

Abstract
The workshop “Bioequivalence, Biopharmaceutics Classification System, and Beyond” was held May 21–23, 2007 in North Bethesda, MD, USA. This workshop provided an opportunity for pharmaceutical scientists to discuss the FDA guidance on the Biopharmaceutics Classification System (BCS), bioequivalence of oral products, and related FDA initiatives such as the FDA Critical Path Initiative. The objective of this Summary Workshop Report is to document the main points from this workshop. Key highlights of the workshop were (a) the described granting of over a dozen BCS-based biowaivers by the FDA for Class I drugs whose formulations exhibit rapid dissolution, (b) continued scientific support for biowaivers for Class III compounds whose formulations exhibit very rapid dissolution, (c) scientific support for a number of permeability methodologies to assess BCS permeability class, (d) utilization of BCS in pharmaceutical research and development, and (e) scientific progress in in vitro dissolution methods to predict dosage form performance.
Keywords: bioavailability, bioequivalence, biopharmaceutics classification system (BCS), oral absorption, permeability, regulatory science, solubility
INTRODUCTION
The workshop “Bioequivalence, Biopharmaceutics Classification System, and Beyond” was held May 21–23, 2007 in North Bethesda, MD, USA. There were 225 workshop participants, including participants from 17 countries. Co-sponsored by the American Association of Pharmaceutical Scientists (AAPS) and the Food and Drug Administration (FDA), this workshop provided an opportunity for pharmaceutical scientists to discuss the FDA guidance on the Biopharmaceutics Classification System (BCS), bioequivalence of oral products, and related FDA initiatives such as the FDA Critical Path Initiative. The BCS guidance was introduced in 2000 (1). Shortly after this introduction, an AAPS/FDA co-sponsored workshop was held (2). The industry now has several years of experience in employing the BCS as a drug discovery, development, and regulatory tool. FDA has observed increased regulatory application of BCS. This workshop highlighted BCS applications to date as well as areas of potential BCS-based extensions for biowaivers. Additionally, bioequivalence issues for highly variable drugs, as well as the FDA Critical Path Initiative, were discussed.
The goals of the workshop were to:
Review and discuss the industrial and regulatory experience and perspective on using the BCS guidance for regulatory applications
Provide a forum to discuss best practices to classify drugs in the BCS
Identify scientific issues related to the extension of biowaivers using BCS, including biorelevant dissolution and applications around the world
Discuss current issues in bioequivalence of oral products, including highly variable drugs
Provide a forum to discuss FDA initiatives such as the Critical Path Initiative
The workshop took place over two and one-half days, and consisted largely of lectures with panel question-and-answer sessions. There also were two debates. Speakers and discussion leaders convened during the workshop to summarize main scientific points raised during the workshop. These summary points were presented during the final session of the workshop and form the basis for this report.
The objective of this Summary Workshop Report is to document these summaries and the main points from the workshop, in order to provide future direction in oral biopharmaceutics and bioequivalence, and related regulatory guidance. The workshop and this workshop report are presented as five topics: Regulatory Significance of BCS, Implementation of BCS and Biowaiver for Class III Drugs, Biorelevant Dissolution and BCS Future Development, Bioequivalence for Highly Variable Drugs, and FDA and International Initiatives.
Key highlights of the workshop were (a) the described granting of several BCS-based biowaivers by the FDA for Class I drugs whose formulations exhibit rapid dissolution (1), (b) continued scientific support for biowaivers for Class III compounds whose formulations exhibit very rapid dissolution (2), (c) scientific support for a number of permeability methodologies to assess BCS permeability class, (d) utilization of BCS in pharmaceutical research and development, and (e) scientific progress in in vitro dissolution methods to predict dosage form performance.
REGULATORY SIGNIFICANCE OF BCS
A preceding AAPS/FDA workshop was held in September 2002, shortly after the introduction of the BCS guidance. The workshop was entitled “Biopharmaceutics Classification System—Implementation Challenges and Extension Opportunities”(2). At that time, there was consensus that “The regulatory impact of the guidance has not been substantial, in part since the guidance was issued less than two years before the workshop. Additionally, with the need among sponsors for certainty of regulatory outcome, the BCS approach at this time was viewed as less familiar and thus less desirable, relative to in vivo bioequivalence studies.”
FDA Activity
In contrast to the September 2002 workshop, there was clear evidence of the regulatory impact of BCS, specifically for BCS-based biowaivers for Class I compounds whose formulations exhibit rapid dissolution. Biowaiver in this workshop report means the utilization of a BCS-based approach to document bioequivalence (e.g. in vitro studies), rather than an in vivo study to document bioequivalence. The FDA’s Critical Path Initiative has been progressing. In order to improve medical product development, two priorities identified in the FDA’s Critical Path Initiative are biomarker development and streamlining clinical trials (3). The comparison of test and reference drug plasma profiles to demonstrate bioequivalence is the most commonly used and successful biomarker. The BCS is an example of a science-driven approach to efficiently demonstrate bioequivalence. Based on a mechanistic understanding of drug absorption, critical drug substance properties and drug product performance requirements that provide a high level of assurance of bioequivalence were identified. When these conditions are met, bioequivalence can be assured without in vivo studies. There was also discussion that in vitro studies are sometimes better than conventional human BE studies in assessing equivalence (4).
A key highlight of the workshop was the described granting of over a dozen BCS-based biowaivers by the FDA for Class I drugs whose formulations exhibit rapid dissolution. A representative of the CDER BCS Committee presented activities of that committee. The CDER BCS Committee was formed in March 2004, previously functioning as the BCS Technical Committee. Objectives of the CDER BCS Committee are (1) to provide expert advice on all BCS review (NDA and ANDA) issues especially those where Class I claim is requested, (2) to serve as the point of contact for BCS related policy, questions, interactions and clarifications within FDA and with external constituents, and (3) to evaluate periodically if there is a need to consider updating the BCS guidance, based on internal and public information. Regarding assessments of Class I claims, committee outcomes are yes, no, or insufficient information.
The CDER BCS Committee met six times each year in 2004, 2005, and 2006. Twenty five drug products were evaluated. Sixteen were classified as BCS Class I. Of the 25 drug products evaluated, 11 were new chemical entities, with seven of those 11 receiving Class I designation. Four of these 11 evaluations were at the IND stage. Two received Class I designation and agreement on biowaivers; one received high solubility and high permeability designation, but dissolution was not rapid; one had insufficient information. Seven of these 11 new drugs evaluations were at the NDA review stage. Five received Class I designation and related regulatory treatment; one was turned down; one had insufficient information. Of the 25 drug products evaluated, the remaining 14 were generics, with nine receiving Class I designation. Numerous ANDAs have received regulatory relief.
Examples of regulatory relief include waiver of in vivo BE studies between clinical and to-be-marketed formulations, waiver of in vivo BE study for a new strength, waiver of in vivo BE study between different strengths of to-be-marketed formulations, and waiver of in vivo BE studies for a new (solution) dosage form NDA based on the BCS knowledge of earlier approved (tablet) NDA. The CDER BCS Committee has observed that proper integration of BCS information during drug development can save time and money. Additionally, the CDER BCS Committee has observed that updating of the BCS guidance, including expanding the biowaiver possibilities, should be undertaken using objective data and efficient process; one such source of data is the FDA NDA BCS Database.
While there have been an increasing number of successful BCS-based biowaiver applications, this progress has been attenuated by lack of international harmonization and implementation barriers within companies, including perception of project delay risk. Future progress in using in vitro biopharmaceutic data as a surrogate for in vivo bioequivalence data would be benefited by perceived greater certainty in regulatory decision-making (e.g. more scientific opportunities to discuss necessary data and successful examples).
BCS in Drug Development
BCS has had a significant impact in drug discovery and development, where there has been a growing recognition to design “drug-like” properties into new chemical entity programs (2). However, it was recognized that disease targets are increasingly behind hydrophobic “barriers”, such that molecular complexity is increasing to overcome these potency and “barrier” issues. It was also recognized that dose uncertainty during early development makes BCS application less precise.
Another more recent development is the consideration of a drug’s biopharmaceutics properties in the context of the review paradigms of Quality-by-Design and Question-based Review. Drug biopharmaceutics properties are being integrated into quantitative and predictive models of dosage form pharmacokinetic performance, guiding the selection of drug candidates, active pharmaceutical ingredient (API) processing and form selection, and dosage form technology. Ku describes the use of the BCS in early drug development (5), where biopharmaceutic characteristics are used for preliminary BCS classification of pipeline compounds. A decision strategy is described to facilitate early development, including a BCS-based animal formulation development decision tree. Compounds are triaged into one of five formulation strategies, with the goal of consistent pharmacokinetic performance and avoiding bridging BA/BE studies.
IMPLEMENTATION OF BCS AND BIOWAIVER FOR CLASS III DRUGS
In this workshop session, current BCS case studies, Class III biowaivers, and permeability methodologies were discussed.
Case Studies
Several case studies of BCS-based biowaivers for both new drugs and generics were presented. In a case study of pregabalin, bioequivalence needed to be studied near the time of submission. Three different formulation series comprised 11 different strengths. A strategy was devised to compare dissolution profiles of the highest and lowest strengths of each series. An in-house educational effort, along with interactions with FDA scientists, allayed in-house concerns about BCS as less familiar compared to the traditional in vivo BE approach. The subsequent BCS Class I biowaiver resulted in filing over one month earlier, with a savings of more than one million dollars compared to a more traditional approach that would have utilized four separate bioequivalence studies.
Cases studies from a generic pharmaceutical company were also presented. In one case, an ANDA for a higher strength was approved, based upon solubility studies and rat perfusion permeability studies. Literature data supported the drug to be BCS Class I. Two lower strengths had previously been submitted to FDA, based upon conventional in vivo BE data. The higher strength dosage form was demonstrated to be stable and rapidly dissolving. In a second case, an ANDA was submitted with a request for a biowaiver. Solubility and Caco-2 permeability indicated the drug to be BCS Class I, which was further supported by pharmacokinetic literature data. The test and reference products are rapidly dissolving. In a third case, an ANDA was submitted with a request for a biowaiver. Solubility and perfusion permeability studies indicated the drug to be BCS Class I, which is supported by reference labeling and pharmacokinetic literature data. The test and reference products are rapidly dissolving.
Cook et al. describe several examples where application of the BCS has been beneficial, including obtaining biowaivers as well as facilitating formulation development during the clinical development cycle (6).
Class III Biowaivers
A key highlight of the workshop was the continued scientific support for biowaivers for Class III compounds whose formulations exhibit very rapid dissolution. Scientific consensus has previously found broad consensus supporting biowaivers for at least some Class III drugs whose formulations exhibit very rapid dissolution (2). The topic of Class III biowaivers was discussed on several occasions during the workshop, and included scientific considerations which may need to be examined to assess risks associated with waiving in vivo bioequivalence studies for Class III drugs (7). However, no actual scientific evidence was presented to deny biowaivers to Class III drugs whose formulations (1) contain excipients common in solid oral products, in usual amounts, and (2) exhibit very rapid dissolution. Observations from FDA and EU scientists supports such biowaivers (4).
A number of potential concerns regarding the allowance of biowaivers of Class III were discussed, with an emphasis on potential excipient effects (e.g. effect of excipient on intestinal permeability including transporters, intestinal motility, and intestinal inflammation). Experience to date suggests that limitations present in the BCS guidance (i.e. common excipients in typical quantities) adequately address these potential concerns. There was also discussion and recognition that a priori assessment of excipient effects on Class III drug permeability and human bioavailability would be welcomed, including excipient dose–response studies. It was also recognized that drug instability in the GI tract and drug metabolism in the brush border and/or gut wall are areas for further research.
As discussed previously, some concerns were described about biowaivers for very poorly permeable drugs (2). The suggestion was made that risk of non-equivalence may vary significantly, depending on the magnitude of the low permeability, such that risk analysis techniques may be pertinent to deciding whether or not a BCS III biowaiver could be granted for any particular drug product. These concerns appeared to be addressed by limiting the lowest acceptable fraction dose absorbed at about 20–40%, in order to be considered for biowaivers (2).
Permeability Methodologies
Another key highlight of the workshop was the scientific support for a number of permeability methodologies to assess BCS permeability class. While it was noted that human pharmacokinetic data, when available, should always be presented, several examples were presented where in vitro permeability was used to classify a drug as either high or low permeability. These examples employed different permeability methods (e.g. Caco-2 cell culture, MDCK cell culture, rat intestinal perfusion). Like prior consensus (2), the BCS guidance’s lack of mandate for any one permeability method or any prescribed set of specific experimental methods was viewed favorably, affording flexibility that promotes the implementation of BCS across laboratories, particularly those laboratories with established permeability methods. Unlike prior consensus which suggested the need for further guidance (2), there was little discussion for the need of further guidance at this workshop. This change in the perceived need for further guidance perhaps reflects the much greater experience by both industry and FDA in applications of BCS, compared to the year 2002.
It was expressed that human data (e.g. human mass balance studies) are generally pivotal for permeability classification of new chemical entities. However, it was also recognized that reduced use of intravenous dosing studies may increase the importance of in vitro or in situ permeability studies. Regarding biowaiver requests for ANDA applications, in vitro or in situ permeability determinations are particularly valuable, especially in the absence of reliable in vivo data.
BIORELEVANT DISSOLUTION AND BCS FUTURE DEVELOPMENT
In this workshop session, the topics of Biorelevant Dissolution and BCS Future Development were discussed. A key highlight of the workshop was broad support for further scientific progress in in vitro dissolution methods to predict dosage form performance. The FDA guidance on in vitro–in vivo correlations is well in place (8). The majority of lectures and discussions concerned strategies and computational approaches to better employ dissolution data to understand product performance, including during product development, where drug biopharmaceutic characteristics are also considered.
In Vitro Dissolution
Three ideal attributes of in vitro dissolution tests are (1) sensitivity to product changes such that in vitro dissolution testing ensures high quality and consistent product performance, (2) predictability of in vivo drug product performance such that human studies can be reduced and product development is accelerated, and (3) recognition by regulatory policies and procedures to support product applications. It was also noted that biorelevant dissolution methodologies can be the same as or different from quality control dissolution methodologies.
For oral solid dosage forms, in vitro dissolution testing is a critical contributor to Quality-by-Design (QbD) implementation. QbD posits the assurance of product quality by designing the product and manufacturing process to meet user needs. The achievement of such assurance requires an understanding of product and process design space (9, 10). Design space is the multidimensional combination and interaction of input variables (e.g. material attributes) and process parameters that have been demonstrated to provide assurance of quality. Working within the design space is not considered as a 539 change. Movement out of the design space is considered to be a change and would normally initiate a regulatory post-approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval. Product quality is clinically relevant quality. QbD challenges pharmaceutical scientists to link product manufacturing parameters to clinical performance. Traditional methods for measuring clinical quality (i.e. clinical pharmacokinetic studies) are not viable, since there are a large number of batches generated during process development. New methods are required. In vitro dissolution testing is clearly one tool for this purpose. The present bioequivalence and BCS guidelines provide a regulatory platform for in vitro dissolution to serve as a surrogate for clinical quality with respect to drug exposure in the body.
Dissolution testing can be a viable approach to assure desired clinical performance with respect to consistent bioavailability for a wide range of drugs via QbD, based on BCS considerations and specific product knowledge (10). However, to support QbD, in vitro dissolution data need to be better integrated into product design to yield a higher level of product understanding. Such understanding should be reflected in regulatory documentation, including an assessment of product risk. In fact, during product development and process establishment, risk assessment should drive the choice of product attributes/process variables to be assessed for impact on in vivo performance.
A QbD development package should identify the rate limiting step(s) in the absorption process, consider the in vivo relevance of in vitro dissolution test conditions, and interpret bioavailability studies involving the relevant formulation/process variables. A goal could be to establish an in vitro/in vivo correlation (IVIVC) as a significant number of drugs in development are Class II compounds. However, failure to establish a classical IVIVC could be a successful outcome of an in vitro/in vivo study in the context of QbD, if all variants (i.e. side batches) produce the same exposure, as assessed via in vivo testing. The understanding conveyed in the QbD development package should allow for regulatory relief, including possibly no need for a “final” bioequivalency study, fewer clinical pharmacokinetic bridging studies between phases, and specifications based upon process understanding and not based on process capability.
Dissolution of Class II Drugs
The potential for BCS-based biowaivers of Class II drugs, particularly weak acids, was also discussed. For weak acids, two hypotheses underpinning BCS Class II biowaivers of IR dosage forms are (1) Class II weak acids in IR dosage forms may be eligible for biowaivers if the dose dissolves completely before reaching mid-jejunum and (2) gastrointestinal simulation technology may be used as a tool to recommend extension of biowaivers for Class II weak acids. From case studies involving available dissolution and pharmacokinetic data, as well as simulation, the risk for bioinequivalence for Cmax was higher than for AUC. Hence, biowaivers for some Class II compounds may require relaxation of some BE criteria (e.g. Cmax confidence interval), with appropriate consideration of consumer risk assessment. However, it was concluded that there remains a need for robust and predictive dissolution methods, along with additional simulation validation, in order to broadly recommend BCS-based biowaivers for Class II drugs.
Tubic-Grozdanis et al. describe gastrointestinal simulation for BCS classification (11). Gastrointestinal simulation technology, using physiological parameters and experimentally determined physicochemical and pharmacokinetic drug properties, was applied to predict biopharmaceutical drug classification of several weak acid and weak base BCS Class II compounds. Their findings indicate that in silico models are useful to identify BCS class II biowaiver candidate drugs, where the risk of bioinequivalence in terms of Cmax is higher than for AUC. However, Class II weak acids and bases may be eligible for biowaivers provided that the dose dissolves completely before reaching the mid-jejunum.
From the meeting, it appeared that a significant portion of new chemical entities, probably a majority, are BCS Class II. This perspective implies that additional research on BCS Class II is merited, including re-considering the solubility class boundary between high solubility and low solubility. The current BCS solubility media exclude surfactant. An additional point of discussion was the question of how to consider, from a regulatory standpoint, situations where the formulation enhances drug solubility, including where BCS II drug is formulated to function as a highly soluble drug in vivo (e.g. change from Class II to Class I). While the current BCS solubility class is based solely upon the API itself, the question was raised as to whether any future changes to BCS could accommodate this “formulation” effect.
Gastrointestinal Physiology and Prediction of Food-Effect
There was significant discussion of gastrointestinal physiology as it relates to oral solid dosage form performance, including the prediction of food-effect. A prerequisite for the strategic development of any product with high and reproducible absorption is an understanding of the gastrointestinal conditions that are responsible for drug dissolution, including the impact of formulation components. This understanding is perhaps most useful for Class II drugs, due to their low solubility yet frequent occurrence in development programs. Studies of in vivo gastrointestinal fluid composition are of value, as well as studies of in vivo gastrointestinal hydrodynamics. From such studies, physiologically-relevant in vitro dissolution models can be developed to mimic the in vivo events within the gastrointestinal tract during digestion and drug absorption.
The dynamic process of lipid digestion can be studied via the in vitro lipolysis model. This model is a tool in the development of lipid-based formulations, as well as prediction of food-effect. Important considerations in the development of biorelevant dissolution media are the levels of bile salt and phospholipid from bile, as well as the presence of solubilizing products from the digestion of dietary or formulation lipid (e.g. free fatty acids and mono-glycerides). Biorevelant media containing different levels of these components have been characterized in terms of micellar and vesicular size and shape. However, it remains that the biopharmaceutics toolbox needs to be expanded with better predictive in vitro methods to simulate events that take place in the gastrointestinal tract.
Animal models have also been employed to predict food-effect. However, few in vivo models exist to predict the magnitude of change in human pharmacokinetic parameters when drug is dosed in the presence of food. In vivo studies in rat, dog, and monkey have been investigated for this purpose with varying success. Most reports are retrospective in that a food-effect is first observed clinically, and then studies are performed in animal models to determine whether the results could have been anticipated, or studies are designed specifically to understand the mechanism of the effect. At the drug discovery/clinical interface, it is desirable to determine the likelihood for the drug substance itself to show potential food-effect. A fed, pentagastin-treated dog was described as a model for predicting human food-effect (12). The model utilizes a test meal consisting of a 50-gram aliquot of the FDA standard breakfast, since meal type/amount for canine was important in order to simulate the human fed state. However, it remains that no perfect model exists for predicting human food-effect. Choice of existing models should rely on specific need or stage of development.
BIOEQUIVALENCE FOR HIGHLY VARIABLE DRUGS
Currently, the bioequivalence statistical analysis involves the following metrics and criteria: AUC and Cmax; log-transformed data; ANOVA model with period, sequence, subject (sequence), and treatment; and 90% confidence intervals must fit between 80–125%. This analysis is applied to highly variable drugs (HVD), as well as non-HVD. HVDs are drugs with high within-subject variabilities (ANOVA-CV ≥30%) in Cmax and/or AUC. It is well appreciated that HVDs often require a greater numbers of subjects than non-HVD. This added complexity in study design has motivated the development of several novel methods and possible alternative acceptance criteria for HVDs.
An FDA perspective was presented. At the April 14, 2004 meeting of the Advisory Committee of Pharmaceutical Science (ACPS), different approaches were discussed, such as expansion of bioequivalence limits and scaled average bioequivalence. The committee favored scaled average bioequivalence over other approaches. An FDA working group was created. A research project to evaluate scaling was initiated and provided preliminary results to the ACPS at its October 6, 2006 meeting. The committee was in favor of using a point estimate constraint with scaled average bioequivalence. Most members favored a minimum sample size of 24.
Simulation results based on scaled average bioequivalence indicated that a partial replicate, 3-way crossover design appears to work well. In practice, reference product (R) would be administered twice and test product (T) administered once, such that sequences are RTR, TRR, and RRT. A point estimate constraint had little impact at lower variability (~30%), but more significant effect at greater variability (~60%). Sample size would need to be determined by the sponsor (e.g. adequate power), with a minimum of 24 subjects. Bioequivalence criteria should be scaled to reference variability (Cmax and AUC), where upper/lower criteria are 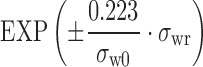 , where σw0 = 0.25.
, where σw0 = 0.25.
σwr is the intrasubject standard deviation for the reference product. σw0 is a constant to be set by the regulatory agency, such that BE acceptance limits are permitted to be broadened by scaling. In simulations, σw0 = 0.25 demonstrated a good balance between a conservative approach and a practical one. Additionally, in the FDA proposal under evaluation, the point estimate (test/reference geometric mean ratio) must fall within (0.80–1.25). This use of point estimate constraint addresses concerns that products with large geometric mean ratio differences may be judged bioequivalent.
Other perspectives were also presented. An industrial viewpoint employed simulations of accumulating and non-accumulating compounds. It was observed that Cmax variance has little effect on steady state conditions and that AUC can have significant effect, such that Cmax/AUC is a better rate measure than Cmax.
Also discussed were observations from ANDA bioequivalence studies submitted to the FDA (13). About 10% of bioequivalence submissions are for drugs that meet the highly variable criteria. Of these, the variability in 70% is attributed to disposition of the drug substance. The remaining 30% might be due to formulation, study conduct, or aberrant subjects.
FDA AND INTERNATIONAL INITIATIVES
In addition to the above discussions, the workshop featured several other initiatives at FDA and at international agencies that impacts on pharmaceutical product quality, including oral biopharmaceutics and bioequivalence.
FDA Initiatives
FDA initiatives include the Critical Path Initiative, the Drug Safety Initiative, and the Quality by Design (QbD) Initiative. The Critical Path Initiative aims to stimulate and facilitate a national effort to modernize the scientific process through which a potential human drug, biological product, or medical device is transformed from a discovery or “proof of concept” into a medical product (3). A Critical Path Opportunities for Generic Drugs document was recently released (14, 15). Bioequivalence plays an important role in this process of developing medications. It is recognized that products change over their lifetime (e.g. clinical formulations evolve on the way to market, major changes to formulation or manufacturing after approval, generic drugs). Bioequivalence studies are conducted to fill gaps in knowledge about product performance. Success along the critical path will reduce knowledge gaps.
The intent of the Drug Safety Initiative is to strengthen and improve the management of drug safety issues. The FDA Safety Initiative impacts bioavailability and bioequivalence in that it calls for a better understanding of bioavailability. Specifically, there is a need to better understand exposure differences between sub-groups. There is also a need to better understand the mechanisms of drug distribution and elimination, with the promise for better predictive models of liver toxicity and other adverse events.
The QbD and cGMPs for the twenty-first century initiatives aim to enhance and modernize the regulation of pharmaceutical manufacturing and product quality—to bring a twenty-first century focus to this critical FDA responsibility. Several workshop presentations referenced these initiatives, particularly in terms of drug biopharmaceutic properties and in vitro dissolution testing serving as surrogates for product quality. Two implementation programs are the Office of New Drug Quality Assessment (ONDQA) pilot program for new drugs and the Question-based Review (QbR) for generic drugs(16, 17). In the ONDQA pilot program, there are six original and two supplemental NDAs, as well as three INDs. Among the seven submitted to date, three were approved, one is approvable, and three remain under review. In the QbR program, over 210 QbR ANDAs have been received; five ANDAs have been approved.
Additionally, FDA scientists within the Center for Veterinary Medicine are investigating veterinary application of the BCS. Reasons for interest in a veterinary BCS (vBCS) parallel those for human pharmaceuticals. However, interspecies diversity in gastrointestinal physiology renders it unlikely to establish a single set of criteria for highly soluble and highly permeable compounds. Therefore, the current focus is on defining the vBCS criteria for canines. The canine is the animal species associated with the majority of FDA-approved solid oral dosage forms. Also, the dog is frequently used as a preclinical species in human drug development, such that advances in the canine model will potentially translate to advances in human drug development.
There are differences between canine and human gastrointestinal physiology. Regarding intestinal permeability, dog permeability is greater than human permeability for small hydrophilic compounds. For highly permeable compounds, the two are approximately equal. However, due to residence time effects, the extent of absorption from sustained release formulations is less from dogs than from humans. Regarding solubility, there are several complications impacting solubility classification in dogs, including large variations in dog size depending on breed, and the fact that most drug doses are administered with <15 ml water. For these reasons (e.g. variability in dose number as a function of dog size), solubility needs to be defined by USP methods in dogs. It is anticipated that fewer drugs will be classified as highly soluble in dogs. It was also noted that lower pH gradient between stomach and intestine of dogs may lead to less risk of drug (weak base) re-precipitation in dog versus human.
World Health Organization
The World Health Organization (WHO) provides guidance to regulatory agencies around the world and recognizes BCS-based biowaivers (18). WHO defines high permeability as extent of absorption is at least 85%, compared to the 90% value used in the current FDA BCS guidance. WHO recognizes BCS-based biowaivers for Class I drugs whose formulations exhibit rapid dissolution, Class III drugs whose formulations exhibit very rapid dissolution, and Class II drugs that are weak acids that are highly soluble at pH 6.8 and whose formulations exhibit rapid dissolution at pH 6.8 (and its dissolution profile is similar to that of the reference product at pH 1.2, 4.5 and 6.8) (16). Compared to the current FDA BCS guidance, which is recognized to be a conservative original effort (2), the WHO BCS framework is broader, as it allows Class III biowaivers, as well as Class II biowaivers for weak acids.
International BCS/BE Initiatives
There were discussions about BCS initiatives through out the world, reflecting the several international workshop speakers and the fact that workshop participants represented 17 different countries. The similarities and differences between the USA and European Union (EU) review processes were discussed. BCS-based biowaiver criteria are very similar between the FDA BCS guidance (1) and the European Medicines Agency (EMEA) Note (19). The EU document does not set a limit of 90% fraction dose absorbed for the high permeability limit, but rather employs linear and complete absorption as the criteria. Currently, for applications for new drug products that move to EU assessment level, biowaiver via BCS is seldom applied for, as larger companies do not risk rejection by one of the national EU regulatory bodies. This circumstance reflects that, in contrast to the USA system where FDA is the single assessor, all assessments and most decisions in the EU are performed by national experts and national regulatory bodies. EMEA functions only as an administrative center. Although the EU has adopted uniform regulatory requirements, regulatory practice remains less transparent as long as applications are reviewed nationally. Meanwhile, biowaiving via BCS is being invoked on the national level. The greatest regulatory impact of BCS concerns post-approval manufacturing changes, where there are typically small differences between pre-and post-change products. No mechanism is in place in Japan for BCS-based biowaivers.
SUMMARY
Key highlights of the workshop were (a) the described granting of a dozen BCS-based biowaivers by the FDA for Class I drugs whose formulations exhibit rapid dissolution, (b) continued scientific support for Class III biowaivers whose formulations exhibit very rapid dissolution, (c) scientific support for a number of permeability methodologies to assess BCS permeability class, (d) utilization of BCS in pharmaceutical research and development, (E) and scientific progress in in vitro dissolution methods to predict dosage form performance.
Footnotes
Opinions expressed in this report are those of the authors (LXY and HNW) and do not necessarily reflect the views or policies of the FDA.
References
- 1.Guidance for Industry, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. August 2000, CDER/FDA. http://www.fda.gov/cder/guidance/3618fnl.htm (accessed 12/15/07)
- 2.Polli J. E., Yu L. X., Cook J. A., Amidon G. L., Borchardt R. T., Burnside B. A., Burton P. S., Chen M.-L., Conner D. P., Faustino J., Hawi A. A., Hussain A. S., Joshi H. N., Kwei G., Lee V. H. L., Lesko L. J., Lipper R. A., Loper A. E., Nerurkar S. G., Polli J. W., Sanvordeker D. R., Taneja R., Uppoor R. S., Vattikonda C. S., Wilding I., Zhang G. Summary workshop report: biopharmaceutics classification system—implementation challenges and extension opportunities. J. Pharm. Sci. 2004;93:1375–1381. doi: 10.1002/jps.20064. [DOI] [PubMed] [Google Scholar]
- 3.Critical Path Opportunities Report. March 2006, FDA. http://www.fda.gov/oc/initiatives/criticalpath/reports/opp_report.pdf (accessed 12/15/07)
- 4.J. E. Polli. In vitro studies are sometimes better than conventional human in vivo studies in assessing bioequivalence for immediate-release solid oral dosage forms. AAPS J. in press (2008). [DOI] [PMC free article] [PubMed]
- 5.M. S. Ku. Use of the biopharmaceutical classification system in early drug development. AAPS J. in press (2008). [DOI] [PMC free article] [PubMed]
- 6.J. A. Cook, W. Addicks, and Y. H. Wu. Application of the biopharmaceutical classification system in clinical drug development—an industrial view. AAPS J. in press (2008). [DOI] [PMC free article] [PubMed]
- 7.S. Stavchansky. Scientific perspectives on extending the provision for waivers of in vivo bioavailability and bioequivalence studies for drug products containing high solubility–low permeability drugs (BCS-Class 3). AAPS J. in press (2008). [DOI] [PMC free article] [PubMed]
- 8.Guidance for Industry, Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. September 1997, CDER/FDA. http://www.fda.gov/cder/guidance/1306fnl.pdf (accessed 12/15/07) [DOI] [PubMed]
- 9.L. X. Yu. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res.25:781–791 (2008). [DOI] [PubMed]
- 10.W. W. Lee, P.Dickinson, P. Stott, A. Townsend, J. Smart, P. Ghahramani, T. Hammett, L. Billett, S. Behn, R. Gibb, and B. Abrahamsson. Clinical relevance of dissolution testing in quality by design. AAPS J. in press (2008). [DOI] [PMC free article] [PubMed]
- 11.M. Tubic-Grozdanis, M. Bolger, and P. Langguth. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J.10:213–216. [DOI] [PMC free article] [PubMed]
- 12.K. A. Lentz. Current methods for predicting human food effect. AAPS J. in press (2008). [DOI] [PMC free article] [PubMed]
- 13.B. Davit, D. P. Conner, B. Fabian-Fritsch, S. H. Haidar, X. Jiang, D. T. Patel, P. R. Seo, K. Suh, and C. L. Thompson. Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPS J. 10:213–226 (2008). [DOI] [PMC free article] [PubMed]
- 14.Critical Path Opportunities for Generic Drugs. May 2007, CDER/FDA.http://www.fda.gov/oc/initiatives/criticalpath/reports/generic.html (accessed 12/15/07)
- 15.R. A. Lionberger. FDA critical path initiatives: opportunities for generic drug development. AAPS J. 10:103–109. [DOI] [PMC free article] [PubMed]
- 16.Yu L. X., Raw A., Lionberger R. A., et al. U.S. FDA Question-based review for generic drugs: A new pharmaceutical quality assessment system. J. Generic Med. 2007;4:239–248. doi: 10.1057/palgrave.jgm.4950073. [DOI] [Google Scholar]
- 17.Lee L., Lionberger R. A., Yu L. X., Mundkur C., Munro G., Johnston G., Famulare J. C. FDA Office of Generic Drugs’ pharmaceutical quality initiative: progress and feedback on question-based review. Pharm. Eng. 2007;27:52–61. [Google Scholar]
- 18.Anonymous. “Annex 7: Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability”. In WHO Expert Committee on Specifications for Pharmaceutical Preparations: Fortieth Report; WHO: Geneva, Switzerland, 2006, pp. 347–390. http://whqlibdoc.who.int/trs/WHO_TRS_937_eng.pdf (accessed 12/15/07)
- 19.Committee for Proprietary Medicinal Products. Note for Guidance on the Investigation of Bioavailability and Bioequivalence. July 2001, EMEA. http://www.emea.europa.eu/pdfs/human/qwp/140198enfin.pdf (accessed 12/15/07)