

Abstract
Induction of drug metabolizing enzymes, such as the cytochromes P450 (CYP) is known to cause drug-drug interactions due to increased elimination of co-administered drugs. This increased elimination may lead to significant reduction or complete loss of efficacy of the co-administered drug. Due to the significance of such drug interactions, many pharmaceutical companies employ screening and characterization models which predict CYP enzyme induction to avoid or attenuate the potential for drug interactions with new drug candidates. The most common mechanism of CYP induction is transcriptional gene activation. Activation is mediated by nuclear receptors, such as AhR, CAR, and PXR that function as transcription factors. Early high throughput screening models utilize these nuclear hormone receptors in ligand binding or cell-based transactivation/reporter assays. In addition, immortalized hepatocyte cell lines can be used to assess enzyme induction of specific drug metabolizing enzymes. Cultured primary human hepatocytes, the best established in vitro model for predicting enzyme induction and most accepted by regulatory agencies, is the predominant assay used to evaluate induction of a wide variety of drug metabolizing enzymes. These in vitro models are able to appropriately predict enzyme induction in patients when compared to clinical drug-drug interactions. Finally, transgenic animal models and the cynomolgus monkey have also been shown to recapitulate human enzyme induction and may be appropriate in vivo animal models for predicting human drug interactions.
Key words: CYP3A4, cytochrome P450, drug interactions, hepatocytes, induction, PXR
INTRODUCTION
Enzyme induction is the process of creating more enzyme than is normally present in a biological system. As early as 1954, the first report of enzyme induction appeared in a manuscript by Brown, et al. who described the enzyme inducing effects of various diets given to rodents; this was followed some time later by the first review on microsomal enzyme induction in 1967 (1,2). The increase in enzyme activity, in effect an increase in intrinsic metabolic clearance, is reflected by increased hepatic clearance of drugs metabolized by the induced enzyme. Pharmacokinetically, the drugs affected by enzyme induction generally demonstrate reduced AUC, Cmax, and half-life as a reflection of increased clearance. For example, the antidiabetic drug rosiglitazone (CYP2C8 substrate) when administered with the inducing agent rifampicin has been reported to cause 65%, 31%, and 62% decreases in AUC, Cmax, and half-life of rosiglitazone, respectively (3). A more dramatic example is illustrated by the interaction between midazolam (CYP3A4 substrate) and the herbal antidepressant St. John’s Wort (CYP3A4 inducer) where the midazolam AUC and Cmax decreased by 79% and 65%, respectively (4). These pharmacokinetic changes have significant pharmacological consequences because increased metabolism reduces the duration and pharmacological effect of co-adminstered drugs. For example, the induction of CYP3A4 results in reduced ethinylestradiol levels from oral contraceptives that can lead to unexpected pregnancies and reduced cyclosporine concentrations in transplant patients can lead to organ rejection (both drugs are predominately metabolized by CYP3A4). In general, the decrease in AUC ranges from 15–98%, depending on the potency of the inducing agent, the fraction of drug metabolized, and the overall elimination of a drug via the induced pathway, as illustrated in Table I with several well known human probe substrates for each CYP.
Table I.
Percent AUC Changes in Humans with Probe Drugs for Various CYP Enzymes
| CYP | Drug Probes | Percent AUC Decrease |
|---|---|---|
| CYP1A | Theophylline | 15–66% |
| CYP2B6 | Efavirenz | 20–30% |
| CYP2C8 | Rosiglitazone/Repaglinide | 32–80% |
| CYP2C9 | Warfarin/Tolbutamide | 16–63% |
| CYP2C19 | Omeprazole | 25–49% |
| CYP3A4 | Midazolam | 20–98% |
Data taken from the University of Washington Metabolism and Transport Drug Interaction database. http://www.druginteractioninfo.org/
In addition to the examples where one drug (perpetrator) causes the induction and a second drug’s (victim) pharmacokinetics are altered, there exists a situation where a single drug acts as both inducer and affected drug, called autoinduction. Carbamazepine and artemisinin are examples of CYP3A4 and CYP2B6 substrates and inducers, respectively, that are known to exhibit autoinduction (5,6). Autoinduction is a phenomenon that can occur anytime a drug induces an enzyme which is also predominately involved in its own metabolic clearance. Therefore, when understanding the overall liability of an enzyme inducer, a complete understanding of the major pathways and enzymes involved in elimination are important to assess autoinduction.
Drug-drug interactions (DDIs) are occurring with increasing frequency due to the rise in multiple prescriptions taken by individual patients. The average older adult uses between two and six prescription medications, and between one and three over-the-counter medications regularly (7). Due to the increase in DDIs, the need to develop compounds lacking the ability to induce metabolizing enzymes has become a significant concern and exercise in developing new drugs. Most pharmaceutical companies have implemented several layers of screening and characterization to assess enzyme induction. In early drug discovery, large numbers of compounds are screened by means of high throughput assays to eliminate compounds with enzyme induction potential or reduce the liability by selecting compounds with lower potencies for the inductive effect. During preclinical development, characterization in more complex human systems helps to further define the induction potential of a compound. Ultimately, the effect or lack of effect of a potential enzyme inducer is evaluated in human volunteers/patients with well characterized CYP probe substrates (Table I) where the actual magnitude of pharmacokinetic change is measured. Therefore, the aim of this review is to describe the assays most commonly employed to assess enzyme induction in the pharmaceutical industry. The focus will be on CYP enzymes induced in the liver, as this is the main emphasis for pharmaceutical companies and encompasses the most significant drug-drug interactions, in particular, CYP3A4 interactions.
NUCLEAR HORMONE RECEPTORS—TRANSCRIPTION FACTORS
There are two general mechanisms by which enzyme induction occurs: (1) stabilization of the mRNA or enzyme (e.g., CYP2E1) (8) and (2) increased gene transcription. The most common mechanism of CYP enzyme induction is transcriptional gene activation. For drug metabolizing enzymes, transcriptional activation is mediated by nuclear receptors that function as transcription factors, such as AhR (aromatic hydrocarbon receptor), CAR (constitutive androstane receptor), and PXR (pregnane X receptor or SXR-steroid X receptor) (9–14). The general concept for nuclear receptor signaling is that in the absence of a ligand (drug), the nuclear receptor is associated with co-repressor complexes, conferring a basal level of transcription. Ligand (drug) binding to the ligand binding domain (LBD) of the nuclear receptors induces conformational changes that lead to the release of co-repressors and recruitment of co-activators. Recruitment of co-activators and a dimerization partner (retinoid X receptor, RXR, for CAR/PXR and the AhR nuclear translocator, ARNT, for AhR) lead to chromatin remodeling and subsequent transcriptional activation. Regulation of gene transcription is achieved through binding of the nuclear receptor DNA binding domain (DBD) to respective DNA response elements present in the promoter region of target genes (drug metabolizing enzymes) (14,15). Figure 1 illustrates the components of both the nuclear receptor and target gene (promoter and gene or reporter gene). Table II lists several nuclear receptors and their target genes (CYP, glucuronyl- and sulfo-transferases: UGT and ST, respectively). It should be noted that the LBDs of many nuclear receptors are different between various animal species and human (14). Therefore, in vitro or in vivo animal models (e.g., rat and dog) of enzyme induction can be misleading and are generally not employed to assess the potential induction effect in humans.
Fig. 1.

Illustration of domains of nuclear receptors (a) and structure of a gene construct with promoter region and target gene or transfection vector for a transactivation assay with promoter region and reporter gene (b)
Table II.
Nuclear Receptors and Their Target Genes
| Nuclear Receptor | Target Genes |
|---|---|
| AhR | CYP1A1, CYP1A2 CYP1B1, CYP2S1, UGT1A1, UGT1A6, SULT2B |
| CAR | CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP3A4, UGT1A1, UGT2B1, SULT2A1 |
| PXR | CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A4, CYP3A5, CYP3A7 UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, SULT2A1 |
PXR-derived nuclear hormone receptor models are the most common high throughput assays to evaluate enzyme induction due to the simplicity of ligand-based activation and importance of target genes, such as CYP3A4, CYP2B6, and CYP2Cs in drug interactions. Ligand binding assays generally consist of genetically expressed and isolated receptors incubated with test compound and a radiolabeled ligand. Competition of the radiolabeled ligand with test compound is measured and an IC50 determined (16). These assays are reasonably straightforward to conduct, however their simplicity can also be a deficit in some situations. Often, only a portion of the receptor is expressed (LBD), not the full length receptor, and there is no cell membrane to act as a potential barrier to drug access (or allow for drug accumulation). Cell-based PXR transactivation assays illustrate a more complex nuclear hormone receptor assay. The PXR transactivation model is comprised of two expression vectors: (1) full length human PXR and (2) a variation of the CYP3A4 5′-promoter region coupled to a reporter gene (17,18) both expressed in an appropriate cell line. Similar transactivation assays also exist for the Ah receptor (19). Here, increased luminescence is typically measured as an indication of enzyme induction potential and EC50 and Emax values are determined. In general, there is a good correlation between EC50 and IC50 values in PXR transactivation and binding assays, respectively (16). However, there are examples where significant binding to the PXR receptor does not lead to transactivation or enzyme induction. These unique situations can be caused by PXR antagonists or ligands which do not elicit the appropriate displacement of co-repressors or recruitment of co-activators [e.g., docetaxel and paclitaxel (20)]. Many companies have replaced the PXR binding assay with a PXR transactivation assay which has equal throughput, less false positives, no need for radiolabeled reagents, and improved correlation to human drug–drug interactions (18).
Interpretation of hPXR data can be as simple as rank ordering of EC50 values and comparison to known CYP3A4 inducers (rifampicin) providing useful information on potency. However, many hPXR transactivation plots yield partial response plots (partial agonists) which make EC50 determinations less useful and incorporation of Emax values along with EC50’s necessary. More accurate predictions of drug interactions incorporate therapeutic drug concentrations, such as therapeutic total drug concentrations (Cmax or Css) along with transactivation data. Combining values and obtaining a ratio of Cmax/EC50 can be predictive in assessing the likelihood of a drug interaction much like the approach used for drug inhibition assessments. More complex calculations include several parameters, such as EC50, Emax, and Cmax and have been used to assess induction effects for multiple induction models (transactivation, immortalized hepatocytes, and primary hepatocytes) (18,21,22).
Equation 1 (where Ceff is the efficacious drug concentration):
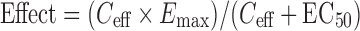 |
1 |
Another approach compares the percent transactivation at the therapeutic drug concentration (Cmax) to a known enzyme inducer. Using this approach, it has been demonstrated that compounds causing >40% transactivation at a therapeutic drug concentrations are predicted to “likely” have drug interactions, compounds with percent transactivation between 15–40% “may” elicit drug interactions, and those with percent transactivation <15% are “not anticipated” to cause drug interactions (18). Figure 2 illustrates how three different efficacious drug concentrations can affect the prediction of CYP3A4 induction based on PXR transactivation data. Concentrations A, B and C (low, moderate, and high effective concentrations) would be predicted to not elicit a PXR response, may elicit a response, and would be expected to elicit a response, respectively. In another example, hyperforin has an EC50 = 0.04 μM in the hPXR transactivation assay, an approximate efficacious plasma concentration of 0.3 μM, and a percent transactivation of 80% at the efficacious concentration. In contrast, pioglitazone has an EC50 = 31 μM in the hPXR transactivation assay, an efficacious plasma concentration of ∼3.8 μM, and a percent transactivation of 10% at the efficacious concentration. Combined, the parameters of these two drugs would predict that hyperforin would cause CYP3A4 drug interactions while pioglitazone should not and this is borne out in actual clinical drug interactions (18).
Fig. 2.

Example of how efficacious drug concentration can affect the predictability of induction potential from a plot of PXR transactivation vs. drug concentration. A, B, and C represent multiple efficacious concentrations for a hypothetical drug
Unfortunately, few examples exist in the literature of successful elimination of PXR-mediated CYP3A4 induction by use of the PXR transactivation assay (23,24). This may be due to the relatively new application of these assays in the pharmaceutical industry and the proprietary nature of the work. What has been learned from in silico modeling of PXR ligands indicate that disruption of key interactions (hydrogen bond acceptor and hydrophobic interactions) between ligand and PXR ligand binding domain are essential to attenuate or eliminate PXR-mediated induction (23).
The mechanisms of CAR and AhR-mediated gene transcription differs somewhat from that of PXR. Both CAR and AhR can be activated by direct ligand binding as described above, however they can also be activated by ligand binding independent mechanisms. Translocation of CAR to the nucleus can be initiated by direct agonist binding to the receptor or through a partially elucidated ligand-independent mechanism involving kinases which dephosphorylate CAR. Phenobarbital is an example of a drug that does not bind to CAR yet causes nuclear translocation and transcriptional activation of the target gene, CYP2B6 (12). Similarly, the inactive AhR resides in the cytoplasm and can be activated upon ligand binding or via protein tyrosine kinases. Omeprazole mediated induction of CYP1A has been shown to proceed through this kinase-mediated pathway (25) although this premise has come under debate (26).
One disadvantage of nuclear hormone receptor based assays is that each assay only evaluates one receptor-mediated process at a time. For example, hPXR transactivation assays will predict induction of PXR target genes; however activation of the same target genes can occur through other nuclear hormone receptors, such as CAR which would not be represented in a PXR assay. This nuclear receptor cross-talk with target genes are best evaluated in more complex cell based models (hepatocytes) where multiple nuclear hormone receptors can work in concert and provide a cumulative effect on target genes (14).
IMMORTALIZED HEPATOCYTES
Immortalized cells have the ability to grow and divide indefinitely and can occur naturally, from tumoral origin, or through oncogenic immortalization of primary cells. The advantages of using immortalized cell lines over transfected cell systems or primary hepatocytes for enzyme induction studies include: (1) potential to simultaneously capture multiple nuclear hormone receptor-mediated pathways; (2) easy access and availability; 3) the ability to propagate (i.e., continual supply); and (4) a more consistent response to inducers. Although immortalized cell lines are used extensively to study various mechanisms of enzyme induction or as the basis for specific nuclear receptor assays (e.g., hPXR transactivation), those will not be described here. The following discussion will describe cell lines that have an innate ability to respond directly to enzyme inducers and produce an induction response, as measured by mRNA expression or enzyme activity.
The most utilized cell system in the pharmaceutical industry is the non-tumorigenic immortalized hepatic cell line Fa2N-4. Fa2N-4 cells demonstrate inducible CYP1A1/2, CYP3A4, CYP2C9, UGT1A and MDR1 mRNA expression and increases in enzyme activity (CYP1A2, CYP2C9 and CYP3A4) when treated with prototypical inducers. The responses to rifampicin and beta-naphthoflavone were shown to be within the range observed for primary human hepatocytes (21,27). This cell line appears promising for discovery-stage induction studies keeping in mind some limitations. For example, the native or basal enzyme activities are low and therefore it is difficult to measure enzyme activities in vehicle treated samples without sensitive analytical methods. Due to this, mRNA is a more routine endpoint when using this cell line. In a recent publication, 24 compounds that have been previously evaluated for induction in primary human hepatocytes were assessed for induction in Fa2N-4 cells using mRNA analysis as the marker for induction (21). The data indicated a majority of the compounds showed comparable induction between primary hepatocytes and Fa2N-4 cells as assessed by using Eq. 1 and developing a Relative Induction Score for each compound. One exception was troglitazone which is a very potent inducer both in the clinic and in primary hepatocytes (28), yet gave a very moderate induction response with the Fa2N-4 cell line, indicating that perhaps all the nuclear receptor pathways are not present or fully functional in Fa2N-4 cells. Indeed, Lyon, et. al., have shown that the Fa2N-4 cells have very low levels of CAR and do not respond to the CAR-specific agonist CITCO (29).
Other cell lines of note, albeit not employed extensively in the pharmaceutical industry, are HepG2, HepaRG, and BC2. These human hepatoma cell lines have been used for induction studies, as some of the CYP enzymes are inducible, even though the expression levels of Phase I and Phase II drug metabolism enzymes are significantly lower than those in human primary hepatocytes. HepG2 and HepaRG cells have been shown to respond, to varying degrees, to CYP1A1/2 and 3A4 inducers (30–32) and BC2 cells have been reported to respond to CYP1A inducers (33). While these cell lines can be used for preliminary studies, they do not maintain all of the phenotypic characteristics of human hepatocytes, such as enzyme or receptor function or expression, and their use may result in erroneous conclusions. Nonetheless, they appear to be gaining more attention as an earlier alternative to primary hepatocyte experiments for assessing CYP1A and 3A induction potential in drug discovery.
PRIMARY HUMAN HEPATOCYTES
Cultured primary human hepatocytes are the most accepted (industry, academia, regulatory) in vitro system for assessing the potential for drug candidates to induce human CYP expression (34). Human hepatocytes are a cellular system comprised of human receptors, co-activators and repressors, target genes and promoters, as well as human drug metabolizing enzymes capable of biotransforming drugs. These properties are analogous to the liver and are essential to effectively model the inducibility of drug candidates and their metabolites. Primary human hepatocyte culture systems have been shown to effectively model human in vivo induction responses and are recognized by the FDA as an effective tool for assessing induction potential (35,36). The enzyme induction data from in vitro methods are known to correlate well with clinical observations, provided the in vitro experiments are performed at pharmacologically relevant concentrations of drug (37). When hepatocytes are cultured using appropriate conditions which facilitate liver-like cell morphology and expression of liver-specific proteins, CYP enzymes are effectively induced in vitro analogous to the in vivo situation in terms of the magnitude and specificity of induction (37,38). For example, omeprazole is an effective in vitro inducer of human CYP1A2 (37,39), but the EC50 of omeprazole induction is much larger than the typical exposure in humans treated for gastric ulcers, thus it rarely causes significant clinical drug interactions because the concentration of omeprazole required to induce CYP1A2 exceeds the concentrations achieved in vivo (37,40,41). In addition to CYP enzymes, numerous studies have been reported using primary hepatocyte culture systems to assess induction of a variety of gene targets from Phase II enzymes and transporters (42–45).
The FDA draft guidance on drug interactions states “A drug that induces a drug-metabolizing enzyme can increase the rate of metabolic clearance of a co-administered drug that is a substrate of the induced pathway. A potential consequence of this type of drug-drug interaction is sub-therapeutic blood concentrations” (36). The draft guidance recommends analysis of catalytic activity of the major drug metabolizing enzymes CYP1A2 and CYP3A4 in freshly isolated or attachable cryopreserved hepatocyte cultures. Since co-induction of CYP2C, CYP2B and P-gp occurs with CYP3A, negative results in vitro with CYP3A may also eliminate the need to address interactions by these CYPs and P-gp. Samples should be analyzed from at least three individual donors, treated with the test compound, vehicle and positive controls (known prototypical inducers) for 2–3 days. Each hepatocyte donor preparation is deemed acceptable if the recommended positive control elicits a greater than 2-fold increase in enzyme activity of the probe substrate. A minimum of three test compound concentrations, based on the expected human plasma drug levels are suggested, one of which should be an order of magnitude greater than this concentration. In the absence of knowledge of human plasma levels, concentrations ranging over at least two orders of magnitude should be studied. The FDA draft guidance allows for enzymatic analysis to be conducted in microsomes prepared from hepatocytes or in situ, using media-based assays and CYP-specific probe substrates, where the metabolite formed can be attributed to one major CYP enzyme.
The data from cultured hepatocyte experiments can take several forms, such as, fold increase, percent increase or EC50 values. More recently, percent increase and EC50 values have been found to be more predictive than fold increase. For example, a drug that produces an increase in probe drug enzyme activity that is equal to or more than 40% of the enzyme activity of positive control inducer will be regarded as an enzyme inducer. EC50 values are often used to rank order test compounds based on inducer potency or used in calculations such as Eq. 1. The interpretation and prediction of in vivo induction (DDI) from human hepatocytes are very similar to those employed in nuclear receptor transactivation assays. When results from in vitro studies demonstrate significant induction by test compound vs. induction with the positive control, the potential for in vivo induction should be assessed in clinical DDI studies.
Additional methods to determine enzyme induction currently being evaluated are quantitation of CYP protein by Western immunoblotting and reverse transcriptase-polymerase chain reaction (RT-PCR) to measure mRNA levels (RNA expression is particularly useful when both enzyme induction and inhibition are occurring). However, measurement of enzyme activities from incubations in primary human hepatocytes is still considered the most reliable method to determine induction potential of a compound.
While fresh human hepatocytes are the standard for evaluating in vitro induction of CYP enzymes, attachable cryopreserved hepatocytes can also be used and data using these are accepted by the FDA. The drug metabolizing enzymes remain inducible after cryopreservation, and due to the significant variation in activities of drug metabolizing enzymes between individual human livers, certain lots of cryopreserved cells can generate results essentially indistinguishable from freshly isolated cells (35,46). mRNA, protein expression and activities of CYP1A2, 2B6, 2C9, 2E1 and 3A4 in cryopreserved hepatocytes are inducible by prototypical inducers (47), as is the activity of various UDP-glucuronosyl-transferases, carboxylesterases, and sulfotransferases (48,49). The advantage cryopreserved cells offer over fresh isolates is that experiments can be planned ahead and are not dependent on the sporadic availability of fresh primary hepatocytes. Unfortunately, the commercial supply of attachable cryopreserved human hepatocyte preparations is somewhat limited, as a majority of cryopreserved hepatocyte preparations lose the ability to attach to extracellular matrices in culture.
CLINICAL DRUG INTERACTION STUDIES
Clinical DDI studies are performed to measure the changes in pharmacokinetic parameters of a test compound by concomitant drug administration. The specific objective of DDI studies is to provide pharmacokinetic recommendations for dose or interval adjustments of the drug itself, or the drugs with which it may be prescribed, and to determine if additional therapeutic monitoring is required (36). DDI studies are usually required in Phase II or III, however they can be planned at any time during drug development if results from in vitro data suggest there is a potential for DDIs. The general design of an in vivo DDI study is to compare the concentrations of test drug with and without a comedicant (36). A cross-over design, in healthy volunteers (both male and female), is usually appropriate in order to reduce variability. In general, the same subjects are dosed initially with a probe substrate (assess baseline pharmacokinetics), followed by ∼2 weeks of dosing with the drug candidate, and finally a dose of the probe substrate. The study design should be such that both drug and comedicant are dosed to exposures that are clinically relevant; the route of administration is the same as planned for clinical use, and given at the highest clinical dose possible using the shortest dosing interval. This should maximize the possibility of a DDI being uncovered. When appropriate, exclusion of dietary supplements including herbal products, over the counter medications, alcohol, fruit juices or other foods (including cruciferous vegetables and charbroiled meats) that may affect metabolizing enzymes and transporters should be implemented to avoid variation in study results. In these experiments, complete plasma concentration time profiles of the probe drug are sampled. Often plasma concentrations of drug candidate (inducer) are also measured to ensure appropriate exposure to inducing agent. Endpoints measured include all relevant pharmacokinetic parameters of parent drug and relevant metabolites. Primary endpoints measured are exposure measures such as AUC and Cmax, as well as other pharmacokinetic parameters (clearance, volume of distribution, tmax, and half-life). No-effect-boundaries (where changes in measurement of systemic exposure have no clinical meaning) should be defined based on clinical relevance. Data evaluation in these studies should include noncompartmental pharmacokinetic analysis, descriptive statistics of the pharmacokinetic parameters, calculation of inter- and intra-individual variability, and calculation of the 90% confidence intervals for AUC and Cmax ratios between treatments. A significant outcome of these studies would be alteration of the pharmacokinetics by concomitant drug administration and the subsequent dosing recommendation and labeling of contraindications.
If the drug is being studied as a potential inducer of CYP enzymes, the choice of probe or substrate depends on which CYP enzymes are affected by the inducer. Common probe substrates include midazolam, theophylline, repaglinide, warfarin, and omeprazole for CYP3A4, 1A2, 2C8, 2C9 and 2C19, respectively. If initial studies with these substrates show potential for DDI then additional studies with other probes may be performed if there is a possibility of co-administration. Table III contains a list of substrates/probes used and known inducers for each CYP. For example, the most sensitive probe for CYP3A4 activity is oral midazolam, a commonly used hypnotic since the 1980s (50). Most of the elimination of this drug is through metabolism by CYP3A4/5 (51,52). Lack of CYP3A4 induction with co-administration of midazolam following multiple doses of investigational drug indicates that the drug is not a CYP3A4 inducer.
Table III.
Commonly Used In Vivo Probes for Clinical Studies and CYP Inducers
| CYP Enzyme | Substrate/Probe | Inducer |
|---|---|---|
| 1A2 | Theophylline, caffeine, acetaminophen, aromatic amines, phenacetin | Cigarette smoke, omeprazole charbroiled meats, cruciferous vegetables |
| 2A6 | Coumarin, butadiene, nicotine | Barbiturates |
| 2B6 | Efavirenz | Rifampin |
| 2C8 | Repaglinide, rosiglitazone | Rifampin |
| 2C9 | S-warfarin, tolbutamide, phenytoin, NSAIDs | Rifampin, barbiturates, |
| 2C19 | Omeprazole, esoprazole, lansoprazole, pantoprazole, citalopram, diazepam, hexobarbital, imipramine, proguanil, propanolol | Rifampin, barbiturates |
| 2D6 | Antidepressants, neuroleptics, beta-blockers, antiarrhytmics, codeine, etylmorphine, desipramine, dextromethorphan, atomoxetine, nicoteine | None identified |
| 2E1 | Chlorzoxazone, acetaminophen, alcohols, caffeine, dapsone, enflurane, theophylline | Ethanol, isoniazid |
| 3A4/3A5 | Midazolam, buspirone, felodipine, lovastatin, eletriptan, sildenafil, simavastatin, triazolam, acetaminophen, carbamezapine, cyclosporin, digitoxin, diazepam, erythromycin, fluoxetine, nifedipine, quinidine, saquinavir, steroids (e.g. cortisol), terfenadine, verapamil, warfarin | Rifampin, carbamazepine, dexamethasone, phenytoin, troleandomycin |
Administering a cocktail of different substrates of CYP enzymes is also conducted in clinical investigations of enzyme induction albeit more common in academic than industrial settings. Choice of substrates is important as they need to be CYP specific with no interactions between any of the substrates in the cocktail. A recent study by Asimus et al. (53), investigated the effect of the antimalarial drug artemisinin and its derivatives on the major human CYP enzymes. Asimus et al. designed a six drug cocktail study consisting of caffeine, coumarin, mephenytoin, metoprolol, chlorzoxazone and midazolam to assess CYP1A2, 2A6, 2C19, 2D6, 2E1 and 3A4 activity, respectively. The investigators found that artemisinin and/or one or more of its derivatives caused induction of CYP3A4, 2C19 and CYP1A2 using this cocktail approach. Hence, such a DDI will need to be considered when selecting appropriate therapies for malaria and combination treatments.
FUTURE MODELS
There are multiple in silico, in vitro and in vivo models of induction being developed with varying degrees of success. All appear promising; however additional evaluation and a greater understanding of how best to employ these models are still necessary.
In Silico
In silico simulations and structure-activity relationships (SAR) are useful tools to help eliminate potentially problematic chemotypes early in drug development. Successful induction models combine the crystal structure of nuclear receptor proteins with in silico structural biology (54). For example, the PXR crystal structure and protein indicate that the LBD is hydrophobic with several key polar residues and this information has been used to identify potential ligands (55,56). Evaluation of AhR ligand structures indicates that substrates are planar lipophilic molecules about 6.8 × 13.7 Å in dimension (57). Ligands for hCAR identified by pharmacophore modeling were relatively planar structures with one hydrogen bond acceptor and three hydrophobic features (58). Potent ligands for PXR are similar to CAR ligands, in that at least one hydrogen bond acceptor feature and one or more hydrophobic interactions are necessary for good binding and activation (23).
Merely docking the compound in the protein structure may not always suffice, as the large ligand-binding domain of hPXR accommodates ligands of all sizes. Ligands of all shapes and sizes have been shown to alter the protein structure after binding indicative of an ‘induced-fit’ mechanism which ultimately is difficult to model (54). Complicating factors in ligand binding pharmacophore models are adjacent sites on the nuclear receptor which can be involved in conformational changes that affect binding. For example, Ekins and Wang have reported that agonists and antagonists bind to distinct regions on PXR and that the effects of ketoconazole were mediated through binding to the AF-2 region of PXR and not the LBD (Fig. 2) (59,60).
Stem Cells
Human stem cells (hSC) could theoretically provide an unlimited source of hepatocytes once culture conditions are optimized to enable differentiation. These stem cells can differentiate into hepatocyte-like cells, albeit often with limited drug metabolizing capability. Due to the random nature in obtaining primary human hepatocytes and the variable results from different donors, multiple laboratories are currently working on developing a hepatocyte-like cell line from hSC. Some examples of cells derived from hSC are HLC cells which exhibit hepatocyte morphology and limited CYP metabolism (61). Two hepatocyte-like cell lines (SA002 and SA167) derived from hSC demonstrate inducible CYP1A1 and CYP3A4/7 mRNA and immunoreactive protein, as well as CYP2C8/9/19 mRNA but no catalytic activity (62). The NeoHep cells developed from terminally differentiated peripheral blood monocytes, exhibit hepatocyte-like morphology, expression of hepatocyte markers, limited drug metabolism (CYP1A and UGT) and CYP3A induction (63). Phenobarbital-inducible CYP mRNA expression and enzymatic activity (64) and inducible CYP1A activity (65) have been reported in other hSC-derived cell lines. While these studies provide hope for hSC-derived hepatocytes being developed for metabolism studies in the future, thus far, no stem-cell derived cell lines (differentiated hepatocytes) are employed in enzyme induction screening or characterization.
Transgenic/Chimeric Animals
Humanized nuclear receptor transgenic mice, knock out mice and chimeric mice with human transplanted hepatocytes, are useful tools for assessing enzyme regulation by drug candidates. These in vivo models provide an advantage over previous in vitro models, as the systems are dynamic, with drug absorption, distribution, metabolism, and elimination occurring concurrently. Drug candidates can be administered over several days at doses/exposures equivalent to those expected in human plasma to enable improved assessments of human induction potential. For example, dose-dependent increases in CYP3A11 mRNA were observed with increasing concentrations of rifampicin in hPXR transgenic mice (66). The limitation of the transgenic/knock out systems are that only one or two genes are “humanized” and the cross-talk between nuclear receptors/enzymes/transporters may be compromised as the human and mouse systems may or may not be working in a similar fashion. Humanized chimeric mice were developed by injecting human hepatocytes into SCID mice (80% of the hepatocytes in the liver were of human origin) (67). CYP1A1/2 and CYP3A4 mRNA, protein content, and enzyme activity have been shown to be induced in chimeric mice treated with known human inducers and in hepatocytes isolated from humanized chimeric mice (68–70).
In Vivo Monkey
Rhesus and cynomolgus monkeys are used routinely for preclinical pharmacological, pharmacokinetic and safety evaluations and more recently for evaluations of in vivo drug interactions. The recent cloning of rhesus monkey CYP3A64 revealed 93% homology in the protein sequence with human CYP3A4 and 83% homology to CYP3A5 (71). More importantly, the nucleotide sequences of ligand binding domains between rhesus and human PXR are 96% similar (11). In vitro studies using monkey hepatocytes showed that rifampicin induced CYP3A64 mRNA with an EC50 = 0.5 μM (72), which is similar (EC50 = 0.2 μM) to that reported for human hepatocytes (28). In rhesus monkeys, the pharmacokinetics of midazolam can be significantly altered with rifampicin co-administration, resulting in reduced systemic exposure and hepatic bioavailability (72), similar to human. The ability to employ an in vivo animal model that replicates human pharmacokinetic changes would be extremely useful in placing in vitro results in the proper context and the similarities between human and monkey suggest that monkeys have the potential to be a predictive tool for human CYP3A-mediated drug interactions.
CHALLENGES AND SUMMARY
One of the biggest challenges in predicting human DDIs from in vitro experiments is the lack of understanding between in vitro drug concentrations and drug concentration at the site of action in the liver. This is complicated by several factors, such as, protein binding or non-specific binding in vitro and in vivo, the use of total vs. free (unbound) efficacious plasma concentrations, or the use of systemic vs. portal vein drug concentrations, and compounds with low or high liver-to-plasma ratios. The two most common approaches in all human in vitro models are: (1) total efficacious systemic Cmax or Css concentration or (2) unbound drug concentration at the portal vein (18,21,22). The former approach does not account for protein binding effects on drug disposition, yet is considered a conservative estimate that uses a high (total) drug concentration after the first-pass effect (metabolism/elimination). The latter approach does account for protein binding on drug disposition and reflects the maximum drug absorbed and presented to the liver.
Extrapolations from in vitro induction studies are challenging when the compound being evaluated is also an inhibitor of CYP enzymes. If the compound is an inhibitor, enzyme induction can be missed when only activity is assessed. For example, CI-1034 (endothelin-A antagonist) is an inducer and metabolism-based inhibitor of CYP3A4 and activity assays revealed no change in enzyme activity (73). Due to the common nature of CYP inhibition, conducting both activity and mRNA analysis are encouraged. Also, when utilizing data on only CYP1A1/2 and CYP3A4 induction, it is possible to under-predict clinical induction. This can also be due to the cross talk between the nuclear receptors e.g. PXR and CAR (74). When evaluating only CYP1A and CYP3A and not CYP2B6, induction via CAR could be misrepresented. Finally, cytotoxicity assessments should be conducted with cell-based systems to ensure that a negative response is not due to cell death. Often this can be as simple as visualizing the cells under a microscope and observing cell morphology up to and including more sophisticated measures of cell membrane permeability or cell death.
Figure 3 illustrates the possible models that can be employed while assessing the CYP induction potential of new drug candidates. From HTS models, which mimic the early steps of the enzyme induction process and have utility in SAR, mechanistic, and prediction, to intermediate assays which incorporate larger numbers of CYP enzymes and their respective induction pathways. Moving eventually to primary human hepatocytes and ultimately to clinical DDI studies where actual effects on pharmacokinetic parameters of co-administered drugs (or inducing agent itself/autoinduction) are measured and subsequent dosing guidelines are adopted. While all of the assays provide useful SAR, mechanistic or confirmatory information pertaining to potential enzyme induction, the most useful and accepted model remains primary human hepatocytes.
Fig. 3.

A representation of in vitro and in vivo induction models in order of decreasing throughput and increasing reliability/predictability (open squares: current models; open circles: future models). Assays such as the transactivation and immortalized hepatocyte assays are extremely high throughput, however they lack the complexity of human hepatocytes which represent the multitude of transcription factors/receptors and their interactions and dynamic drug exposure which occurs in patients
Acknowledgements
The authors wish to thank Drs S. Ferguson, K. Santone, J. Silva, and D. Tweedie for their useful contributions and comments regarding this manuscript.
Abbreviations
- AhR
aromatic hydrocarbon receptor
- AUC
area under the plasma concentration-time curve
- CAR
constitutive androstane receptor
- CITCO
(6-(4-chlorophenyl)imidazo[2,1-β][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorbenzyl)oxime)
- Cmax
maximum plasma concentration
- Css
drug concentration at steady state
- CYP
cytochrome P450
- EC50
concentration of an agonist that produces 50% of the maximal response
- DBD
DNA binding domain
- DDI
drug-drug interaction
- Emax
maximal response (effect)
- hSC
human stem cells
- IC50
concentration of drug that produces 50% inhibition
- LDB
ligand binding domain
- PXR
pregnane X receptor
- Pgp
P-glycoprotein
- STs
sulfo-transferases
- SXR
steroid and xenobiotic receptor
- UGTs
glucuronosyl-transferases
References
- 1.Conney A. H. Pharmacological implications of microsomal enzyme induction. Pharmacol. Rev. 1967;19(3):317–366. [PubMed] [Google Scholar]
- 2.Brown R. R., Miller J. A., Miller E. C. The metabolism of methylated aminoazo dyes. IV. Dietary factors enhancing demethylation in vitro. J. Biol. Chem. 1954;209(1):211–222. [PubMed] [Google Scholar]
- 3.Park J. Y., Kim K. A., Kang M. H., Kim S. L., Shin J. G. Effect of rifampin on the pharmacokinetics of rosiglitazone in healthy subjects. Clin. Pharmacol. Ther. 2004;75(3):157–162. doi: 10.1016/j.clpt.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Mueller S. C., Majcher-Peszynska J., Uehleke B., et al. The extent of induction of CYP3A by St. John’s wort varies among products and is linked to hyperforin dose. Eur. J. Clin. Pharmacol. 2006;62(1):29–36. doi: 10.1007/s00228-005-0061-3. [DOI] [PubMed] [Google Scholar]
- 5.Bertilsson L., Hojer B., Tybring G., Osterloh J., Rane A. Autoinduction of carbamazepine metabolism in children examined by a stable isotope technique. Clin. Pharmacol. Ther. 1980;27(1):83–88. doi: 10.1038/clpt.1980.13. [DOI] [PubMed] [Google Scholar]
- 6.Simonsson U. S., Jansson B., Hai T. N., Huong D. X., Tybring G., Ashton M. Artemisinin autoinduction is caused by involvement of cytochrome P450 2B6 but not 2C9. Clin. Pharmacol. Ther. 2003;74(1):32–43. doi: 10.1016/S0009-9236(03)00092-4. [DOI] [PubMed] [Google Scholar]
- 7.Multiple Prescriptions. Medical Edge Mayo Clinic; 2005.
- 8.Gonzalez F. J. The 2006 Bernard B. Brodie award lecture. Cyp2e1. Drug Metab. Dispos. 2007;35(1):1–8. doi: 10.1124/dmd.106.012492. [DOI] [PubMed] [Google Scholar]
- 9.Honkakoski P., Sueyoshi T., Negishi M. Drug-activated nuclear receptors CAR and PXR. Ann. Med. 2003;35(3):172–182. doi: 10.1080/07853890310008224. [DOI] [PubMed] [Google Scholar]
- 10.Mandal P. K. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J. Comp. Physiol. [B] 2005;175(4):221–230. doi: 10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- 11.Moore L. B., Maglich J. M., McKee D. D., et al. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol. Endocrinol. 2002;16(5):977–986. doi: 10.1210/me.16.5.977. [DOI] [PubMed] [Google Scholar]
- 12.Qatanani M., Moore D. D. CAR, the continuously advancing receptor, in drug metabolism and disease. Curr. Drug Metab. 2005;6(4):329–339. doi: 10.2174/1389200054633899. [DOI] [PubMed] [Google Scholar]
- 13.Tirona R. G., Kim R. B. Nuclear receptors and drug disposition gene regulation. J. Pharm. Sci. 2005;94(6):1169–1186. doi: 10.1002/jps.20324. [DOI] [PubMed] [Google Scholar]
- 14.Wang H., LeCluyse E. L. Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin. Pharmacokinet. 2003;42(15):1331–1357. doi: 10.2165/00003088-200342150-00003. [DOI] [PubMed] [Google Scholar]
- 15.Lemaire G., Delescluse C., Pralavorio M., Ledirac N., Lesca P., Rahmani R. The role of protein tyrosine kinases in CYP1A1 induction by omeprazole and thiabendazole in rat hepatocytes. Life Sci. 2004;74(18):2265–2278. doi: 10.1016/j.lfs.2003.09.056. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Z., Kim S., Chen T., et al. Correlation of high-throughput pregnane X receptor (PXR) transactivation and binding assays. J. Biomol. Screen. 2004;9(6):533–540. doi: 10.1177/1087057104264902. [DOI] [PubMed] [Google Scholar]
- 17.Yueh M. F., Kawahara M., Raucy J. High volume bioassays to assess CYP3A4-mediated drug interactions: induction and inhibition in a single cell line. Drug Metab. Dispos. 2005;33(1):38–48. doi: 10.1124/dmd.104.001594. [DOI] [PubMed] [Google Scholar]
- 18.Sinz M., Kim S., Zhu Z., et al. Evaluation of 170 xenobiotics as transactivators of human pregnane X receptor (hPXR) and correlation to known CYP3A4 drug interactions. Curr. Drug Metab. 2006;7(4):375–388. doi: 10.2174/138920006776873535. [DOI] [PubMed] [Google Scholar]
- 19.Yueh M. F., Kawahara M., Raucy J. Cell-based high-throughput bioassays to assess induction and inhibition of CYP1A enzymes. Toxicol In Vitro. 2005;19(2):275–287. doi: 10.1016/j.tiv.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Harmsen S., Meijerman I., Beijnen J. H., Schellens J. H. The role of nuclear receptors in pharmacokinetic drug-drug interactions in oncology. Cancer Treat Rev. 2007;33(4):369–380. doi: 10.1016/j.ctrv.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Ripp S. L., Mills J. B., Fahmi O. A., et al. Use of immortalized human hepatocytes to predict the magnitude of clinical drug-drug interactions caused by CYP3A4 induction. Drug Metab. Dispos. 2006;34(10):1742–1748. doi: 10.1124/dmd.106.010132. [DOI] [PubMed] [Google Scholar]
- 22.Hewitt N. J., de Kanter R., LeCluyse E. Induction of drug metabolizing enzymes: a survey of in vitro methodologies and interpretations used in the pharmaceutical industry—do they comply with FDA recommendations? Chem. Biol. Interact. 2007;168(1):51–65. doi: 10.1016/j.cbi.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 23.Gao Y. D., Olson S. H., Balkovec J. M., et al. Attenuating pregnane X receptor (PXR) activation: a molecular modelling approach. Xenobiotica. 2007;37(2):124–138. doi: 10.1080/00498250601050412. [DOI] [PubMed] [Google Scholar]
- 24.Harper S., Avolio S., Pacini B., et al. Potent inhibitors of subgenomic hepatitis C virus RNA replication through optimization of indole-N-acetamide allosteric inhibitors of the viral NS5B polymerase. J. Med. Chem. 2005;48(14):4547–4557. doi: 10.1021/jm050056+. [DOI] [PubMed] [Google Scholar]
- 25.Backlund M., Ingelman-Sundberg M. Regulation of aryl hydrocarbon receptor signal transduction by protein tyrosine kinases. Cell Signal. 2005;17(1):39–48. doi: 10.1016/j.cellsig.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Hu W., Sorrentino C., Denison M. S., Kolaja K., Fielden M. R. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol. Pharmacol. 2007;71(6):1475–1486. doi: 10.1124/mol.106.032748. [DOI] [PubMed] [Google Scholar]
- 27.Mills J. B., Rose K. A., Sadagopan N., Sahi J., de Morais S. M. Induction of drug metabolism enzymes and MDR1 using a novel human hepatocyte cell line. J. Pharmacol. Exp. Ther. 2004;309(1):303–309. doi: 10.1124/jpet.103.061713. [DOI] [PubMed] [Google Scholar]
- 28.Sahi J., Hamilton G., Sinz M., et al. Effect of troglitazone on cytochrome P450 enzymes in primary cultures of human and rat hepatocytes. Xenobiotica. 2000;30(3):273–284. doi: 10.1080/004982500237668. [DOI] [PubMed] [Google Scholar]
- 29.Lyon K., Faris R., Liu J., et al. Improving lot-to-lot variability and dynamic range of CYP3A4 indution in immortalized human hepatocytes (Fa2N-4 cells) Maui, HI: Internation Society for the Study of Xenobiotics; 2005. [Google Scholar]
- 30.Aninat C., Piton A., Glaise D., et al. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab. Dispos. 2006;34(1):75–83. doi: 10.1124/dmd.105.006759. [DOI] [PubMed] [Google Scholar]
- 31.Guillouzo A., Corlu A., Aninat C., Glaise D., Morel F., Guguen-Guillouzo C. The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007;168(1):66–73. doi: 10.1016/j.cbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 32.W. M. Westerink and W. G. Schoonen. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol In Vitro. 21:1581–1591. [DOI] [PubMed]
- 33.Gomez-Lechon M. J., Donato T., Jover R., et al. Expression and induction of a large set of drug-metabolizing enzymes by the highly differentiated human hepatoma cell line BC2. Eur. J. Biochem. 2001;268(5):1448–1459. doi: 10.1046/j.1432-1327.2001.02011.x. [DOI] [PubMed] [Google Scholar]
- 34.LeCluyse E. L., Alexandre E., Hamilton G. A., et al. Isolation and culture of primary human hepatocytes. Methods Mol. Biol. 2005;290:207–229. doi: 10.1385/1-59259-838-2:207. [DOI] [PubMed] [Google Scholar]
- 35.Hewitt N. J., Lechon M. J., Houston J. B., et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab. Rev. 2007;39(1):159–234. doi: 10.1080/03602530601093489. [DOI] [PubMed] [Google Scholar]
- 36.FDA. Guidance for industry. Drug interaction studies—study design, data analysis, and implications for dosing and labeling. Draft Guidance. 2006. [DOI] [PubMed]
- 37.LeCluyse E., Madan A., Hamilton G., Carroll K., DeHaan R., Parkinson A. Expression and regulation of cytochrome P450 enzymes in primary cultures of human hepatocytes. J. Biochem. Mol. Toxicol. 2000;14(4):177–188. doi: 10.1002/(SICI)1099-0461(2000)14:4<177::AID-JBT1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 38.Runge D., Michalopoulos G. K., Strom S. C., Runge D. M. Recent advances in human hepatocyte culture systems. Biochem. Biophys. Res. Commun. 2000;274(1):1–3. doi: 10.1006/bbrc.2000.2912. [DOI] [PubMed] [Google Scholar]
- 39.Shih H., Pickwell G. V., Guenette D. K., Bilir B., Quattrochi L. C. Species differences in hepatocyte induction of CYP1A1 and CYP1A2 by omeprazole. Hum. Exp. Toxicol. 1999;18(2):95–105. doi: 10.1191/096032799678839699. [DOI] [PubMed] [Google Scholar]
- 40.Daujat M., Peryt B., Lesca P., Fourtanier G., Domergue J., Maurel P. Omeprazole, an inducer of human CYP1A1 and 1A2, is not a ligand for the Ah receptor. Biochem. Biophys. Res. Commun. 1992;188(2):820–825. doi: 10.1016/0006-291X(92)91130-I. [DOI] [PubMed] [Google Scholar]
- 41.Dilger K., Zheng Z., Klotz U. Lack of drug interaction between omeprazole, lansoprazole, pantoprazole and theophylline. Br. J. Clin. Pharmacol. 1999;48(3):438–444. doi: 10.1046/j.1365-2125.1999.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kodama S., Koike C., Negishi M., Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol. Cell. Biol. 2004;24(18):7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raucy J. L. Regulation of CYP3A4 expression in human hepatocytes by pharmaceuticals and natural products. Drug Metab. Dispos. 2003;31(5):533–539. doi: 10.1124/dmd.31.5.533. [DOI] [PubMed] [Google Scholar]
- 44.Raucy J. L., Lasker J., Ozaki K., Zoleta V. Regulation of CYP2E1 by ethanol and palmitic acid and CYP4A11 by clofibrate in primary cultures of human hepatocytes. Toxicol. Sci. 2004;79(2):233–241. doi: 10.1093/toxsci/kfh126. [DOI] [PubMed] [Google Scholar]
- 45.Raucy J. L., Mueller L., Duan K., Allen S. W., Strom S., Lasker J. M. Expression and induction of CYP2C P450 enzymes in primary cultures of human hepatocytes. J. Pharmacol. Exp. Ther. 2002;302(2):475–482. doi: 10.1124/jpet.102.033837. [DOI] [PubMed] [Google Scholar]
- 46.Schehrer L., Regan J. D., Westendorf J. UDS induction by an array of standard carcinogens in human and rodent hepatocytes: effect of cryopreservation. Toxicology. 2000;147(3):177–191. doi: 10.1016/S0300-483X(00)00190-6. [DOI] [PubMed] [Google Scholar]
- 47.Roymans D., Annaert P., Van Houdt J., et al. Expression and induction potential of cytochromes P450 in human cryopreserved hepatocytes. Drug Metab. Dispos. 2005;33(7):1004–1016. doi: 10.1124/dmd.104.003046. [DOI] [PubMed] [Google Scholar]
- 48.Reinach B., de Sousa G., Dostert P., Ings R., Gugenheim J., Rahmani R. Comparative effects of rifabutin and rifampicin on cytochromes P450 and UDP-glucuronosyl-transferases expression in fresh and cryopreserved human hepatocytes. Chem. Biol. Interact. 1999;121(1):37–48. doi: 10.1016/S0009-2797(99)00089-7. [DOI] [PubMed] [Google Scholar]
- 49.Nishimura M., Imai T., Morioka Y., Kuribayashi S., Kamataki T., Naito S. Effects of NO-1886 (Ibrolipim), a lipoprotein lipase-promoting agent, on gene induction of cytochrome P450s, carboxylesterases, and sulfotransferases in primary cultures of human hepatocytes. Drug Metab. Pharmacokinet. 2004;19(6):422–429. doi: 10.2133/dmpk.19.422. [DOI] [PubMed] [Google Scholar]
- 50.Chung E., Nafziger A. N., Kazierad D. J., Bertino J. S., Jr. Comparison of midazolam and simvastatin as cytochrome P450 3A probes. Clin. Pharmacol. Ther. 2006;79(4):350–361. doi: 10.1016/j.clpt.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 51.Gorski J. C., Hall S. D., Jones D. R., VandenBranden M., Wrighton S. A. Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem. Pharmacol. 1994;47(9):1643–1653. doi: 10.1016/0006-2952(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 52.Kronbach T., Mathys D., Umeno M., Gonzalez F. J., Meyer U. A. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol. Pharmacol. 1989;36(1):89–96. [PubMed] [Google Scholar]
- 53.Asimus S., Elsherbiny D., Hai T. N., et al. Artemisinin antimalarials moderately affect cytochrome P450 enzyme activity in healthy subjects. Fundam. Clin. Pharmacol. 2007;21(3):307–316. doi: 10.1111/j.1472-8206.2007.00471.x. [DOI] [PubMed] [Google Scholar]
- 54.Handschin C., Podvinec M., Meyer U. A. In silico approaches, and in vitro and in vivo experiments to predict induction of drug metabolism. Drug News Perspect. 2003;16(7):423–434. doi: 10.1358/dnp.2003.16.7.829354. [DOI] [PubMed] [Google Scholar]
- 55.Lemaire G., Benod C., Nahoum V., et al. Discovery of a highly active ligand of human pregnane X receptor: a case study from pharmacophore modeling and virtual screening to “in vivo” biological activity. Mol. Pharmacol. 2007;72(3):572–581. doi: 10.1124/mol.106.033415. [DOI] [PubMed] [Google Scholar]
- 56.Watkins R. E., Wisely G. B., Moore L. B., et al. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292(5525):2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 57.Crivori P., Poggesi I. Computational approaches for predicting CYP-related metabolism properties in the screening of new drugs. Eur. J. Med. Chem. 2006;41(7):795–808. doi: 10.1016/j.ejmech.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Xiao L., Cui X., Madison V., White R. E., Cheng K. C. Insights from a three-dimensional model into ligand binding to constitutive active receptor. Drug Metab. Dispos. 2002;30(9):951–956. doi: 10.1124/dmd.30.9.951. [DOI] [PubMed] [Google Scholar]
- 59.Ekins S., Chang C., Mani S., et al. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 2007;72(3):592–603. doi: 10.1124/mol.107.038398. [DOI] [PubMed] [Google Scholar]
- 60.Wang H., Huang H., Li H., et al. Activated pregnenolone X-receptor is a target for ketoconazole and its analogs. Clin. Cancer Res. 2007;13(8):2488–2495. doi: 10.1158/1078-0432.CCR-06-1592. [DOI] [PubMed] [Google Scholar]
- 61.Hay D. C., Zhao D., Ross A., Mandalam R., Lebkowski J., Cui W. Direct differentiation of human embryonic stem cells to hepatocyte-like cells exhibiting functional activities. Cloning Stem Cells. 2007;9(1):51–62. doi: 10.1089/clo.2006.0045. [DOI] [PubMed] [Google Scholar]
- 62.Ek M., Soderdahl T., Kuppers-Munther B., et al. Expression of drug metabolizing enzymes in hepatocyte-like cells derived from human embryonic stem cells. Biochem. Pharmacol. 2007;74(3):496–503. doi: 10.1016/j.bcp.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 63.Ruhnke M., Nussler A. K., Ungefroren H., et al. Human monocyte-derived neohepatocytes: a promising alternative to primary human hepatocytes for autologous cell therapy. Transplantation. 2005;79(9):1097–1103. doi: 10.1097/01.TP.0000157362.91322.82. [DOI] [PubMed] [Google Scholar]
- 64.Schwartz R. E., Linehan J. L., Painschab M. S., Hu W. S., Verfaillie C. M., Kaufman D. S. Defined conditions for development of functional hepatic cells from human embryonic stem cells. Stem Cells Dev. 2005;14(6):643–655. doi: 10.1089/scd.2005.14.643. [DOI] [PubMed] [Google Scholar]
- 65.Rambhatla L., Chiu C. P., Kundu P., Peng Y., Carpenter M. K. Generation of hepatocyte-like cells from human embryonic stem cells. Cell Transplant. 2003;12(1):1–11. doi: 10.3727/000000003783985179. [DOI] [PubMed] [Google Scholar]
- 66.Xie W., Barwick J. L., Downes M., et al. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406(6794):435–439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 67.Tateno C., Yoshizane Y., Saito N., et al. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am. J. Pathol. 2004;165(3):901–912. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katoh M., Matsui T., Nakajima M., et al. In vivo induction of human cytochrome P450 enzymes expressed in chimeric mice with humanized liver. Drug Metab. Dispos. 2005;33(6):754–763. doi: 10.1124/dmd.104.002600. [DOI] [PubMed] [Google Scholar]
- 69.Katoh M., Watanabe M., Tabata T., et al. In vivo induction of human cytochrome P450 3A4 by rifabutin in chimeric mice with humanized liver. Xenobiotica. 2005;35(9):863–875. doi: 10.1080/00498250500296231. [DOI] [PubMed] [Google Scholar]
- 70.Nishimura M., Yokoi T., Tateno C., et al. Induction of human CYP1A2 and CYP3A4 in primary culture of hepatocytes from chimeric mice with humanized liver. Drug Metab. Pharmacokinet. 2005;20(2):121–126. doi: 10.2133/dmpk.20.121. [DOI] [PubMed] [Google Scholar]
- 71.Carr B., Norcross R., Fang Y., et al. Characterization of the rhesus monkey CYP3A64 enzyme: species comparisons of CYP3A substrate specificity and kinetics using baculovirus-expressed recombinant enzymes. Drug Metab. Dispos. 2006;34(10):1703–1712. doi: 10.1124/dmd.106.009977. [DOI] [PubMed] [Google Scholar]
- 72.Prueksaritanont T., Kuo Y., Tang C., et al. In vitro and in vivo CYP3A64 induction and inhibition studies in rhesus monkeys: a preclinical approach for CYP3A-mediated drug interaction studies. Drug Metab. Dispos. 2006;34(9):1546–1555. doi: 10.1124/dmd.106.009878. [DOI] [PubMed] [Google Scholar]
- 73.Sahi J., Sinz M. W., Campbell S., et al. Metabolism and transporter-mediated drug-drug interactions of the endothelin-A receptor antagonist CI-1034. Chem. Biol. Interact. 2006;159(2):156–168. doi: 10.1016/j.cbi.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 74.Faucette S. R., Sueyoshi T., Smith C. M., Negishi M., Lecluyse E. L., Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J. Pharmacol. Exp. Ther. 2006;317(3):1200–1209. doi: 10.1124/jpet.105.098160. [DOI] [PubMed] [Google Scholar]
- 75.Handschin C., Meyer U. A. Induction of drug metabolism: the role of nuclear receptors. Pharmacol. Rev. 2003;55(4):649–673. doi: 10.1124/pr.55.4.2. [DOI] [PubMed] [Google Scholar]
- 76.Nakata K., Tanaka Y., Nakano T. Nuclear receptor-mediated transcriptional regulation in Phase I, II, and III xenobiotic metabolizing systems. Drug Metab Pharmacokinet. 2006;21(6):437–457. doi: 10.2133/dmpk.21.437. [DOI] [PubMed] [Google Scholar]
- 77.Puga A., Maier A., Medvedovic M. The transciptional signature of dioxin in human hepatoma HepG2 cells. Biochem. Pharmacol. 2000;60(8):1129–1142. doi: 10.1016/S0006-2952(00)00403-2. [DOI] [PubMed] [Google Scholar]
- 78.Rivera S. P., Saarikoski S. T., Hankinson O. Identification of a novel dioxin-inducible cytochrome P450. Mol. Pharmacol. 2002;61(2):255–259. doi: 10.1124/mol.61.2.255. [DOI] [PubMed] [Google Scholar]
- 79.Runge-Morris M., Kocarek T. Regulation of sulfotransferases by xenobiotic receptors. Curr Drug Metab. 2005;6(4):299–307. doi: 10.2174/1389200054633871. [DOI] [PubMed] [Google Scholar]
- 80.Zhou J., Zhang J., Xie W. Xenobiotic nuclear receptor-mediated regulation of UDP-glucuronosyl-transferases. Curr. Drug Metab. 2005;6(4):289–298. doi: 10.2174/1389200054633853. [DOI] [PubMed] [Google Scholar]
- 81.Tucker G. T., Houston J. B., Huang S. M. Optimizing drug development: strategies to assess drug metabolism/transporter interaction potential--toward a consensus. Pharm. Res. 2001;18(8):1071–1080. doi: 10.1023/A:1010994022294. [DOI] [PubMed] [Google Scholar]