce GABA, and fetal GABA-ergic cells into the epileptic foci have been shown to transiently reduce seizures in a variety of animal models (Kokaia et al., 1994; Löscher et al., 1998; Gernert et al., 2002; Thompson, 2005; Castillo et al., 2006). However, it is unknown whether grafting of GABA-ergic cells shortly after an IPI would be efficacious for restraining SRMS on a long-term basis. Studies on this issue have been hampered by the poor survival of grafted GABA-ergic cells in the epileptic brain (Zaman et al., 2000). Thus, for prolonged anti-seizure effects, apart from grafting cells that are capable of differentiating into GABA-ergic interneurons, it will be necessary to graft these cells using strategies that promote their enduring survival in the epileptic brain.
We hypothesize that bilateral grafting of appropriately treated striatal precursor cells into hippocampi shortly after SE is efficacious for diminishing SE-induced chronic epilepsy through long-term survival and differentiation into GABA-ergic neurons. We addressed this hypothesis through studies in a SE-model of chronic TLE using Fischer 344 rats (Rao et al., 2006). Status epilepticus was induced via graded intraperitoneal injections of kainic acid (KA), which triggered continuous stages III–V seizures (i.e. SE) for over 3 hrs. Grafting was performed bilaterally into hippocampi at 4 days after SE using striatal precursors from the embryonic day (E) 15 lateral ganglionic eminence as donor cells. The donor cells were pre-treated and grafted with fibroblast growth factor-2 (FGF-2) and a caspase inhibitor Ac-YVAD-cmk, to enhance their survival following grafting. The frequency of SRMS in SE-rats receiving grafts was measured at 9–12 months post-grafting and compared with SE-rats that underwent sham-grafting surgery or SE-rats that served as epilepsy-only controls. The survival and differentiation of grafted cells into GABA-ergic and distinct subtypes of GABA-ergic neurons were quantitatively assessed after 12 months of grafting.
Materials and Methods
Animals and induction of status epilepticus using kainic acid
Young adult (4–5 months old) Fischer 344 (F344) rats acquired from Harlan Sprague-Dawley (Indianapolis, IN) were employed in these experiments. All animal experiments performed in this study have been approved by the Duke University Institutional Animal Care and Use Committee and animal studies subcommittee of the Durham Veterans Affairs Medical Center. In addition, all efforts were made to lessen animal suffering and to use only the number of animals necessary to produce a reliable scientific data. The schematic of major experiments performed in this study is illustrated in figure 1.
Figure 1.
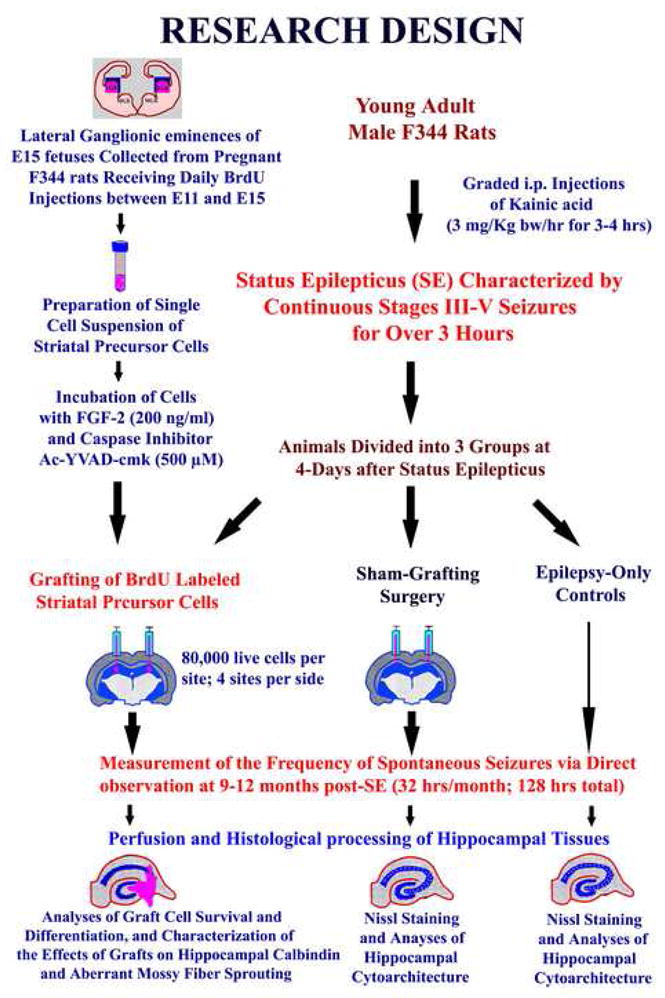
Schematic of major experiments performed in this study. The top left half illustrates the preparation of donor cells for grafting. The steps include dissection of lateral ganglionic eminences from embryonic day 15 fetuses, preparation of a single cell suspension of striatal precursor cells, and pre-treatment of donor cells with fibroblast growth factor and a caspase inhibitor Ac-YVAD-cmk. The donor cells were pre-labeled with 5′-bromodeoxyuridine (BrdU) via intraperitoneal injections of BrdU between embryonic days 11 and 15 to pregnant mothers of the fetuses used in this study. The top right half describes the preparation of young adult male rats for striatal precursor cell grafting, sham-grafting surgery and epilepsy-alone groups. The status epilepticus (SE) was induced through graded intraperitoneal injections of the kainic acid (KA) in all rats. In rats designated for grafting, ~80,000 live striatal precursor cells were placed into each of 4 sites in the hippocampus on both sides, whereas in rats designated for sham-grafting surgery, grafting needles were introduced to all sites of hippocampi chosen for grafting. Another group of rats which received neither grafts nor sham grafting surgery served as epilepsy-only controls. The frequency of spontaneous recurrent motor seizures (SRMS) was measured at 9–12 months post-SE and animals were perfused for histological analyses at one-year after grafting.
The protocol for inducing SE in F344 rats using KA was adapted from the procedure developed by Hellier et al. (Hellier et al., 1998) for Sprague-Dawley rats, and seizures were scored as per the modified Racine’s scale (Racine, 1972; Ben-Ari, 1985; Rao et al., 2006). Rats received graded intraperitoneal injections of KA (3.0 mg/Kg b.w./hr) until they exhibited SE. On an average, induction of SE required each rat to receive a total KA dose of ~10.5 mg/Kg b.w., which is consistent with our earlier study (Rao et al., 2006). The motor seizures were characterized by unilateral forelimb clonus with lordotic posture (stage III seizures), bilateral forelimb clonus and rearing (stage IV seizures) and bilateral forelimb clonus with rearing and falling (stage V seizures). All animals that received a total KA dose of 10.5 mg/Kg b.w. exhibited >10 stages IV-V seizures during the 3-hr of observation after the onset of SE. The motor seizures subsided gradually thereafter and were not apparent at 5–6 hours after SE. Rats were given moistened rat chow and subcutaneous injections of lactated Ringer’s solution (10 ml/day) for 3–5 days after SE.
In vivo labeling of striatal precursor cells
The donor striatal cells were labeled with BrdU in vivo by employing daily intraperitoneal injections of BrdU (50 mg/kg body weight) into timed pregnant rats between embryonic days 11 and 15. Our previous study has shown that this labeling protocol gives a reliable and consistent labeling of vast majority of striatal precursor cells (Zaman et al., 2000).
Preparation of striatal precursor cells for grafting
On embryonic day 15, fetuses were removed from deeply anesthetized pregnant rats by cesarean section and collected in a culture medium comprising Dulbecco’s modified Eagle’s medium/F12, L-glutamine, and B-27 nutrient mixture containing vitamins, essential fatty acids, hormones and antioxidants (Life Technologies, Grand Island, NY). The brains were carefully removed and by cutting through the cortical plate, the medial and lateral ganglionic eminences were identified. Following this, the lateral ganglionic eminence tissue in every hemisphere was carefully dissected out, collected in 2 ml of fresh culture medium and triturated (Zaman et al., 2000). The resulting cell suspension was diluted with 8 ml of fresh medium, sieved through a cell strainer (pore size 70 μm) and centrifuged at 800 rpm for 8 minutes, and the pellet was re-suspended in 10 ml of fresh medium and washed twice through centrifugation. The final pellet was re-suspended in a fresh culture medium and viability was assessed using the trypan blue exclusion test. Both live and dead cells were counted and the percentage of live cells relative to the total number of cells was measured to ascertain cell viability in the cell suspension. Cell suspensions where viability was >75% were used for treatment with FGF-2 and caspase inhibitor.
Pre-treatment of striatal precursor cells with FGF-2 and caspase inhibitor and BrdU labeling index
Striatal precursor cell suspension containing >75% viable cells was re-suspended in 2 ml culture medium and incubated with the neurotrophic factor FGF-2 (200ng/ml; Peprotech, Rocky Hill, NJ), and a caspase inhibitor Ac-YVAD-cmk (500μM; Calbiochem, La Jolla, CA) for 3 hours at 37°C in a water bath with gentle shaking. Following this, the cell suspension was washed twice with fresh culture medium through centrifugation. The tissue pellet was re-suspended in 40μl of the proliferation medium and viability was again assessed using the trypan blue exclusion test. The viability of cells in the suspension did not change with 3-hour incubation of cells in a culture medium containing FGF-2 and caspase inhibitor. The cell density was adjusted 1 × 105 viable cells per μl. To ascertain BrdU labeling index at the time of grafting in the cell suspension, samples from the cell suspension were plated onto poly-lysine coated 35 mm culture dishes and incubated in the culture medium for an hour. The adhered cells were then fixed using 2% paraformaldehyde solution and the dishes were processed for rapid BrdU immunocytochemical staining (Shetty et al., 1994). Quantification of BrdU positive cells relative to the total cells revealed that 93% (Mean ± SEM = 93.4 ± 0.4, n = 4) of striatal precursor cells were immunopositive for BrdU at the time of grafting.
Neural grafting into hippocampi of rats that underwent status epilepticus
Striatal precursor cells pre-treated with FGF-2 and caspase inhibitor were transplanted bilaterally into hippocampi of rats that underwent SE, at 4 days post-SE (n=5). Grafting protocol was similar to that described in our earlier reports (Hattiangady et al., 2006; Rao et al., 2007). Eighty thousand live cells in 0.8μl of the cell suspension were injected into each of the four sites in every hippocampus using the following stereotaxic co-ordinates: (i) antero-posterior (AP) = 3.0 mm from bregma, lateral (L) = 1.8 mm from midline, and ventral (V) = 3.5 mm from the surface of the brain; (ii) AP = 3.6 mm, L = 2.5 mm, V = 3.5 mm; (iii) AP = 4.2 mm, L = 3.0 mm, V = 3.5 mm; (iv) AP = 4.8 mm, L = 3.5 mm, V = 4.0 mm. These co-ordinates were specifically chosen to place the grafts in the CA3 subfield through injection of the cell suspension at the end of hippocampal fissure. Each hippocampus received ~320,000 live striatal fetal cells. Out of these cells, ~298,880 cells were BrdU labeled, based on the BrdU labeling index measured at the time of grafting. For elucidation of the specific effects of striatal precursor cell grafting (rather than the grafting procedure) on SE-induced chronic epilepsy, a second group of rats that underwent SE received sham-grafting surgery (n=5). These rats went through the entire grafting procedure except that cells were not injected into hippocampi. However, grafting needles were introduced into all sites of hippocampi chosen for grafting in SE-rats that received striatal precursor cell grafts. For further comparison with grafted and sham-grafted groups, a third group of rats that underwent SE (n=5) were used as epilepsy-only controls. Neither grafting nor surgery was performed on these rats.
Analyses of spontaneous recurrent motor seizures (SRMS)
The behavior of rats in different groups (SE-rats receiving striatal cell grafts, SE-rats receiving sham-grafting surgery, and epilepsy-only controls) were observed at 9–12 months post-SE for measuring the frequency of SRMS. For this, rats were observed for eight hours every week (4 hrs/session, two sessions per week) during the day light period (total = 32 hrs/month, and 128 hours for the entire duration of 4 months). The scoring of SRMS was similar to that described earlier (Racine, 1972; Ben-Ari, 1985). Only stages III–V motor seizures were scored, as the occurrence of these seizures is very apparent to the observer due to their conspicuous features. The recorded seizures had one or more of the following features: unilateral forelimb clonus (stage III seizure), bilateral forelimb clonus with rearing (stage IV seizure), and bilateral forelimb clonus with rearing and falling (stage V seizure). While the observer was aware of the broad treatment status in different groups (i.e. SE-alone group, SE-rats with sham-grafting surgery, SE-rats with grafts), the observer did not know about the type of cell transplants the grafted group received, as SE rats that received striatal precursor cell transplants in this study were observed with SE rats that received other types of cell transplants belonging to additional studies in the laboratory. Thus, the observer was blind to the specific type of cells the grafted group received.
Tissue Processing, Nissl staining, and selection of grafts for BrdU immunostaining and quantification
Rats were perfused transcardially with 4% paraformaldehyde at one-year after SE. The brains were cryoprotected in sucrose and sliced coronally (30-μm-thick sections) through the hippocampus using a cryocut, and collected serially in phosphate buffer (PB). Every 10th section through the entire hippocampus was stained for Nissl and the sections were scanned to identify the presence and location of transplants in relation to different cell layers of the hippocampus in the grafted group or to identify the extent of lesion in other groups. In rats that received striatal precursor cells, grafts were mostly located partly inside the hippocampus and partly below the hippocampus in the lateral ventricle. For quantitative analyses of the yield of surviving grafted cells, every 10th section through transplants was processed for BrdU immunostaining, as detailed in our earlier reports (Hattiangady et al., 2006; Rao et al., 2007).
Quantification of graft cell survival using the optical fractionator method
For analyses of graft cell survival, 20 transplants (in five different hippocampi) were analyzed. In each transplant, BrdU+ cells were counted in every 10th section through the entire antero-posterior extent of transplants using the StereoInvestigator system (Microbrightfield Inc., Williston, VT). The StereoInvestigator system consisted of a color digital video camera (Optronics Inc., Muskogee, OK) interfaced with a Nikon E600 microscope. In each transplant, BrdU+ cells were counted from 50–100 randomly and systematically selected frames using the 100X oil immersion objective lens. The numbers and densities of frames were determined by entering the parameter grid size in the optical fractionator component of the StereoInvestigator system. Details of the counting procedure are described in our previous reports (Rao and Shetty, 2004; Hattiangady et al., 2006; Rao et al., 2007). Briefly, the contour of the transplant area was first delineated in every section using the tracing function and the optical fractionator component was activated. The number and location of counting frames, and the counting depth was determined by entering parameters such as the grid size (75 × 75 μm), the thickness of top guard zone (4–μm) and the optical dissector height (i.e. 6 μm). Based on the above parameters and cell counts, the StereoInvestigator program calculated the total number of BrdU+ cells per transplant by utilizing the optical fractionator formula (Rao and Shetty, 2004).
Phenotypic analyses of grafted cells using dual immunofluorescence and confocal microscopy
To measure percentages of neurons, GABA-ergic neurons and subtypes of GABA-ergic interneurons among surviving BrdU+ grafted cells, representative sections through transplants (4 transplants per animal for each antigen) were processed for identification of BrdU and one of markers of neurons such as neuron-specific nuclear antigen (NeuN), gamma-amino butyric acid (GABA), calbindin, parvalbumin, calretinin and neuropeptide Y, using dual immunofluorescence methods. A sequential immunofluorescence method was employed and BrdU was visualized first. Immunofluorescence staining for BrdU mainly comprised treatment of sections for various BrdU staining pre-treatment protocols (Rao and Shetty, 2004), overnight incubation of sections in a rat anti-BrdU solution (1:200; Serotech, Raleigh, NC), and treatment of sections with the anti-rat IgG conjugated to Alexa Fluor 594 (Molecular Probes) for 60 minutes. Following confirmation of BrdU immunofluorescence, separate sets of sections comprising BrdU immunofluorescence in grafted cells were processed for identification of different neural antigens using specific primary antibodies. The antibodies include mouse anti-NeuN (1:1000; Millipore, CA), rabbit anti-GABA, mouse anti-calbindin, mouse anti-parvalbumin (1:1000; Sigma), rabbit anti-calretinin (1:1000; Millipore) or rabbit anti-neuropeptide Y (1:1000; Peninsula lab). For this, the sections were incubated overnight in the primary antibody solution at 4°C, washed in tris-buffered saline (TBS), incubated in appropriate secondary antibody solution (biotinylated anti-mouse or anti-rabbit IgG; 1:200; Vector labs Burlingame, CA) for 60 minutes, rinsed in TBS and treated with streptavidin fluorescein (1:200; Molecular Probes, Eugene, OR) solution for 60 minutes. The sections were then washed and mounted on clean glass slides using a slow-fade mounting medium (Molecular Probes Eugene, OR). The above protocol facilitated visualization of BrdU as red fluorescence and other markers as green fluorescence. The sections processed were examined using a confocal microscope (LSM-510, Carl Zeiss) and Z-stacks were taken at 1-μm intervals through the transplant area. From the multiple z-section stacks, percentages of BrdU positive cells (i.e. grafted cells) expressing NeuN, GABA, calbindin, parvalbumin, calretinin or neuropeptide Y were measured in different transplants using the LSM image browser.
Calbindin immunostaining
To identify the effect of striatal precursor cell grafting early after SE on the expression of calbindin in the host hippocampus, we processed serial sections (every 15th) through the hippocampus from both grafted and epilepsy-only groups for calbindin immunostaining, as described in our earlier report (Shetty and Hattiangady, 2007). Briefly, the sections were blocked in 10% normal horse serum, incubated overnight in a mouse anti-calbindin antibody (1:1000; Sigma), washed in PBS, treated with biotinylated anti-mouse IgG (Vector lab) for 60 minutes, washed in PBS, and incubated with ABC reagent (Vector labs) for 60 minutes. The sections were then thoroughly washed in PBS, developed using diaminobenzidine as the chromogen, dehydrated, and cover slipped using Permount.
Neuropeptide Y immunostaining
Because neuropeptide Y identifies mossy fiber terminals in the epileptic hippocampus, we used neuropeptide Y immunostaining to visualize aberrant mossy fiber sprouting in the dentate supragranular layer (DSGL) of epilepsy-only rats and SE-rats that received striatal precursor cell grafts. In brief, serial sections (every 15th) through the hippocampus were treated with 0.1 M Tris buffer (TB) containing 1% hydrogen peroxide for 30 minutes, washed in TB, and treated consecutively with TB containing 0.1% Triton X-100 (Tris A), TB containing 0.1% Triton X-100 and 0.005% bovine serum albumin (Tris B), and 10% normal goat serum in Tris B. Sections were then treated sequentially with Tris A and Tris B and incubated in rabbit anti-neuropeptide Y antibody for 48 hours at 4°C. Sections were then washed consecutively in Tris A and Tris B solutions, and incubated in biotinylated goat anti-rabbit IgG (1:1000; Vector) for 45 minutes, washed again in Tris A and Tris D (0.5M TB containing 0.1% Triton X-100 and 0.005% bovine serum albumin) solutions, and incubated in ABC reagent diluted in Tris D (1:1000) for 60 minutes, as described elsewhere (Scharfman et al., 2002; Hattiangady et al., 2005). The tissue-bound peroxidase was then developed using diaminobenzidine (Vector DAB kit, Vector) as chromogen.
Statistical analyses
All comparisons for different parameters used one-way analysis of variance (ANOVA) with Student’s Newman-Keuls multiple comparisons post-hoc tests. The differences were considered significant if the p value was found to be less than 0.05.
Results
Extent of chronic epilepsy in SE-rats receiving striatal precursor cell grafts
To ascertain whether striatal precursor cell grafting at 4-days after SE restrains SRMS on a long-term basis, we quantified the frequency of SRMS in grafted rats for four months at extended time-points after SE (i.e. at 9–12 months after SE) (Fig. 1). For comparison, we also quantified SRMS in age-matched SE-rats that received sham grafting surgery at 4 days post-SE and age-matched SE-rats that remained as epilepsy-only controls (Fig. 1). The measurement of SRMS comprised 8 hours of direct observation per week (4 hrs/session, 2 sessions/week, 32 hours/month, and 128 hours in total). Only stages III–V motor seizures were carefully scored, in view of the fact that an episode of these seizures during surveillance is obvious to the observer due to their striking features. All rats that underwent SE alone exhibited robust SRMS; the average frequency of SRMS varied from 2.96–3.24 per hour (Fig. 2) suggesting that, in the absence of any therapeutic intervention, the frequency of SRMS remains stable. Similarly, all SE-rats that received sham-grafting surgery exhibited robust SRMS; the average frequency of SRMS varied from 2.76–3.18 per hour (Fig. 2), which is highly comparable to the frequency observed in rats that underwent SE alone. This suggests that sham-grafting surgery after SE does not interfere with the development of chronic epilepsy. Only minor variations in the frequency of SRMS (per month) over the recording period within the above two groups further suggests that 4 hrs of continuous observation per session (with two sessions of observation per week) is adequate for reliably quantifying the frequency of SRMS in the F344 model of TLE, which is consistent with our earlier study (Rao et al., 2006).
Figure 2.
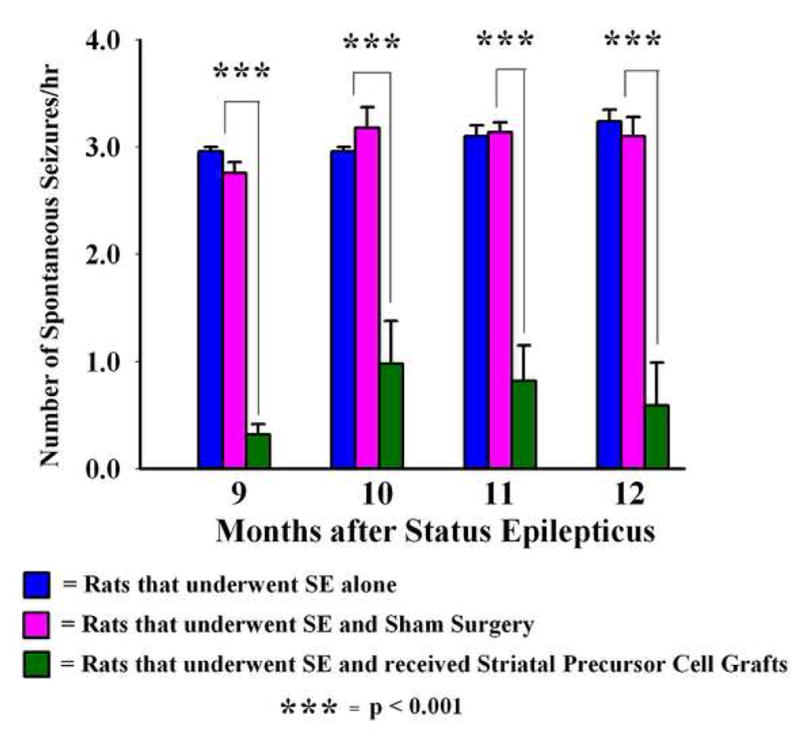
Effects of striatal precursor cell grafting shortly after status epilepticus (SE) on the extent of chronic epilepsy. The bar chart illustrates the frequency of spontaneous recurrent motor seizures (SRMS) at 9–12 months post-SE in different groups of rats that underwent SE. Values in different groups represent means and standard errors. Both rats receiving no grafts (i.e. epilepsy-only rats, n = 5) and rats receiving sham grafting surgery (n = 5) exhibit ~3.0 SRMS per hour during this period. In sharp contrast, epileptic rats receiving striatal precursor cell grafts (treated with fibroblast growth factor-2 and a caspase inhibitor Ac-YVAD-cmk, n = 5) exhibit considerably diminished frequency of SRMS during this period. Overall, there was 67–89% decrease in seizure frequency in the grafted group, in comparison to epilepsy-only and sham grafting surgery groups (p <0.001).
All SE-rats that received striatal precursor cell grafts treated and transplanted with FGF-2 and caspase inhibitor also exhibited some SRMS. However, the frequency of SRMS was dramatically less than that observed in epilepsy-only rats and SE-rats that received sham grafting surgery at all post-SE months examined in this study (Fig. 2). The overall reductions in the frequency of SRMS in comparison to epilepsy-only rats and SE-rats that received sham grafting surgery were equivalent to 89% at 9 months post-SE (p < 0.001), 67–69% at 10 months post-SE (p < 0.001), 74% at 11 months post-SE (p < 0.001) and 71–72% at 12 months post-SE (p < 0.001; Fig. 2). Thus, bilateral placement of striatal precursor cell grafts (treated and transplanted with FGF-2 and caspase inhibitor) shortly after SE into hippocampi is efficacious for considerably reducing the frequency of SRMS on a long-term basis in the chronic epilepsy period.
Hippocampal neurodegeneration in different groups, and cytoarchitecture and location of grafts
The cytoarchitecture of the hippocampus in epilepsy-only rats was conspicuous through bilateral loss of neurons in the dentate hilus, the CA3c subregion and the CA1 pyramidal cell layer (Fig. 3 [B1–B3]), in comparison to the age-matched naive hippocampus (Fig. 3 [A1–A3]). In addition, there was thinning of the CA3 pyramidal cell layer throughout the antero-posterior extent of the hippocampus. The loss of pyramidal neurons in CA3a and CA3b subregions was extensive in some rats however (data not illustrated). In contrast, in grafted animals, the overall neurodegeneration particularly in the CA1 region (Fig. 3 [D2]) appeared less than that observed in epilepsy-only rats (Fig. 3 [B1]). Examination of serial sections (every 10th) stained for Nissl or immunostained for BrdU from grafted animals demonstrated the presence of grafts along the entire septo-temporal axis of the hippocampus. Visualization with BrdU immunostaining demonstrated that grafted cells remained in clusters at the grafted site, as BrdU+ cells were densely packed in the graft core (Fig. 3 [C3]) and no BrdU+ cells were observed away from the graft core in the hippocampus. Majority of BrdU+ cells within grafts exhibited dense BrdU immunoreaction product (Fig. 3 [C3]) suggesting that transplanted cells did not undergo significant cell division after grafting. Examination of the Nissl stained sections revealed the presence of a large number of neurons within the graft core (Fig. 3 [D1–D3]).
Figure 3.
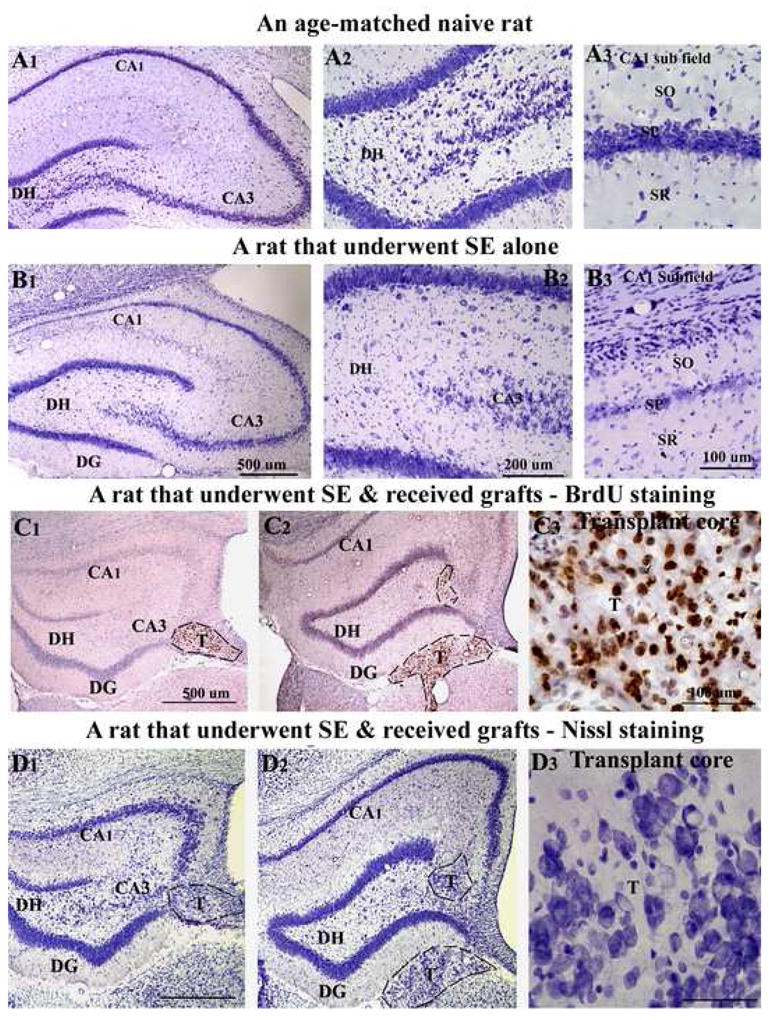
Hippocampal cytoarchitecture and location of grafts. The top two panels illustrate the hippocampal cytoarchitecture in a naive age-matched intact rat (A1–A3), and a rat that exhibited status epilepticus (SE) induced chronic epilepsy and euthanized at 12 months post-SE (B1–B3). The figures A2–A3 and B2–B3 are magnified views of dentate and CA1 regions from A1 and B1. Note a significant loss of neurons in the dentate hilus (B2), the CA1 subfield (B3) and the CA3c subregion (B2). The third and fourth panels show the location of striatal precursor cell grafts, as revealed by 5′-bromodeoxyuridine (BrdU) immunostaining (C1–C3) and Nissl staining (D1–D3) of adjacent sections. Note that, transplants are located partly inside the hippocampus and partly project into the lateral ventricle below. The figures C3 and D3 are magnified views of transplant regions from C2 and D2 showing the morphology of grafted cells and neurons. Scale bar, A1, B1, C1–C2, D1–D2 = 500μm; A2, B2 = 200μm; A3, B3, C3, D3 = 100μm. DG, dentate gyrus; DH, dentate hilus; SO, stratus oriens; SP, stratum pyramidale; SR, stratum radiatum; T, transplant.
In all grafted animals, grafts were located either inside the hippocampus or in close vicinity of the hippocampus with clear attachments to the hippocampus at all septo-temporal levels examined. Because we aimed to place the grafts in the CA3 region by injecting the cell suspension at the end of the hippocampal fissure, grafts were found either in the CA3b subregion (i.e. close to the lateral ends of the granule cell layer) or on the ventral surface of the CA3b subregion with projections into the lateral ventricle below (Fig. 3 [C1, C2, D1, D2]). The location of grafts in serial sections through the hippocampus of a rat that received striatal precursor cell grafts is illustrated in Figure 4, via tracing of different hippocampal cell layers, subfields and grafts using Neurolucida (Microbrightfield Inc., Williston, VT). The location of grafts with respect to the hippocampal cell layers and subfields shown in this figure is a good example of transplant location found in all grafted animals.
Figure 4.
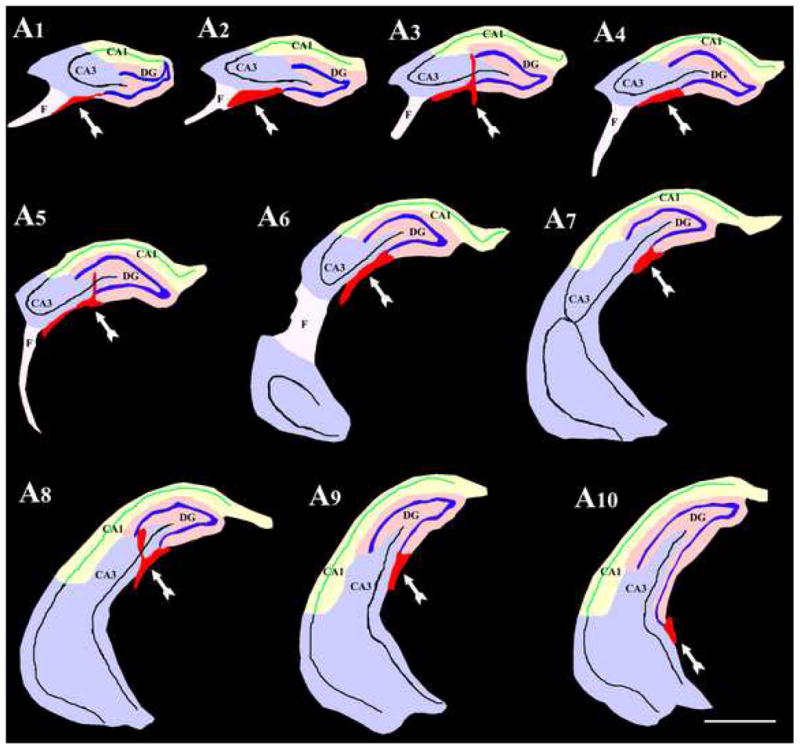
Location of transplants with respect to hippocampal cell layers and subfields. The figure illustrates drawings of serial sections through the hippocampus of a rat that received striatal precursor cell grafts. The tracings of different hippocampal cell layers, subfields and grafts were done using the Neurolucida software. Note that, grafts (the red masses denoted by arrows in all drawings) were located either in the CA3b subregion close to the lateral ends of the granule cell layer (A3, A5, A8) or on the ventral surface of the CA3b subregion with projections into the lateral ventricle below (A1–A10). Scale bar, 1000μm.
Neuronal phenotypes within transplants
Examination of grafted cells using BrdU and NeuN dual immunofluorescence and confocal microscopy demonstrated the presence of neurons within grafts (Fig. 5 [upper panels]). Grafted cells that were immunoreactive for both BrdU and NeuN displayed BrdU immunofluorescence in the nucleus and NeuN expression in both nucleus and cytoplasm of the cell body (Fig. 5). Quantification of the percentages of BrdU+ grafted cells expressing NeuN in different groups demonstrated that 87% (Mean ± S.E.M. = 87.2 ± 3.4%) of surviving grafted cells are neurons. Although BrdU+ cells counted at the time of grafting likely comprised both neuronal and glial precursors, E15 lateral ganglionic eminence (the age of striatal precursors used in this study for collecting donor cells) mostly comprise neuronal precursors based on earlier cell culture analyses (Zaman et al., 2000). This may be one of the reasons for most of the BrdU cells encountered within grafts are neurons. In addition, neuronal precursors (i.e. cells from E15 lateral ganglionic eminence) being post-mitotic at the time of grafting retain the BrdU label permanently, whereas glia (even though fewer in number at the time of grafting) likely divide after grafting and lose their BrdU label over time. Assessment of GABA-ergic neurons via BrdU and GABA double immunofluorescence methods and confocal microscopy revealed the presence of a large number of GABA-ergic neurons within grafts (Fig. 5 [lower panels]). Quantification revealed that 69% (68.8 ± 3.1%) of surviving grafted cells are GABA-ergic neurons, suggesting that a vast majority of neurons derived from striatal precursor cell grafts are GABA-ergic neurons.
Figure 5.
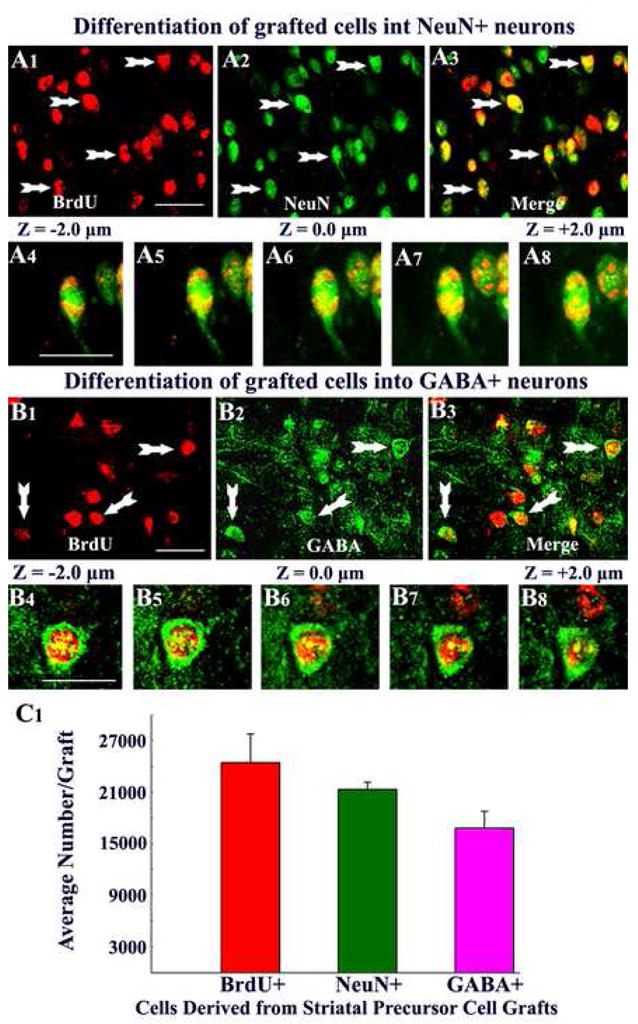
Differentiation of grafted striatal precursor cells into neurons and gamma-amino butyric acid (GABA) positive neurons. Figures A1–A3 show examples of 5′-bromodeoxyuridine (BrdU) positive grafted cells that differentiated into neuron-specific nuclear antigen (NeuN) positive neurons (arrows), using BrdU and NeuN dual immunofluorescence. Figures A4–A8 illustrate Z-section analyses of grafted cells that are positive both BrdU and NeuN, using confocal microscopy. Figures B1–B3 show examples of grafted cells that differentiated into GABA-positive neurons (arrows), using BrdU and GABA dual immunofluorescence. Figures B4–B8 illustrates Z-section analyses of a grafted cell that is positive for both BrdU and GABA using confocal microscopy. The bar chart in C1 depicts average numbers of BrdU positive cells, NeuN positive neurons, and GABA-ergic neurons derived from individual striatal precursor cell grafts in the hippocampus. A1–A3, B1–B3 = 20μm; A4–A8, B4–B8 = 10μm.
Yields of surviving cells, total neurons, and GABA-ergic neurons from grafts
We transplanted 80,000 live cells into each of the 4 locations in every hippocampus. This amounts to 320,000 live cells per hippocampus and 640,000 live cells per animal. However, based on the BrdU labeling index (93.4%), the average number of BrdU labeled live cells implanted per site was ~74,700 (or ~298,800 cells per hippocampus). We quantified the total yield of surviving grafted cells (i.e. BrdU+ cells) in individual transplants within hippocampi of different grafted animals (n=20 transplants in 5 hippocampi of 5 different animals) using serial sections (every 10th) immunostained for BrdU and the optical fractionator cell counting method. The yield of BrdU+ cells was equivalent to ~24,500 ± 3,300 cells per transplant (Fig. 5 [C1]), ~98,000 ± 13,200 per hippocampus, which amounts to ~196,000 cells per animal. In comparison to the number of BrdU-labeled cells initially grafted per site (i.e. 74,700 live cells), the average yield of surviving grafted cells was equivalent 33% of injected cells. Thus, the overall long-term survival of striatal precursors treated and grafted with FGF-2 and caspase inhibitor into the epileptic hippocampus is moderate in comparison to similarly treated fetal hippocampal cells in the epileptic hippocampus (98% of injected cells; (Rao et al., 2007) but is clearly enhanced in comparison to the standard striatal precursor cell grafts placed into the injured hippocampus (Zaman et al., 2000).
Through extrapolation of numbers of BrdU+ cells recovered from different grafts with the percentages of NeuN expressing neurons among BrdU+ cells (87%), we obtained the yield of neurons per graft. The average yield of neurons per graft was ~21,300 ± 800 (Fig. 5 [C1]), which amounts to addition of ~85,200 new neurons to each hippocampus. In comparison to the number of BrdU-labeled cells initially grafted (i.e. 74,700 live cells), the yield of surviving neurons was equivalent to 29% of injected cells. Furthermore, extrapolation of the total numbers of BrdU+ cells recovered from different transplants with fractions of GABA-ergic interneurons among BrdU+ cells (68.8%), we obtained the yield of GABA-ergic neurons (Fig. 5 [C1]). The average yield of GABA-ergic neurons per graft was ~16,800 ± 2,000 (Fig. 5 [C1]), which amounts to addition of ~67,200 new GABA-ergic neurons to each hippocampus. In comparison to the number of BrdU-labeled cells initially grafted (i.e. 74,700 live cells), the average yield of surviving GABA-ergic neurons in each graft was equivalent to 23% of injected cells. Collectively, our results suggest that a considerable number of new GABA-ergic neurons get incorporated into the circuitry of the epileptic hippocampus following striatal precursor cell grafting at 4 days post-SE.
Subtypes of GABA-ergic phenotypes within transplants
Since a vast majority of surviving neurons within striatal precursor cell grafts were GABA-ergic neurons, we examined whether this population comprises different subtypes of GABA-ergic neurons through dual immunofluorescence and confocal microscopic analyses of cells positive for BrdU and markers of subtypes of GABA-ergic neurons such as calbindin, parvalbumin, calretinin and neuropeptide Y. Interestingly, all of these subtypes of GABA-ergic neurons were present in grafts and representative samples are illustrated in Figure 6. Quantification revealed that, among surviving grafted cells, 30% (30.4 ± 4.9%) are calbindin+ neurons, 29% (28.9 ± 3.1%) are parvalbumin+ neurons, 31% (30.8 ± 2.2%) are calretinin+ neurons, and 8% (7.8 ± 3.1%) are neuropeptide Y+ neurons. Some overlap between these populations likely exists, as addition of percentages of different subclasses of GABA-ergic neurons amounts to more than 100% and a greater percentage than the percentage of GABA-ergic neurons in the graft. Extrapolation of the total numbers of BrdU+ cells recovered from different transplants with fractions of different subtypes of GABA-ergic interneurons among BrdU+ cells, we obtained the overall yields of these subtypes of GABA-ergic neurons (Fig. 6 [E1]). The average yield per graft was ~7,300 ± 1,200 for calbindin+ neurons, ~7,100 ± 700 for parvalbumin+ neurons, ~7,600 ± 500 for calretinin+ neurons, and ~2,000 ± 700 for neuropeptide Y+ neurons (Fig. 6 [E1]).
Figure 6.
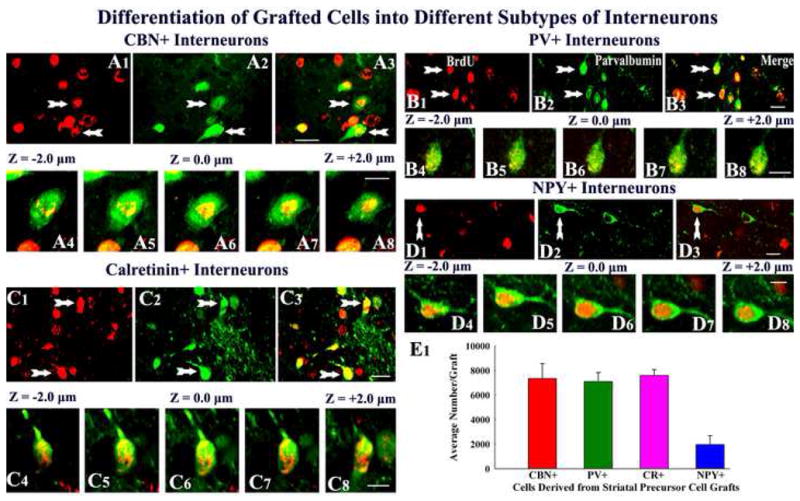
Differentiation of grafted striatal precursor cells into subclasses of gamma-amino butyric acid (GABA) positive neurons, visualized through dual immunofluorescence and confocal microscopy. The figures show examples of 5′-bromodeoxyuridine (BrdU) positive grafted cells that differentiate into interneurons positive for calbindin (A1–A3), parvalbumin (PV, B1–B3), calretinin (C1–C3) and neuropeptide Y (NPY, D1–D3). Figures A4–A8, B4–B8, C4–C8 and D4–D8 illustrate Z-section analyses of grafted cells that are positive BrdU & calbindin, BrdU & parvalbumin, BrdU & calretinin, and BrdU & neuropeptide Y respectively. The bar chart in E1 depicts average numbers of interneurons positive for calbindin, parvalbumin, calretinin and neuropeptide Y in individual striatal precursor cell grafts. Scale bar, A1–A3, B1–B3, C1–C3, D1–D3 = 10μm; A4–A8, B4–B8, C4–C8, D4–D8 = 5μm. CBN, calbindin, PV, parvalbumin, CR, calretinin, and NPY, neuropeptide Y.
Effects of grafting on hippocampal calbindin immunoreactivity
Calbindin plays an important role in controlling abnormal discharges in hippocampal neurons via its function as an intracellular facilitator of calcium diffusion with a high affinity and selectivity for calcium in the micromolar range (Baimbridge et al., 1992). A sustained loss of the calcium binding protein calbindin has been observed in major fractions of dentate granule and CA1 pyramidal cells in animal models of TLE (Shetty and Turner, 1995; Shetty and Hattiangady, 2007), which is consistent with the loss of calbindin observed in the surviving dentate granule cells of TLE patients (Magloczky et al., 1997). Although the precise reason for calbindin loss during TLE is still unclear, studies suggest that the loss of calbindin in dentate granule cells and CA1 pyramidal cells after the KA-induced CA3-region injury implies the existence of hyperexcitability (Nagerl et al., 2000; Kim et al., 2006; Selke et al., 2006). Considering these, we investigated whether striatal precursor cell grafting early after SE would prevent or diminish the loss of calbindin in dentate granule cells and CA1 pyramidal neurons.
In the naive control hippocampus, calbindin is robustly expressed in dentate granule cells and superficial cells of the CA1 pyramidal cell layer (Fig. 7 [A1–A3]). However, in epilepsy-only rats (i.e. rats exhibiting robust SRMS), there was dramatic down-regulation of calbindin in both dentate granule cells and CA1 pyramidal neurons (Fig. 7 [B1–B3]), consistent with previous studies in other models of TLE (Shetty and Turner, 1995; Shetty and Hattiangady, 2007). Interestingly, in SE rats that received striatal precursor cell grafts at 4 days post-SE, calbindin was preserved in both dentate granule cells and CA1 pyramidal neurons (Fig. 7 [C1–C3]). The overall expression of calbindin in the hippocampus of grafted rats was dramatically greater than epilepsy-only rats, though the extent of immunoreactivity was less than that in the age-matched hippocampus of naive rats. Overall, it is clear that the expression of calbindin is robust when no seizures are present (i.e. naive control rats), moderate with lower frequency of seizures (i.e. SE-rats that received striatal precursor cell grafts), and scarce with much greater frequency of seizures (i.e. epilepsy only rats). Thus, it appears that there is a link between the extent of SRMS and hippocampal calbindin expression.
Figure 7.
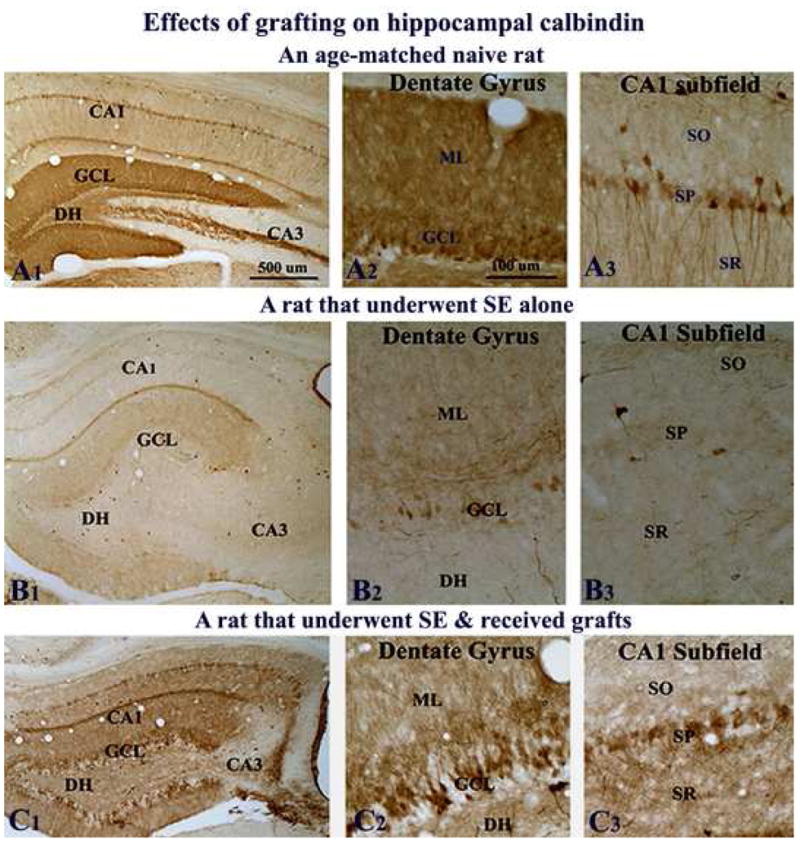
Effects of grafting on calbindin immunoreactivity. Figures show calbindin immunoreactivity in the hippocampus of a naive adult rat (A1–A3), a rat that exhibited SE-induced chronic epilepsy (B1–B3), and a rat that underwent SE and received grafts of striatal precursor cells (C1–C3). A2–A3, B2–B3 and C2–C3 are magnified views of dentate and CA1 regions from A1, B1, and C1. Note that calbindin immunoreactivity in the hippocampus diminishes dramatically in both dentate granule cells and CA1 pyramidal neurons during SE-induced chronic epilepsy (B1–B3), in comparison to the naive hippocampus. Contrastingly, when rats that underwent SE receive striatal precursor cell grafts, substantial amount of calbindin is preserved in both dentate gyrus and CA1 subfield of the hippocampus during the chronic phase (C1–C3). Scale bar, A1, B1, C1 = 500μm; A2–A3, B2–B3, C2–C3 = 100μm. DH, dentate hilus; GCL, granule cell layer; ML, molecular layer; SO, stratus oriens; SP, stratum pyramidale; SR, stratum radiatum.
Effects of grafting on aberrant mossy fiber sprouting
To examine whether reduced frequency of seizures in SE-rats that received striatal precursor cell grafts was linked to a diminished abnormal mossy fiber sprouting, we examined the extent of mossy fiber sprouting using NPY immunostaining. The NPY immunostaining in the hippocampus of naive rats reveals mainly the NPY+ interneurons and NPY+ positive axon terminals (Hattiangady et al., 2005). However, in the epileptic hippocampus, because of the up-regulation of NPY expression in dentate mossy fibers, NPY immunostaining has been found to be useful for ascertaining aberrant mossy fiber sprouting into the dentate supragranular layer (Rao et al., 2007). As expected, NPY immunostaining revealed a large number of interneurons in the dentate gyrus of naive rats (Fig. 8 [A1–A2]). The NPY immunostaining in epilepsy only rats on the other hand revealed reduced number of interneurons in the dentate hilus but robust NPY expression in mossy fibers (Fig. 8 [B1]). Aberrant sprouting of mossy fibers into the dentate supragranular layer could be visualized very clearly (Fig. 8 [B2]). Rats that received grafts after SE also exhibited aberrant mossy fiber sprouting into the dentate supragranular layer (Fig. 8 [C1–C2]), suggesting that the suppression of aberrant mossy fiber sprouting is not the mechanism underlying the reduced frequency of SRMS in SE-rats receiving grafts.
Figure 8.
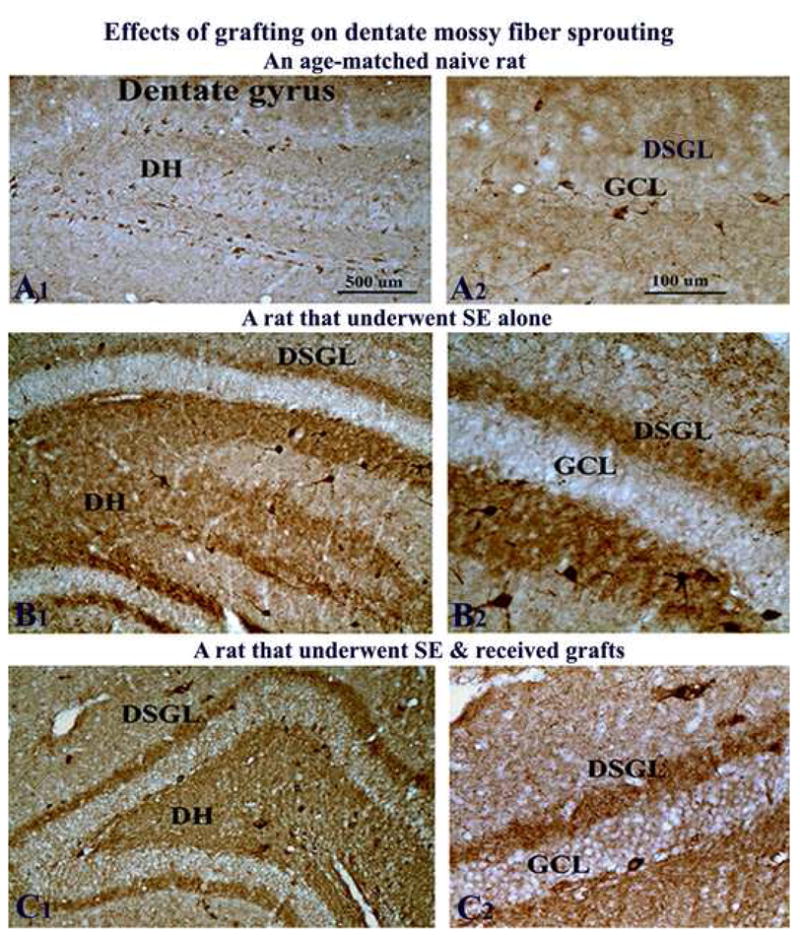
Effects of grafting on abnormal mossy fiber sprouting in the host hippocampus. Figures illustrate neuropeptide Y (NPY) immunostaining in the hippocampus of a naive adult rat (A1–A2), a rat that exhibited SE-induced chronic epilepsy (B1–B2), and a rat that underwent SE and received grafts of striatal precursor cells (C1–C2). A2, B2 and C2 are magnified views of a dentate gyrus region from A1, B1, and C1. In naive rat (A1–A2), NPY immunostaining visualizes mainly the interneurons in the dentate hilus, whereas in the rat that underwent SE-induced chronic epilepsy (B1–B2), NPY expression is found in mossy fibers including those mossy fibers that sprout aberrantly into the dentate supragranular layer (DSGL). Note that the extent of aberrant sprouting in the DSGL is comparable between the rat that underwent SE alone (B1–B2) and the rat that underwent SE and received striatal precursor cell grafts (C1–C2). Scale bar, A1, B1, C1 = 500μm; A2, B2, C2 = 100μm. DH, dentate hilus; GCL, granule cell layer; ML, molecular layer.
Discussion
This study, for the first time, demonstrates that bilateral grafting of appropriately treated striatal precursor cells into hippocampi shortly after SE is efficacious for considerably reducing the frequency of SRMS on a long-term basis in the chronic epilepsy period. A reduced frequency of SRMS after striatal precursor cell grafting in SE rats was associated with long-term (one-year) survival of a sizable fraction grafted cells with differentiation of a vast majority of grafted cells into GABA-ergic neurons. Additional analyses revealed the presence of multiple subclasses of GABA-ergic neurons within grafts, including neurons positive for calbindin, parvalbumin, calretinin and neuropeptide Y. Furthermore, significantly reduced frequency of SRMS in SE rats that received striatal precursor cell grafts was coupled with substantial preservation of the hippocampal calbindin, in contrast to its dramatic loss in rats that underwent SE alone. Additionally, substantially diminished frequency of SRMS in SE rats that received striatal precursor cell grafts appeared to be specific to the presence of grafts, as sham grafting surgery had no effects on the frequency of SRMS in the chronic epilepsy period. These results have importance towards developing an apt cell transplantation therapy for restraining chronic epilepsy after SE or brain injury.
Links between decreased frequency of SRMS and graft-derived new GABA-ergic neurons
Analyses of the frequency of SRMS revealed ~3 seizures per hour during 9–12 months post-SE in both SE-rats receiving no grafts (i.e. epilepsy-only rats) and SE-rats receiving sham grafting surgery, suggesting that sham-grafting surgery has no influence on the evolution of SE into chronic epilepsy. On the contrary, in SE-rats that received striatal precursor cell grafts, the frequency of SRMS was 67–89% less than that observed in both epilepsy-only rats and SE-rats that received sham grafting surgery. This suggests that a remarkably reduced frequency of SRMS observed after striatal precursor cell grafting is linked to graft-derived neurons in the hippocampus rather than effects of the grafting surgery. However, there are certain issues that need to be addressed rigorously in future studies to validate the efficacy of striatal precursor cell grafting for substantially diminishing the SE-induced chronic epilepsy. First, as only the frequency of major motor seizures (stages III–V seizures) was scored in this study, it remains to be seen whether striatal precursor cell grafting is efficacious for reducing both frequency and intensity of electrographic seizures. Second, as SRMS were recorded only at 9–12 months after SE and grafting, it is unclear whether striatal precursor cell grafting into hippocampi delays the evolution of SE into chronic epilepsy. Third, as no control cells, dead cells or medium was injected in SE rats that underwent sham grafting surgery, it remains to be validated whether the beneficial effects observed in this study were specific to striatal precursor cell grafts. From these perspectives, continuous EEG recordings for prolonged periods after grafting of striatal precursor cells, control cells or dead cells in different groups of SE rats are needed in future to address these issues.
Nevertheless, histological analyses at one-year post-grafting in this study revealed long-term survival of sizable fraction (31%) of grafted cells with differentiation of a vast majority of them into GABA-ergic neurons, which supports the premise that decreased frequency of SRMS observed in SE rats after striatal precursor cell grafting is likely linked to graft-derived GABA-producing neurons. Based on the yield of surviving cells, GABA-ergic neurons among surviving cells and the number of grafts placed per hippocampus, the average number of graft-derived GABA-ergic neurons added to each hippocampus was ~67,300, which is equivalent to addition of ~134,600 new GABA-ergic neurons to hippocampi of each animal. One-year survival of graft-derived GABA-ergic neurons reflects their integration into the host hippocampal circuitry, as lack of integration of grafted cells typically leads to their degeneration over time after grafting. It is likely that a decreased frequency of SRMS observed in SE-rats receiving striatal precursor cell grafts is influenced by these newly added GABA-ergic neurons because, in the absence of these new GABA-ergic neurons (i.e. in rats receiving sham-grafting surgery), the frequency of SRMS remains comparable to epilepsy-only rats. This possibility is also supported by the following observations. First, it emerges that a reduced frequency of seizures following striatal precursor cell grafting is not due to the suppression of aberrant mossy fiber synaptic reorganization, an epileptogenic change that is believed to contribute to the occurrence or extent of SRMS (Cronin and Dudek, 1988; Mathern et al., 1993; Okazaki et al., 1995; Buhl et al., 1996; Lynch et al., 2000; Wuarin and Dudek, 2001; Shetty et al., 2005). This is because aberrantly sprouted mossy fibers could be clearly observed in the dentate supragranular layer of both SE-rats receiving no grafts and SE-rats receiving striatal precursor cell grafts. Second, it is unlikely that striatal precursor cell grafts mediate repair of the disrupted tri-synaptic hippocampal circuitry as observed with grafts of fetal hippocampal cells (Shetty and Turner, 1997, 2000; Shetty et al., 2005) because a vast majority of striatal precursor cells differentiate into GABA-ergic neurons rather than hippocampal CA1 or CA3 pyramidal neurons. Considering the above, and based on yields of new GABA-ergic neurons derived from grafts, it is likely that strengthening of the inhibitory GABA-ergic neurotransmission in the host hippocampus underlies the beneficial effects mediated by grafts. Thus, addition of a significant number of new GABA-ergic neurons into the hippocampus after SE appears to be a useful approach for restraining chronic epilepsy development after SE or brain injury.
Would increased addition of new GABA-ergic neurons to the hippocampus prevent SE-induced chronic epilepsy?
While the frequency of SRMS in SE rats receiving striatal precursor cell grafts was considerably less than both SE-rats receiving no grafts and SE-rats receiving sham grafting surgery, striatal precursor cell grafting did not completely prevent SE-induced chronic epilepsy. This could be due to addition of an insufficient number of new GABA-ergic interneurons into the hippocampal circuitry, as seizure-induced hippocampal injury is associated with dramatic depletions in the number of GABA-ergic interneurons, reduced granule cell inhibition and reduced excitatory drive onto interneurons (Sloviter, 1987; Shetty and Turner, 2000, 2001; Doherty and Dingledine, 2001; Kobayashi and Buckmaster, 2003; Sloviter et al., 2003; Ben-Ari, 2006). Previous studies have also suggested that survival of grafted neurons in the hippocampus is an important pre-requisite for reducing the frequency of SRMS through grafts. For instance, Hasegawa and colleagues (Hasegawa et al., 2004) demonstrated that grafting of 40,000–80,000 Layton Bioscience (LBS) neurons per hippocampus is inefficient for diminishing seizures in a KA-model of TLE because of poor survival of grafted cells. Similarly, a recent study (Rao et al., 2007) reveals that seizure-reducing effects of fetal hippocampal cell grafts is directly linked to their survival in the chronically epileptic hippocampus. Grafting of standard fetal hippocampal cells (i.e. without pre-treatment with graft augmentation factors) led to their poor survival and no seizure-reducing effects whereas grafting of fetal hippocampal cells treated with distinct neurotrophic factors and caspase inhibitor resulted in robust graft cell survival and considerable seizure-reducing effects. In this context, it is plausible that an improved survival of grafted striatal precursor cells might prevent the occurrence of chronic epilepsy after SE. Alternatively, grafting of increased number of precursor cells per site or increasing the number of grafts per hippocampus might be useful approaches for future studies. Additionally, as SE also leads to hyperexcitability in the entorhinal cortex due to loss of inhibitory interneurons (Kobayashi and Buckmaster, 2003; Kumar and Buckmaster, 2006) bilateral grafting of striatal precursor cells into hippocampi as well as entorhinal cortices may be important for completely preventing the chronic epilepsy development after SE.
The survival of striatal precursor cells following placement into the injured hippocampus is generally poor (Zaman et al., 2000). However, the survival of striatal precursor cells in the hippocampus after SE in this study is much improved with recovery at one-year post-grafting equaling 33% of injected cells. This is likely due to the pre-treatment and grafting of donor cells with FGF-2 and caspase inhibitor. The selection of combined neurotrophic supplementation and caspase inhibition for improving the yields of surviving cells and neurons in the hippocampus was based on results of multiple previous studies on graft augmentation in the injured adult brain (Schierle et al., 1999; Hansson et al., 2000; Helt et al., 2001; Cicchetti et al., 2002; Zaman and Shetty, 2002; Hattiangady et al., 2006; Rao et al., 2007). Considering the functions of neurotrophic factors and caspase inhibitors, it appears that enhanced yields of surviving grafted neurons observed after combined neurotrophic supplementation and caspase inhibition are a result of rapid differentiation of grafted cells and rescue of grafted cells from grafting trauma-related apoptosis (Esclapez and Trottier, 1989; Maisonpierre et al., 1990; Friedman et al., 1991; Ernfors et al., 1992; Friedman et al., 1998; Zawada et al., 1998; Sortwell et al., 2001). Nevertheless, in comparison to the survival of similarly treated fetal hippocampal cells in the chronically injured or epileptic hippocampus (with recovery of over 98% of injected cells; Hattiangady et al., 2006; Rao et al., 2007) the overall long-term survival of striatal precursors treated and grafted with FGF-2 and caspase inhibitor in this study is moderate. Thus, graft augmentation strategies that are specific for increasing the survival of striatal precursor cells in the epileptic hippocampus are needed in future.
Significance of reduced frequency of SRMS in SE-rats receiving striatal precursor cell grafts
The efficacy of transplantation of different GABA-producing cells into different regions of the brain for restraining seizures has been examined in several earlier studies. However, the usefulness of these cell grafts for reducing chronic SRMS could not be ascertained, as seizure analyses were performed only during the early post-grafting period, the survival of grafted cells was limited and the beneficial effects were transient. For instance, bilateral grafting of conditionally immortalized neurons engineered to produce GABA into the substantia nigra of rats at 45–65 days after SE in a pilocarpine model of epilepsy results in fewer spontaneous seizures at 1–10 days post-grafting than epileptic rats receiving only control cells (Thompson and Suchomelova, 2004). Additional studies have examined the effects of grafting of GABA-producing cells into the dentate gyrus (Thompson, 2005) or the substantia nigra (Löscher et al., 1998; Castillo et al., 2006) on the development of seizures. Transplantation of these cells were associated with increased GABA levels, enhanced local electrical seizure threshold and delayed onset of behavioral seizures in the kindling model of epilepsy (Thompson, 2005), and diminished extent of seizures with KA administration (Castillo et al., 2006). Although these results are interesting and provide the proof of principle for suppressing seizures using GABA-producing cells, it remains an interesting question to verify whether cells engineered to produce GABA are capable of exhibiting enduring survival and maintaining GABA release on a long-term basis in the epileptic brain. Apart from GABA producing cells, a series of studies have ascertained the effects of grafts of encapsulated fibroblasts engineered to release the brain’s endogenous anticonvulsant adenosine on seizures (Boison et al., 1999, 2002; Boison, 2005; Huber et al., 2001). An initial study reported that transplantation of these cells into the brain ventricles of rats kindled in the hippocampus is associated with only transient reductions (lasting ~2 weeks) in behavioral seizures and afterdischarges. Use of mouse myoblasts engineered to release adenosine also demonstrated similar transient suppressing effects on seizures (Fedele et al., 2004; Guttinger et al., 2005). Recent studies by using embryonic stem (ES) cell derived neural precursors that are engineered to release adenosine as donor cells report sustained protection against developing generalized seizures in a kindling model of TLE (Li et al., 2007) and suppression of spontaneous seizures in mice when implanted after the epileptogenesis-precipitating brain injury (Li et al., 2008). These results are certainly promising for future development of stem cell-mediated adenosine delivery for treating epilepsy. Yet, it remains to be determined whether these cells have the ability to deliver adenosine on a long-term basis.
Thus, to understand the usefulness of distinct grafting approaches for preventing SE-induced chronic epilepsy, it is imperative that prolonged effects of distinct cell transplants placed into the hippocampus or other regions such as the substantia nigra after the induction SE be tested rigorously in appropriate animal models. From this perspective, the finding of the present study that appropriately treated striatal precursor cell grafts placed bilaterally into hippocampi shortly after SE reduce the frequency of SRMS on a long-term basis in the chronic epilepsy period has clinical significance. More importantly, a reduced frequency of SRMS observed after striatal precursor cell grafting was associated with one-year survival of ~33% of grafted cells with differentiation of a vast majority of grafted cells into GABA-ergic neurons. Additionally, these beneficial effects of grafts were associated with substantial preservation of the hippocampal calbindin. Because a dramatic loss of calbindin in hippocampi during epileptic conditions is believed to be due to hyperexcitability (Magloczky et al., 1997; Nagerl et al., 2000; Kim et al., 2006; Selke et al., 2006; Shetty and Hattiangady, 2007), preservation of calbindin in the hippocampus of SE-rats receiving striatal precursor cell grafts and exhibiting reduced frequency of SRMS likely suggests a reduced hippocampal hyperexcitability with grafting of striatal precursor cells. This is presumably mediated through an increased GABA-ergic inhibitory neurotransmission on principal hippocampal neurons by the new GABA-ergic interneurons derived from grafts. However, additional electrophysiological studies of both graft-derived GABA-ergic neurons and hippocampal principal neurons are required in future to confirm the presence of this mechanism. In conclusion, the results obtained with striatal precursor cell grafting in this long-term study are promising towards developing cell based therapies that are potentially capable of restraining chronic epilepsy development after SE or brain injury.
Acknowledgments
This research was supported by grants from the National Institute of Neurological Disorders and Stroke (RO1 NS054780 and RO1 NS043507 to A.K.S.) and Department of Veterans Affairs (VA Merit Review Award to A.K.S.). We thank K. S. Rai for scoring spontaneous seizures in a group of rats and B. Shuai for excellent technical assistance in this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baimbridge KG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992;15:303–308. doi: 10.1016/0166-2236(92)90081-i. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Seizures beget seizures: the quest for GABA as a key player. Crit Rev Neurobiol. 2006;18:135–144. doi: 10.1615/critrevneurobiol.v18.i1-2.140. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist. 2005;11:25–36. doi: 10.1177/1073858404269112. [DOI] [PubMed] [Google Scholar]
- Boison D, Huber A, Padrun V, Deglon N, Aebischer P, Mohler H. Seizure suppression by adenosine-releasing cells is independent of seizure frequency. Epilepsia. 2002;43:788–796. doi: 10.1046/j.1528-1157.2002.33001.x. [DOI] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Tseng JL, Aebischer P, Mohler H. Seizure suppression in kindled rats by intraventricular grafting of an adenosine releasing synthetic polymer. Exp Neurol. 1999;160:164–174. doi: 10.1006/exnr.1999.7209. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- Castillo CG, Mendoza S, Freed WJ, Giordano M. Intranigral transplants of immortalized GABAergic cells decrease the expression of kainic acid-induced seizures in the rat. Behav Brain Res. 2006;171:109–115. doi: 10.1016/j.bbr.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Costantini L, Belizaire R, Burton W, Isacson O, Fodor W. Combined inhibition of apoptosis and complement improves neural graft survival of embryonic rat and porcine mesencephalon in the rat brain. Exp Neurol. 2002;177:376–384. doi: 10.1006/exnr.2002.8007. [DOI] [PubMed] [Google Scholar]
- Cornish SM, Wheal HV. Long-term loss of paired pulse inhibition in the kainic acid-lesioned hippocampus of the rat. Neuroscience. 1989;28:563–571. doi: 10.1016/0306-4522(89)90005-5. [DOI] [PubMed] [Google Scholar]
- Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. 1988;474:181–184. doi: 10.1016/0006-8993(88)90681-6. [DOI] [PubMed] [Google Scholar]
- Detour J, Schroeder H, Desor D, Nehlig A. A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium-pilocarpine model in adult rats. Epilepsia. 2005;46:499–508. doi: 10.1111/j.0013-9580.2005.38704.x. [DOI] [PubMed] [Google Scholar]
- Devinsky O. Therapy for neurobehavioral disorders in epilepsy. Epilepsia. 2004;45(Suppl 2):34–40. doi: 10.1111/j.0013-9580.2004.452003.x. [DOI] [PubMed] [Google Scholar]
- Doherty J, Dingledine R. Reduced excitatory drive onto interneurons in the dentate gyrus after status epilepticus. J Neurosci. 2001;21:2048–2057. doi: 10.1523/JNEUROSCI.21-06-02048.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Ryder KM, Spencer DD. Hippocampal GABA transporter function in temporal-lobe epilepsy. Nature. 1995;376:174–177. doi: 10.1038/376174a0. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Merlio JP, Persson H. Cells expressing mRNA for neurotrophins and their receptors during embryonic rat development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Trottier S. Changes in GABA-immunoreactive cell density during motor focal epilepsy induced by cobalt in the rat. Exp Brain Res. 1989;76:369–385. doi: 10.1007/BF00247895. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Scheurer L, Simpson EM, Mohler H, Brustle O, Boison D. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett. 2004;370:160–165. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, Spencer DD. Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol. 1993;34:774–780. doi: 10.1002/ana.410340604. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Black IB, Kaplan DR. Distribution of the neurotrophins brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 in the postnatal rat brain: an immunocytochemical study. Neuroscience. 1998;84:101–114. doi: 10.1016/s0306-4522(97)00526-5. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Olson L, Persson H. Cells that express brain-derived neurotrophic factor mRNA in the developing postnatal rat brain. Eur J Neurosci. 1991;3:688–697. doi: 10.1111/j.1460-9568.1991.tb00854.x. [DOI] [PubMed] [Google Scholar]
- Gernert M, Thompson KW, Löscher W, Tobin AJ. Genetically engineered GABA-producing cells demonstrate anticonvulsant effects and long-term transgene expression when transplanted into the central piriform cortex of rats. Exp Neurol. 2002;176:183–192. doi: 10.1006/exnr.2002.7914. [DOI] [PubMed] [Google Scholar]
- Guttinger M, Padrun V, Pralong WF, Boison D. Seizure suppression and lack of adenosine A1 receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts. Exp Neurol. 2005;193:53–64. doi: 10.1016/j.expneurol.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Hansson O, Castilho RF, Kaminski Schierle GS, Karlsson J, Nicotera P, Leist M, Brundin P. Additive effects of caspase inhibitor and lazaroid on the survival of transplanted rat and human embryonic dopamine neurons. Exp Neurol. 2000;164:102–111. doi: 10.1006/exnr.2000.7406. [DOI] [PubMed] [Google Scholar]
- Hasegawa T, Kondziolka D, Choi SJ, Balzer J, Dixon EC, Fellows-Mayle W, Elder E. Hippocampal neurotransplantation evaluated in the rat kainic acid epilepsy model. Neurosurgery. 2004;55:191–198. doi: 10.1227/01.neu.0000126881.40748.93. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty AK. Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol Dis. 2004;17:473–490. doi: 10.1016/j.nbd.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty GA, Shetty AK. Brain-derived neurotrophic factor, phosphorylated cyclic AMP response element binding protein and neuropeptide Y decline as early as middle age in the dentate gyrus and CA1 and CA3 subfields of the hippocampus. Exp Neurol. 2005;195:353–371. doi: 10.1016/j.expneurol.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Zaman V, Shetty AK. Incorporation of embryonic CA3 cell grafts into the adult hippocampus at 4-months after injury: effects of combined neurotrophic supplementation and caspase inhibition. Neuroscience. 2006;139:1369–1383. doi: 10.1016/j.neuroscience.2006.01.058. [DOI] [PubMed] [Google Scholar]
- Hellier JL, Patrylo PR, Buckmaster PS, Dudek FE. Recurrent spontaneous motor seizures after repeated low-dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Res. 1998;31:73–84. doi: 10.1016/s0920-1211(98)00017-5. [DOI] [PubMed] [Google Scholar]
- Helt CE, Hoernig GR, Albeck DS, Gerhardt GA, Ickes B, Reyland ME, Quissell DO, Stromberg I, Granholm AC. Neuroprotection of grafted neurons with a GDNF/caspase inhibitor cocktail. Exp Neurol. 2001;170:258–269. doi: 10.1006/exnr.2001.7709. [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D. Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci USA. 2001;98:7611–7616. doi: 10.1073/pnas.131102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Kwak SE, Kim DS, Won MH, Kwon OS, Choi SY, Kang TC. Reduced calcium binding protein immunoreactivity induced by electroconvulsive shock indicates neuronal hyperactivity, not neuronal death or deactivation. Neuroscience. 2006;137:317–326. doi: 10.1016/j.neuroscience.2005.08.052. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M, Aebischer P, Elmer E, Bengzon J, Kalen P, Kokaia Z, Lindvall O. Seizure suppression in kindling epilepsy by intracerebral implants of GABA- but not by noradrenaline-releasing polymer matrices. Exp Brain Res. 1994;100:385–394. doi: 10.1007/BF02738399. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Buckmaster PS. Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2006;26:4613–4623. doi: 10.1523/JNEUROSCI.0064-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DV. Losing neurons: selective vulnerability and mesial temporal sclerosis. Epilepsia. 2005;46(Suppl 7):39–44. doi: 10.1111/j.1528-1167.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain. 2007;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest. 2008;118:571–582. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litt B, Esteller R, Echauz J, D’Alessandro M, Shor R, Henry T, Pennell P, Epstein C, Bakay R, Dichter M, Vachtsevanos G. Epileptic seizures may begin hours in advance of clinical onset: a report of five patients. Neuron. 2001;30:51–64. doi: 10.1016/s0896-6273(01)00262-8. [DOI] [PubMed] [Google Scholar]
- Lloyd KG, Bossi L, Morselli PL, Munari C, Rougier M, Loiseau H. Alterations of GABA-mediated synaptic transmission in human epilepsy. Adv Neurol. 1986;44:1033–1044. [PubMed] [Google Scholar]
- Löscher W, Ebert U, Lehmann H, Rosenthal C, Nikkhah G. Seizure suppression in kindling epilepsy by grafts of fetal GABAergic neurons in rat substantia nigra. J Neurosci Res. 1998;51:196–209. doi: 10.1002/(SICI)1097-4547(19980115)51:2<196::AID-JNR8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Löscher W, Gernert M, Heinemann U. Cell and gene therapies in epilepsy - promising avenues or blind alleys? Trends Neurosci. 2008;31:62–73. doi: 10.1016/j.tins.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sayin U, Bownds J, Janumpalli S, Sutula T. Long-term consequences of early postnatal seizures on hippocampal learning and plasticity. Eur J Neurosci. 2000;12:2252–2264. doi: 10.1046/j.1460-9568.2000.00117.x. [DOI] [PubMed] [Google Scholar]
- Magloczky Z, Halasz P, Vajda J, Czirjak S, Freund TF. Loss of Calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience. 1997;76:377–385. doi: 10.1016/s0306-4522(96)00440-x. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Manford M, Hart YM, Sander JW, Shorvon SD. National General Practice Study of Epilepsy (NGPSE): partial seizure patterns in a general population. Neurology. 1992;42:1911–1917. doi: 10.1212/wnl.42.10.1911. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Leite JP, Pretorius K, Yeoman KM, Kuhlman PA. The pathogenic and progressive features of chronic human hippocampal epilepsy. Epilepsy Res. 1996;26:151–161. doi: 10.1016/s0920-1211(96)00052-6. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Cifuentes F, Leite JP, Pretorius JK, Babb TL. Hippocampal EEG excitability and chronic spontaneous seizures are associated with aberrant synaptic reorganization in the rat intrahippocampal kainate model. Electroencephalogr Clin Neurophysiol. 1993;87:326–339. doi: 10.1016/0013-4694(93)90186-y. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Babb TL. Influence of the type of initial precipitating injury and at what age it occurs on course and outcome in patients with temporal lobe seizures. J Neurosurg. 1995;82:220–227. doi: 10.3171/jns.1995.82.2.0220. [DOI] [PubMed] [Google Scholar]
- McKeown MJ, McNamara JO. When do epileptic seizures really begin? Neuron. 2001;30:1–3. doi: 10.1016/s0896-6273(01)00253-7. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399:A15–22. doi: 10.1038/399a015. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca(2+) influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J Neurosci. 2000;20:1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- Pirttila TJ, Lukasiuk K, Hakansson K, Grubb A, Abrahamson M, Pitkanen A. Cystatin C modulates neurodegeneration and neurogenesis following status epilepticus in mouse. Neurobiol Dis. 2005;20:241–253. doi: 10.1016/j.nbd.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Rai KS, Shetty AK. Strategies for promoting anti-seizure effects of hippocampal fetal cells grafted into the hippocampus of rats exhibiting chronic temporal lobe epilepsy. Neurobiol Dis. 2007;27:117–132. doi: 10.1016/j.nbd.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Reddy DS, Shetty AK. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J Neurosci Res. 2006;83:1088–1105. doi: 10.1002/jnr.20802. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Hunt CA, Bakay RA, Oertel WH. A decrease in the number of GABAergic somata is associated with the preferential loss of GABAergic terminals at epileptic foci. Brain Res. 1986;363:78–90. doi: 10.1016/0006-8993(86)90660-8. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Smith KL, Jackson MB, Goodman JH. Structural and functional asymmetry in the normal and epileptic rat dentate gyrus. J Comp Neurol. 2002;454:424–439. doi: 10.1002/cne.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierle GS, Hansson O, Leist M, Nicotera P, Widner H, Brundin P. Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat Med. 1999;5:97–100. doi: 10.1038/4785. [DOI] [PubMed] [Google Scholar]
- Selke K, Muller A, Kukley M, Schramm J, Dietrich D. Firing pattern and calbindin-D28k content of human epileptic granule cells. Brain Res. 2006;1120:191–201. doi: 10.1016/j.brainres.2006.08.072. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B. Restoration of calbindin after fetal hippocampal CA3 cell grafting into the injured hippocampus in a rat model of temporal lobe epilepsy. Hippocampus. 2007;17:943–956. doi: 10.1002/hipo.20311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Madison RD, Bradley J, Turner DA. Quantitative graft integration of fetal hippocampal transplants labeled with 5′ bromodeoxyuridine into normal adult hippocampus. Exp Neurol. 1994;126:205–224. doi: 10.1006/exnr.1994.1059. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Intracerebroventricular kainic acid administration in adult rat alters hippocampal calbindin and non-phosphorylated neurofilament expression. J Comp Neurol. 1995;363:581–599. doi: 10.1002/cne.903630406. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Development of long-distance efferent projections from fetal hippocampal grafts depends upon pathway specificity and graft location in kainate-lesioned adult hippocampus. Neuroscience. 1997;76:1205–1219. doi: 10.1016/s0306-4522(96)00413-7. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Fetal hippocampal grafts containing CA3 cells restore host hippocampal glutamate decarboxylase-positive interneuron numbers in a rat model of temporal lobe epilepsy. J Neurosci. 2000;20:8788–8801. doi: 10.1523/JNEUROSCI.20-23-08788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Glutamic acid decarboxylase-67-positive hippocampal interneurons undergo a permanent reduction in number following kainic acid-induced degeneration of ca3 pyramidal neurons. Exp Neurol. 2001;169:276–297. doi: 10.1006/exnr.2001.7668. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Hattiangady B. Repair of the injured adult hippocampus through graft-mediated modulation of the plasticity of the dentate gyrus in a rat model of temporal lobe epilepsy. J Neurosci. 2005;25:8391–8401. doi: 10.1523/JNEUROSCI.1538-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, Frotscher M. “Dormant basket cell” hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J Comp Neurol. 2003;459:44–76. doi: 10.1002/cne.10630. [DOI] [PubMed] [Google Scholar]
- Sortwell CE, Camargo MD, Pitzer MR, Gyawali S, Collier TJ. Diminished survival of mesencephalic dopamine neurons grafted into aged hosts occurs during the immediate postgrafting interval. Exp Neurol. 2001;169:23–29. doi: 10.1006/exnr.2001.7644. [DOI] [PubMed] [Google Scholar]
- Strine TW, Kobau R, Chapman DP, Thurman DJ, Price P, Balluz LS. Psychological distress, comorbidities, and health behaviors among U.S. adults with seizures: results from the 2002 National Health Interview Survey. Epilepsia. 2005;46:1133–1139. doi: 10.1111/j.1528-1167.2005.01605.x. [DOI] [PubMed] [Google Scholar]
- Thompson KW. Genetically engineered cells with regulatable GABA production can affect afterdischarges and behavioral seizures after transplantation into the dentate gyrus. Neuroscience. 2005;133:1029–1037. doi: 10.1016/j.neuroscience.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Thompson KW, Suchomelova LM. Transplants of cells engineered to produce GABA suppress spontaneous seizures. Epilepsia. 2004;45:4–12. doi: 10.1111/j.0013-9580.2004.29503.x. [DOI] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol. 2001;85:1067–1077. doi: 10.1152/jn.2001.85.3.1067. [DOI] [PubMed] [Google Scholar]
- Zaman V, Shetty AK. Combined neurotrophic supplementation and caspase inhibition enhances survival of fetal hippocampal CA3 cell grafts in lesioned CA3 region of the aging hippocampus. Neuroscience. 2002;109:537–553. doi: 10.1016/s0306-4522(01)00478-x. [DOI] [PubMed] [Google Scholar]
- Zaman V, Turner DA, Shetty AK. Survival of grafted fetal neural cells in kainic acid lesioned CA3 region of adult hippocampus depends upon cell specificity. Exp Neurol. 2000;161:535–561. doi: 10.1006/exnr.1999.7304. [DOI] [PubMed] [Google Scholar]
- Zawada WM, Zastrow DJ, Clarkson ED, Adams FS, Bell KP, Freed CR. Growth factors improve immediate survival of embryonic dopamine neurons after transplantation into rats. Brain Res. 1998;786:96–103. doi: 10.1016/s0006-8993(97)01408-x. [DOI] [PubMed] [Google Scholar]