

Abstract
Introduction
It is widely believed that acceptable bioequivalence studies of drugs with high within-subject pharmacokinetic variability must enroll higher numbers of subjects than studies of drugs with lower variability. We studied the scope of this issue within US generic drug regulatory submissions.
Materials and Methods
We collected data from all in vivo bioequivalence studies reviewed at FDA’s Office of Generic Drugs (OGD) from 2003–2005. We used the ANOVA root mean square error (RMSE) from bioequivalence statistical analyses to estimate within-subject variability. A drug was considered highly variable if its RMSE for Cmax and/or AUC was ≥0.3. To identify factors contributing to high variability, we evaluated drug substance pharmacokinetic characteristics and drug product dissolution performance.
Results and Discussion
In 2003–2005, the OGD reviewed 1,010 acceptable bioequivalence studies of 180 different drugs, of which 31% (57/180) were highly variable. Of these highly variable drugs, 51%, 10%, and 39% were either consistently, borderline, or inconsistently highly variable, respectively. We observed that most of the consistent and borderline highly variable drugs underwent extensive first pass metabolism. Drug product dissolution variability was high for about half of the inconsistently highly variable drugs. We could not identify factors causing variability for the other half. Studies of highly variable drugs generally used more subjects than studies of lower variability drugs.
Conclusion
About 60% of the highly variable drugs we surveyed were highly variable due to drug substance pharmacokinetic characteristics. For about 20% of the highly variable drugs, it appeared that formulation performance contributed to the high variability.
Key words: bioequivalence, generic drugs, highly variable drugs, presystemic drug metabolism, variable drug product dissolution
INTRODUCTION
The topic of bioequivalence evaluation of highly variable drugs is one that has been intensely debated in many recent articles, conferences and meetings, held both nationally and internationally (1). This topic is pertinent to generic drug development because bioequivalence studies are the pivotal clinical studies submitted to regulatory agencies in support of marketing applications of new generic drug products. A bioequivalence study is conducted to determine whether the new generic drug and corresponding reference listed drug (RLD) have the same rate and extent of absorption (2). The U.S. Food and Drug Administration (FDA) uses peak drug concentrations (Cmax) in plasma or other appropriate biological fluid as an index of drug rate of absorption and the area under the drug plasma concentration versus time curve (AUC) as an index of a drug’s extent of absorption (2). The FDA concludes that a generic drug (test product) and its corresponding RLD (reference-listed-drug or reference product) are bioequivalent if, in the pivotal bioequivalence studies, the 90% confidence intervals of the geometric mean test/reference ratios for both pharmacokinetic parameters Cmax and AUC fall within the bioequivalence limits of 80–125% (3,4). Thus, the FDA considers pharmacokinetic bioequivalence both as a surrogate for therapeutic equivalence and as a comparator of the pharmaceutical quality of the test and reference products.
In bioequivalence evaluation, highly variable drugs are generally defined in the context of within-subject variability in bioequivalence parameters Cmax and AUC. The most often-used definition of a highly variable drug is a drug which has a within-subject (synonymous with “intra-subject”) variability of 30% or more in these two bioequivalence parameters (5,6). As illustrated in Fig. 1, because of this high variability, larger numbers of subjects may be needed in bioequivalence studies to give adequate statistical power to meet FDA bioequivalence limits (5–9). The FDA is currently investigating bioequivalence study design proposals that can reduce the number of subjects needed for a bioequivalence study (9–11).
Fig. 1.

A visual representation of some possible results of the statistical analyses of bioequivalence studies. The three bars represent the widths of hypothetical 90% confidence intervals from bioequivalence studies of drugs with normal variability (green bar), low variability (blue bar), and high variability (red bar). A bell-shaped curve is superimposed over green bar, representing the 90% confidence interval, distributed around the geometric mean test/reference ratio (“point estimate”), for the normal variability drug. For simplification, blue and red bars, respectively, are used in this diagram to represent confidence interval widths of low variability and highly variable drugs. The blue and red bars also actually represent the 90% confidence intervals of the bioequivalence study C max or AUC test/reference ratios normally distributed about the point estimate. The FDA concludes that a test and reference product are bioequivalent if the 90% confidence intervals (expressed as a percent) of the geometric mean C max and AUC test/reference ratios fall within the bioequivalence limits of 80–125%. In this illustration, the 90% confidence interval of the normal variability drug (green bar) meets bioequivalence limits. The 90% confidence interval of the drug with low variability meets bioequivalence limits although the point estimate deviates from 1.00. For a highly variable drug, the 90% confidence interval can exceed bioequivalence limits solely because of the variability. Using more subjects in the bioequivalence study will cause the 90% confidence interval of a highly variable drug to become narrower
A number of factors can contribute to high variability in bioequivalence parameters (12,13). Some of the factors in the gastrointestinal (GI) tract contributing to variability include gastric emptying, intestinal transit, luminal pH, luminal surfactant (i.e. phospholipid, bile acid) concentrations, and/or presence or absence of food. High variability in bioequivalence parameters can be due to extensive presystemic metabolism, occurring either within the lumen of the GI tract or intestinal cell mucosa, or due to hepatic first-pass. In addition, low systemic availability of the parent drug can be accompanied by high variability if drug plasma concentrations are difficult to measure.
The factors described above influence bioequivalence parameter variability due to the characteristics of the drug substance, rather than those of the drug product. Drug product formulation can also contribute to high variability in bioequivalence parameters. For example, if the rate of drug release from the dosage form is highly variable, this factor may cause high variability in bioequivalence parameters and may signify a product with lower product quality. Figure 2 diagrams the steps involved in bioequivalence evaluation of oral dosage form performance and illustrates ways in which high within-subject variability in bioequivalence measures can arise from either the drug substance or the drug product.
Fig. 2.

A diagram relating solid oral dosage form performance to the in vivo system in a bioequivalence study. Once ingested, a solid oral dosage form disintegrates, then dissolves into solution (formulation stage). The dissolved drug is absorbed through the gut wall, enters the liver through the portal vein, and from the liver goes into the systemic circulation, where pharmacokinetic measurement is possible. From the systemic circulation, the drug reaches the site of activity from which one observes a clinical response, where pharmacodynamic or therapeutic measurement is possible. Although the most accurate way of determining bioequivalence would be to compare test and reference product performance at the formulation stage, this is nearly always not possible. Consequently, most bioequivalence studies of systemically absorbed drugs rely on pharmacokinetic measures, as drug blood concentrations are thought to directly relate to the amount of drug released from the dosage form. Therefore, a properly designed in vivo study with pharmacokinetic endpoints can accurately determine whether a test and reference product are bioequivalent. As the drug moves from the formulation to the systemic circulation to the site of activity, the pharmacokinetic or pharmacodynamic response becomes increasingly variable with increasing numbers of steps between the formulation, pharmacokinetic measurement stage, and pharmacodynamic measurement stage. For example, for drugs that undergo extensive presystemic metabolism, the effects of the various biotransformation(s) brought about by various gut wall and/or hepatic metabolism steps contribute to the variability observed in drug pharmacodynamic measurements. This figure also illustrates the two sources of variability in bioequivalence measures—variability due to drug substance pharmacokinetics versus variability due to drug product performance. If high variability exists due to drug substance pharmacokinetics, it may be necessary to use large numbers of subjects to achieve an acceptable bioequivalence study. However, if the high variability is due to the formulation or dosage form performance, this may reflect either a poor quality test or reference product
As the FDA is currently investigating the feasibility of using alternative designs for bioequivalence studies of highly variable drugs, we undertook the following study to determine the scope of this issue within regulatory submissions for new generic products. First, we wanted to understand the prevalence of highly variable drugs in generic drug development; thus, we sought to determine what percentage of all of the applications for new generic drug products received by the OGD encompasses highly variable drugs. We also attempted to characterize the degree and consistency of variability of these highly variable drugs. Next, as it is thought that studies of highly variable drugs must enroll more subjects to demonstrate bioequivalence, we compared the numbers of subjects used for acceptable studies of highly variable drugs versus numbers used for acceptable studies of drugs with lower within-subject variability. Finally, we determined the characteristics of drugs and drug formulations that may be responsible for high variability. In particular, we sought to verify that, in applications for new generic products, the high variability in most cases was due to dispositional characteristics of the drug substance, rather than due to a poor quality formulation or poor study conduct.
MATERIALS AND METHODS
Collection of Data from In Vivo Bioequivalence Studies of Generic Drugs
We collected data from all in vivo bioequivalence studies submitted to the OGD, Center for Drug Evaluation and Research (CDER), FDA, under Abbreviated New Drug Applications (ANDAs) and reviewed within the Division of Bioequivalence (DBE) from 2003 to 2005. Acceptable bioequivalence studies were defined as studies for which the maximum plasma concentration (Cmax), area under the concentration vs. time curve to the last measured time-point (AUC0-t), and AUC extrapolated to infinite time (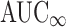 ) met bioequivalence limits. The following data were collected for each bioequivalence study reviewed during that time period: submission number, generic name of drug product, dosage form, number of subjects completing the study, and whether the study was under fasting or fed conditions. The 90% confidence intervals and root mean square error (RMSE) values for Cmax, AUC0-t, and
) met bioequivalence limits. The following data were collected for each bioequivalence study reviewed during that time period: submission number, generic name of drug product, dosage form, number of subjects completing the study, and whether the study was under fasting or fed conditions. The 90% confidence intervals and root mean square error (RMSE) values for Cmax, AUC0-t, and 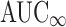 from each study were collected from the analysis of variance (ANOVA) bioequivalence statistical outputs. The 90% confidence interval data were collected to determine if the study was acceptable, i.e., met FDA bioequivalence limits.
from each study were collected from the analysis of variance (ANOVA) bioequivalence statistical outputs. The 90% confidence interval data were collected to determine if the study was acceptable, i.e., met FDA bioequivalence limits.
Identification of Highly Variable Drugs
The RMSE values of the bioequivalence parameters Cmax and AUC0-t was used as an estimate of within-subject variability. Since most of the studies submitted to the DBE used a two-way crossover design, it was not possible to determine the true within-subject variability. Therefore, the RMSE was used as an estimate of within-subject variability. Since highly variable drugs are defined as drugs with within-subject variability of 30% or more in bioequivalence parameters, we considered a drug to have high within-subject variability if the RMSE for either AUC0-t or Cmax was ≥0.3.
Although the FDA evaluates 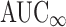 in bioequivalence studies, we did not define a highly variable drug as one for which the
in bioequivalence studies, we did not define a highly variable drug as one for which the 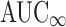 RMSE ≥ 0.3 because the calculations necessary to extrapolate to infinity contribute to the variability of this measure. Therefore, we consider AUC0-t to be a better indicator of variability due to drug substance and/or drug product than
RMSE ≥ 0.3 because the calculations necessary to extrapolate to infinity contribute to the variability of this measure. Therefore, we consider AUC0-t to be a better indicator of variability due to drug substance and/or drug product than 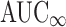 .
.
Numbers of Subjects Enrolled in Studies of Highly Variable Drugs
To test the hypothesis that bioequivalence studies of highly variable drugs must enroll more subjects than studies of drugs with lower variability, we determined the numbers of subjects completing bioequivalence studies of highly variable drugs (drugs with RMSE values ≥0.3 for and Cmax and/or AUC0-t) in the 2003–2005 data set. We compared these values with the numbers of subjects enrolled in bioequivalence studies, reviewed during the 2003–2005, of drugs with lower variability (RMSE values < 0.3 for and Cmax and/or AUC0-t.
Investigation of the Degree of Variability of Drugs Categorized as Highly Variable
We investigated the degree of variability in bioequivalence parameters in bioequivalence studies of highly variable drugs, reviewed from 2003–2005. We determined the number of studies in which the RMSE for AUC0-t and Cmax fell within the following ranges: from >0.30 to 0.35, >0.35 to 0.40, >0.40 to 0.45, >0.45 to 0.50, >0.50 to 0.55, 0.55 to 0.60.
Determination of Whether High Variability in Bioequivalence Parameters Was Consistent
Once the studies of highly variable drugs were identified, we investigated these studies further to determine (1) which drugs were consistently highly variable; (2) which drugs were borderline highly variable; and (3) which drugs were inconsistently highly variable. We defined consistently highly variable drugs as drugs for which the RMSE for Cmax and/or AUC0-t ≥ 0.3 in all bioequivalence studies of that drug reviewed during the 2003–2005 period. We defined borderline highly variable drugs as those for which, in any one bioequivalence study, the RMSE was either slightly greater than or slightly less than 0.3, and averaged approximately 0.3 across all bioequivalence studies of that drug. We classified inconsistently highly variable drugs as drugs that were highly variable in some drug product bioequivalence studies but not in others.
Identification of Drug Substance Characteristics Contributing to High Variability
We hypothesized that the variability observed in the consistent and borderline highly variable drugs was due to characteristics of the drug substance. To test this hypothesis, we characterized the physicochemical and dispositional properties of these drug substances, with an emphasis on drug absorption and metabolism. Characteristics that we evaluated included aqueous solubility, stability in the gastrointestinal tract, existence/extent of presystemic metabolism, food effects, oral bioavailability, and route of administration. We also classified these drugs according to pharmaceutical class to determine if any particular class was consistently associated with high variability.
Identification of Drug Product Characteristics Contributing to Inconsistent High Variability
We defined inconsistent high variability as drugs which met the highly variable criteria in only a small minority of studies on that drug. For these inconsistent highly variable drugs, we evaluated additional factors to determine if the variability was due to either conduct of the in vivo studies or formulation performance.
Bioequivalence Study Performance and High Variability
To investigate ways in which conduct of the in vivo studies might influence variability in bioequivalence parameters, we evaluated bioequivalence study design as well as the bioanalytical methods used to determine drug concentrations in plasma samples from the in vivo studies. We evaluated plasma sampling times and bioanalytical method sensitivity. We also identified the bioanalytical Contract Research Organizations (CROs) responsible for assaying plasma drug concentrations from the bioequivalence study, in order to determine if inconsistent high variability in bioequivalence parameters was associated with any bioanalytical CRO(s) in particular.
Relationship of Variability in Drug Product Dissolution Rate In Vitro to Variability in Bioequivalence Parameters In Vivo
To determine whether drug product formulation performance contributed to high in vivo variability, we compared multipoint dissolution profile variability with bioequivalence parameter variability for each of the drugs with inconsistent variability. We conducted these comparisons as follows. We calculated the average coefficient of variation (CV, standard deviation divided by the mean) for each drug product’s dissolution profile. For each drug product, we then compared rank-ordered dissolution profile CV data with rank-ordered average RMSE data. The average RMSE values for each study were estimated by averaging the RMSE values for Cmax and AUC0-t.
RESULTS AND DISCUSSION
Identification of Highly Variable Drugs
Table I shows the number of bioequivalence studies, drug products, and drugs reviewed by the DBE in 2003–2005. During this time period, the DBE found acceptable 1,010 bioequivalence studies. These 1,010 bioequivalence studies investigated a total of 524 different drug products, for 180 different drugs. Frequently, there are at least several generic versions of any one reference listed drug under review at the OGD during the same time period. Each new generic drug product line is usually the subject of a separate ANDA. Most ANDAs contain at least two bioequivalence studies, one under fasting conditions and one under fed conditions. A minority of ANDAs contain either one fasting bioequivalence study or one fed bioequivalence study.
Table I.
Number of Bioequivalence Studies of Highly Variable Drugs Reviewed by the Division of Bioequivalence in the Office of Generic Drugs from 2003–2005
| Description | Bioequivalence Studies | Different Drug Products | Different Drugs | |||
|---|---|---|---|---|---|---|
| Number | % of Total | Number | % of Total | Number | % of Total | |
| RMSE of AUC0-t and/or C max ≥ 0.3 | 111 | 11 | 101 | 19 | 57 | 32 |
| RMSE of AUC0-t and/or C max < 0.3 | 899 | 89 | 423 | 81 | 123 | 68 |
| Total number of drugs studied | 1,010 | 100 | 524 | 100 | 180 | 100 |
In 111 of these 1,010 acceptable studies, the RMSE was ≥0.3 for either Cmax and/or AUC0-t. As our criteria for classification as a highly variable drug was that the RMSE ≥ 0.3 for Cmax and/or AUC0-t, we concluded that 111, or 11% of these studies were of drug products that showed high variability in bioequivalence parameters. These 111 studies of highly variable drugs were of 101 different drug products, representing 57 different drugs.
Numbers of Subjects Enrolled in Studies of Highly Variable Drugs
We investigated the numbers of subjects completing bioequivalence studies of highly variable drugs in the 2003–2005 data set, to test our hypothesis that, in order to have adequate power to demonstrate bioequivalence, studies of highly variable drugs must use higher numbers of subjects than studies of drugs with lower variability. Figure 3 and 4 present Box-Whisker plots showing the number of subjects completing acceptable bioequivalence studies when the RMSE was either ≥0.3 (high variability) or <0.3 (lower variability) for Cmax and AUC0-t, respectively. We observed that, generally, studies of highly variable drugs (RMSE ≥ 0.3) used more subjects than studies of drugs with lower variability (RMSE < 0.3). For drugs that were highly variable in Cmax, the number of study subjects ranged from 18 to 134, with an average of 46 subjects/study. For drugs with lower variability in Cmax, the number of study subjects ranged from 12 to 113, with an average of 31 subjects/study. For drugs that were highly variable in AUC0-t, the number of study subjects ranged from 24 to 134, with an average of 55 subjects/study. For drugs with lower variability in AUC0-t, the number of study subjects ranged from 12 to 113, with an average of 32 subjects/study.
Fig. 3.

The number of subjects used in bioequivalence studies in which the C max RMSE < 0.3 (lower variability drug group) compared to the number of subjects used in bioequivalence studies in which the C max RMSE ≥ 0.3 (highly variable drug group). The mean number of subjects and distribution of data about the mean for each of the two drug variability groups are depicted via Box-and-Whisker plots
Fig. 4.

The number of subjects used in bioequivalence studies in which the AUC0-t RMSE < 0.3 (lower variability drug group) compared to the number of subjects used in bioequivalence studies in which the AUC0-t RMSE ≥ 0.3 (highly variable drug group). The mean number of subjects and distribution of data about the mean for each of the two drug variability groups are depicted via Box-and-Whisker plots
As shown in Figs. 3 and 4, some bioequivalence studies of drugs with lower variability (RMSE < 0.3) used large numbers of subjects. There appear to be several explanations for this observation. Some lower variability drugs were components in combination products which included a highly variable drug. In these cases, it seems likely that the investigators powered the study to show bioequivalence for the more highly variable of the two or more active ingredients. Some of the lower variability drugs fell into the category of “borderline” highly variable drugs. As previously stated, we defined borderline highly variable drugs as those for which, in any one bioequivalence study, the RMSE was either slightly greater than or slightly less than 0.3, and averaged approximately 0.3 across all bioequivalence studies of that drug. Thus, for borderline drugs, the investigators may have anticipated high variability (RMSE ≥ 0.3) and powered the bioequivalence study accordingly. For some studies of lower variability drugs, it was not clear why the investigators used large numbers of subjects, in particular because other studies of the same drug conducted by different investigators used smaller numbers of subjects. With these exceptions, in general, our data confirmed the hypothesis that higher numbers of subjects are needed to show that highly variable generic drugs are bioequivalent to their corresponding reference drug products.
Investigation of the Degree of Variability of Drugs Categorized as Highly Variable
We investigated the degree of variability in bioequivalence parameters in the 111 bioequivalence studies of highly variable drugs. Fig. 5 shows ranges of RMSE values, from ≥0.30 to <0.60, for the 111 bioequivalence studies of highly variable drugs reviewed by the DBE from 2003–2005. In approximately 50% of these studies, the RMSE values ranged from >0.3 to 0.35 for both Cmax and AUC0-t. The RMSE values ranged from >0.35 to 0.4 for Cmax and AUC0-t in approximately 30% and 17% of the studies, respectively. The highest RMSE value observed ranged from 0.55 to 0.60, for Cmax in one bioequivalence study. Thus, overall, in most of the bioequivalence studies of highly variable drugs reviewed by the DBE from 2003–2005, the RMSE values ranged from 0.30 to 0.40 for both Cmax and AUC0-t, suggesting that the high variability was relatively modest. It is not known why generic applicants submitted relatively few acceptable bioequivalence studies of highly variable drugs with RMSE values >0.4 (corresponding to within-subject variability of approximately 40%). It is possible that there are very few drug substances or drug products with variability this high. Another possible explanation is that generic drug applicants submit fewer ANDAs for drugs with very high variability because, as variability in bioequivalence parameters increases, the study size must also increase, with the result that bioequivalence studies of such drug products become too expensive.
Fig. 5.

The degree of variability in C max and AUC0-t observed in bioequivalence studies of highly variable drugs, reviewed at the Division of Bioequivalence in the Office of Generic Drugs, CDER/FDA, from 2003–2005. We determined the number of bioequivalence studies in which the RMSE values of C max or AUC0-t ranged from ≥0.30–0.35, >0.35–0.40, >0.40–0.45, >0.45–0.50, >0.50–0.55, and >0.55–0.60. The numbers of bioequivalence studies within each RMSE value range are expressed as a percentage of the total number of bioequivalence studies of highly variable drugs (C max or AUC0-t ≥ 0.3) reviewed from 2003–2005. The percentages of studies falling within each C max RMSE range is represented by light blue bars. The percentages of studies falling within each AUC0-t RMSE range is represented by dark blue bars
Determination of Whether High Variability in Bioequivalence Parameters Was Consistent
We further classified drugs for which the RMSE for Cmax and/or AUC0-t ≥ 0.3 as consistently highly variable, borderline highly variable, or inconsistently highly variable. Table II shows the number of bioequivalence studies, drug products, and drugs corresponding to consistently highly variable, borderline highly variable, or inconsistently highly variable. As shown in Table II, combined, the consistently highly variable and borderline highly variable drugs totaled 35, representing 61% (35/57) of all the highly variable drugs reviewed during 2003–2005.
Table II.
Classification of Variability in Bioequivalence Parameters of Drugs Reviewed by the Division of Bioequivalence in the Office of Generic Drugs from 2003–2005
| Description | Bioequivalence Studies | Different Drug Products | Different Drugs | |||
|---|---|---|---|---|---|---|
| Number | % of Total | Number | % of Total | Number | % of Total | |
| Consistently highly variable drugs | 73 | 66 | 62 | 61 | 29 | 51 |
| Borderline highly variable drugs | 12 | 11 | 10 | 10 | 6 | 11 |
| Inconsistently highly variable drugs | 26 | 23 | 29 | 29 | 22 | 39 |
| Total for which C max and/or AUC0-t ≥ 0.3 | 111 | 100 | 101 | 100 | 57 | 100 |
Since the borderline highly variable drugs consistently had Cmax and/or AUC0-t RMSE values either slightly greater than or less than 0.3, we concluded that the borderline high variability occurred consistently. Therefore, for the purpose of evaluating drug substance characteristics, we included borderline highly variable drugs with consistently highly variable drugs.
Identification of Drug Substance Characteristics Contributing to High Variability
We hypothesized that, for the consistently and borderline highly variable drugs, characteristics of the drug substance contributed significantly to the high variability. To validate this hypothesis, we evaluated physicochemical, absorption, and metabolism characteristics of each of these 35 (29 consistent and 6 borderline highly variable) drugs. As shown in Table III, we observed that one had low aqueous solubility, one was acid labile, two had low oral bioavailability (less than 1% of an oral dose), for six food increased variability, one was given by the subcutaneous route of administration in its pivotal bioequivalence studies, and 29 (83% of these 35 drugs) were subject to extensive first pass metabolism (by contrast, about 21% of the 123 drugs with RMSE values <0.3 were subject to extensive first pass metabolism). As these are properties of, or factors influencing, the disposition of the drug substance, we concluded that 61% of the highly variable drugs reviewed in 2003–2005 were likely highly variable due to drug substance characteristics.
Table III.
Characteristics of highly variable drug substances reviewed by the Office of Generic Drugs in 2003–2005
| Drug Therapeutic Class | RMSE Range | Drug Substance has Poor Aqueous Solubility | Drug Substances is Acid Labile | Low Oral Bioavailability | Food Increases Variability | Undergoes Extensive Presystemic Metabolism | Given by Subcutaneous Route |
|---|---|---|---|---|---|---|---|
| ACEa inhibitor 1b | 0.30 | √ | |||||
| ACE inhibitor 2b | 0.30–0.40 | √ | |||||
| ACE inhibitor 3b | 0.30–0.40 | √ | |||||
| ACE inhibitor 4b | 0.30–0.40 | √ | |||||
| ACE inhibitor 5b | 0.30 | √ | |||||
| Alpha1 agonistb | 0.30 | √ | |||||
| Angiotensin II receptor antagonist 1b | 0.30–0.40 | √ | √ | ||||
| Antiarrhythmic | 0.30–0.40 | √ | |||||
| Antibiotic 1 | 0.30–0.40 | √ | |||||
| Antibiotic 2 | 0.30 | √ | |||||
| Anticonvulsantb | 0.30–0.40 | √ | |||||
| Antineoplastic agent | 0.40–0.50 | √ | |||||
| Antiplatelet agent | 0.30–0.40 | √ | |||||
| Antispasmodic agent | 0.30–0.40 | √ | |||||
| Bisphosphonate 1 | 0.40–0.50 | √ | |||||
| Bisphosphonate 2 | 0.40–0.50 | √ | |||||
| Calcium channel blocker 1 | 0.30–0.40 | √ | |||||
| Calcium channel blocker 2 | 0.30–0.40 | √ | |||||
| Dopamine receptor agonist | 0.30–0.40 | √ | |||||
| H1-receptor antagonist | 0.30–0.40 | √ | |||||
| HIV-1c protease inhibitor | 0.30–0.40 | √ | √ | ||||
| HMG-CoA reductased inhibitor 1 | 0.30–0.40 | √ | |||||
| HMG-CoA reductase inhibitor 2b | 0.30–0.40 | √ | |||||
| HMG-CoA reductase inhibitor 3 | 0.30–0.40 | √ | |||||
| 5-Hydroxytryptamine1A receptor agonist | 0.30–0.40 | √ | √ | ||||
| 5-Hydroxytryptamine1B/1D receptor agonist | 0.30–0.40 | √ | |||||
| Peptide hormone analog 1 | 0.30–0.40 | √ | |||||
| Platelet aggregation inhibitorb | 0.30–0.40 | √ | √ | ||||
| Prostaglandin E analogb | 0.30–0.40 | √ | |||||
| Reverse transcriptase inhibitor | 0.30–0.40 | √ | |||||
| Reverse transcriptase inhibitor | 0.30 | √ | |||||
| Skeletal muscle relaxant 1 | 0.40–0.50 | √ | √ | ||||
| Skeletal muscle relaxant 2 | 0.30–0.40 | √ | |||||
| Synthetic androgen | 0.30–0.40 | √ | |||||
| Synthetic Vitamin D analog | 0.30 | √ |
The borderline highly variable drugs are indicted by blue lettering. We define borderline highly variable drugs as those drugs for which, in any one bioequivalence study, the RMSE was either slightly less than or slightly greater than 0.3, with an average of 0.3 across all bioequivalence studies of the drug
aAngiotensin converting enzyme
bThe parent prodrug is inactive
c3-Hydroxy-3-methylglutaryl-coenzyme A reductase
dHuman immunodeficiency virus
Notably, several drugs in each of the following classes were in the consistent and borderline highly variable groups: angiotensin converting enzyme (ACE) inhibitors, calcium channel blockers, 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) inhibitors, and bisphosphonates. All of the ACE inhibitors reviewed during the 2003–2005 period are inactive prodrugs that undergo extensive first-pass metabolism. The calcium channel blockers and HMG-CoA inhibitors reviewed during this period are also known to undergo extensive first pass metabolism. The bisphosphonate drugs reviewed during this period are reported to have absolute oral bioavailability averaging less than 1%. Thus, for some potential generic drug products, it may be possible to predict whether variability in bioequivalence parameters will be high based on what is known about the physicochemical and dispositional characteristics of the drug class in general.
Identification of Drug Product Characteristics Contributing to Inconsistent High Variability
About 40% (22/57) of the drugs that we classified as highly variable demonstrated such high variability only inconsistently. For this class of drug products, the RMSE values of AUC and/or Cmax were <0.3 in most of the bioequivalence studies. Table IV shows, for these inconsistently highly variable drug products, the total number of ANDAs submitted, the total number of bioequivalence studies conducted, the number of bioequivalence studies in which the RMSE for Cmax and/or AUC0-t ≥ 0.3, and the number of studies in which Cmax and/or AUC0-t was <0.3. In most cases, for each of these drugs, the number of studies in which the RMSE values <0.3 was several-fold greater than the number of studies in which the RMSE values were ≥0.3. We speculated that possible factors contributing to this inconsistent variability could be the manner in which the in vivo study was conducted or drug product formulation effects.
Table IV.
Incidence of Sporadic High Variability in Bioequivalence Parameters in Studies of Generic Drug Products
| Drug ID | Total Number of ANDAs Submitted | Total Number of Studies | Number of Studies in Which C max and/or AUC RMSE ≥ 0.3 | Number of Studies in Which C max and/or AUC RMSE < 0.3 |
|---|---|---|---|---|
| 1 | 6 | 8 | 1 | 7 |
| 2 | 2 | 4 | 1 | 3 |
| 3 | 2 | 6 | 1 | 5 |
| 4 | 4 | 8 | 1 | 7 |
| 5 | 2 | 3 | 1 | 2 |
| 6 | 8 | 18 | 2 | 16 |
| 7 | 2 | 5 | 1 | 4 |
| 8 | 4 | 10 | 1 | 9 |
| 9 | 5 | 5 | 1 | 4 |
| 10 | 2 | 2 | 1 | 1 |
| 11 | 3 | 3 | 1 | 2 |
| 12 | 2 | 3 | 2 | 1 |
| 13 | 17 | 35 | 2 | 33 |
| 14 | 2 | 5 | 1 | 4 |
| 15 | 2 | 4 | 1 | 3 |
| 16 | 6 | 12 | 2 | 10 |
| 17 | 4 | 8 | 2 | 6 |
| 18 | 3 | 9 | 1 | 8 |
| 19 | 4 | 8 | 2 | 6 |
| 20 | 8 | 14 | 1 | 13 |
| 21 | 3 | 6 | 1 | 5 |
| 22 | 5 | 10 | 4 | 6 |
Bioequivalence Study Performance and High Variability
We were unable to show that there was a relationship between high variability and either sampling times in the in vivo studies or assay limits of quantitation in the bioanalytical studies. We found that 20 bioanalytical CROs conducted studies in which the bioequivalence parameters of these drugs showed inconsistent high variability. However, each of these CROs also conducted a number of studies in which these same drugs showed lower variability in bioequivalence parameters. Most likely, we could not identify definitive trends due to the relatively small number of studies showing high variability (29/186 studies conducted at 20 CROs). Thus, we were unable to determine factors related to the conduct of either the in vivo or bioanalytical study that could be influencing the degree of variability in bioequivalence parameters.
Relationship of Variability in Drug Product Dissolution Rate In Vitro to Variability in Bioequivalence Parameters In Vivo
To explore the possibility that drug product formulation contributed to high variability, we related dissolution performance variability to bioequivalence parameter variability by comparing the rank orders of dissolution profile % CV values with the rank orders of RMSE values within each inconsistently highly variable drug product line. Table V shows these rankings for the 22 inconsistently highly variable drugs. Based on these comparisons, for 11/22 drugs, the highest variability in dissolution profiles correlated to the highest RMSE value. There was no correlation between dissolution profile variability and in vivo variability for the other 11 drugs. These results suggest that, for 50% of the inconsistently highly variable drugs (representing 19.5% of all the highly variable drugs evaluated by the OGD from 2003–2005), drug formulation performance could be contributing to the high variability. These results also suggest that, for some drug formulations, variability in dissolution performance may be predictive of variability in bioequivalence parameters. Future studies will provide a more detailed analysis of the relationship between dissolution performance and bioequivalence parameter variability.
Table V.
Comparison of In Vitro Dissolution Profile Variability with In Vivo Study Variability for Generic Drugs Having Inconsistent High Variability
| Drug No. | Drug Product No. | Rank by Dissolution % CV | Rank by Average BE Study RMSE | Does Product with Highest Dissolution % CV also have Highest RMSE in BE Studies |
|---|---|---|---|---|
| 1 | 1A | 1 | 1 | Y |
| 1B | 2 | 2 | ||
| 1C | 3 | 3 | ||
| 2 | 2A | 1 | 1 | Y |
| 2B | 2 | 2 | ||
| 3 | 3A | 1 | 1 | Y |
| 3B | 2 | 2 | ||
| 4 | 4A | 1 | 1 | Y |
| 4B | 2 | 2 | ||
| 5 | 5A | Data not available | 1 | N/A |
| 5B | Data not available | 2 | ||
| 6 | 6A | 1 | 2 | N |
| 6B | 2 | 1 | ||
| 6C | 3 | 4 | ||
| 6D | 4 | 6 | ||
| 6E | 5 | 3 | ||
| 6F | 6 | 5 | ||
| 7 | 7A | 1 | 1 | Y |
| 7B | 2 | 2 | ||
| 8 | 8A | 1 | 3 | N |
| 8B | 2 | 1 | ||
| 8C | 3 | 2 | ||
| 9 | 9A | 1 | 1 | Y |
| 9B | 2 | 3 | ||
| 9C | 3 | 5 | ||
| 9D | 4 | 2 | ||
| 9E | 5 | 4 | ||
| 10 | 10A | 1 | 2 | N |
| 10B | 2 | 1 | ||
| 11 | 11A | 1 | 1 | Y |
| 11B | 2 | 2 | ||
| 12 | 12A | 1 | 2 | N |
| 12B | 2 | 1 | ||
| 13 | 13A | 1 | 14 | N |
| 13B | 3 | 12 | ||
| 13C | 4 | 2 | ||
| 13D | 5 | 11 | ||
| 13E | 6 | 16 | ||
| 13F | 7 | 1 | ||
| 13G | 8 | 5 | ||
| 13H | 9 | 13 | ||
| 13I | 10 | 9 | ||
| 13J | 11 | 15 | ||
| 13K | 12 | 8 | ||
| 13L | 13 | 4 | ||
| 13M | 14 | 3 | ||
| 13N | 15 | 7 | ||
| 13O | 16 | 6 | ||
| 13P | 17 | 10 | ||
| 14 | 14A | 1 | 1 | Y |
| 14B | 2 | 2 | ||
| 15 | 15A | 1 | 1 | Y |
| 15B | 2 | 2 | ||
| 16 | 16A | 1 | 2 | N |
| 16B | 2 | 1 | ||
| 17 | 17A | 1 | 1 | Y |
| 17B | 2 | 2 | ||
| 18 | 18A | 1 | 3 | N |
| 18B | 2 | 1 | ||
| 18C | 3 | 2 | ||
| 19 | 19A | 1 | 1 | Y |
| 19B | 2 | 4 | ||
| 19C | 3 | 2 | ||
| 19D | 4 | 3 | ||
| 20 | 20A | 1 | 3 | N |
| 20B | 2 | 2 | ||
| 20C | 3 | 4 | ||
| 20D | 4 | 1 | ||
| 21 | 21A | 1 | 3 | N |
| 21B | 2 | 2 | ||
| 21C | 3 | 1 | ||
| 22 | 22A | 1 | 4 | N |
| 22B | 2 | 3 | ||
| 22C | 3 | 1 | ||
| 22D | 4 | 5 | ||
| 22E | 5 | 2 |
1 corresponds to the highest variability
CONCLUSION
We confirmed that bioequivalence studies of highly variable drugs submitted to the OGD from 2003–2005 generally enrolled more study subjects than studies of drugs with lower variability in the bioequivalence parameters Cmax and AUC. Most likely, 61% of the highly variable drugs evaluated by OGD during this time were highly variable due to drug substance characteristics influencing variability in drug product rate and/or extent of absorption. For 19.5% of the highly variable drugs, the observation that high dissolution performance variability corresponded to high variability in bioequivalence parameters suggested that the high variability could be due to drug product formulation. It was not possible to identify factors contributing to the variability in the remaining 19.5% of the highly variable drugs.
Acknowledgments
The authors thank LCDR Sheryl D. Gunther, Pharm.D., for her assistance in collecting data for this study.
Footnotes
The views expressed in this paper are those of the authors and do not necessarily represent the policy of the Food and Drug Administration.
References
- 1.L. X. Yu. Bioequivalence of highly variable drugs: issues and challenges. Advisory Committee for Pharmaceutical Sciences Meeting Transcript, April 14, 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.pdf (accessed 8/22/07).
- 2.Title 21, Code of Federal Regulations. Section 320.1. U.S. Government Printing Office, Washington, D.C., revised 2008.
- 3.U.S. Dept. of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Considerations. March 2003, BP. Revision 1. http://www.fda.gov/cder/guidance/5356fnl.pdf (accessed 8/13/07).
- 4.U.S. Dept. of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Statistical Approaches to Establishing Bioequivalence, January 2001, BP. http://www.fda.gov/cder/guidance/3616fnl.pdf (accessed 8/13/07).
- 5.C. E. Diliberti. Why bioequivalence of highly variable drugs is an issue. Advisory Committee for Pharmaceutical Sciences Meeting Transcript, April 14, 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.pdf (accessed 8/12/07).
- 6.Midha K. K., Rawson M. J., Hubbard J. W. The bioequivalence of highly variable drugs and drug products. Int. J. Clin. Pharmacol. Ther. 2005;43:485–498. doi: 10.5414/cpp43485. [DOI] [PubMed] [Google Scholar]
- 7.El-Tahtawy A. A., Tozer T. N., Harrison F., Lesko L., Williams R. Evaluation of bioequivalence of highly variable drugs using clinical trial simulations. II: comparison of single and multiple-dose trials using AUC and Cmax. Pharm. Res. 1998;15:98–104. doi: 10.1023/A:1011961006297. [DOI] [PubMed] [Google Scholar]
- 8.Tothfalusi L., Endrenyi L. Limits for the scaled average bioequivalence of highly variable drugs and drug products. Pharm. Res. 2003;20:382–389. doi: 10.1023/A:1022695819135. [DOI] [PubMed] [Google Scholar]
- 9.Haidar S. H., Davit B., Chen M. L., Conner D., Lee L. M., Li Q. H., Lionberger R., Makhlouf F., Patel D., Schuirmann D. J., Yu L. X. Bioequivalence approaches to highly variable drugs and drug products. Pharm. Res. 2008;25:237–241. doi: 10.1007/s11095-007-9434-x. [DOI] [PubMed] [Google Scholar]
- 10.S. H. Haidar. Evaluation of a scaling approach for highly variable drugs. Advisory Committee for Pharmaceutical Sciences Meeting Transcript, October 6, 2006. http://www.fda.gov/ohrms/dockets/ac/06/transcripts/2006–421t2–01.pdf (accessed 8/12/07).
- 11.B. M. Davit. Highly variable drugs – bioequivalence issues: FDA proposal under consideration. Advisory Committee for Pharmaceutical Sciences Meeting Transcript, October 6, 2006. http://www.fda.gov/ohrms/dockets/ac/06/transcripts/2006–421t2–01.pdf (accessed 8/12/07).
- 12.G. L. Amidon. Sources of variability: physicochemical and gastrointestinal. Advisory Committee for Pharmaceutical Sciences Meeting Transcript, April 14, 2004. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4034T2.pdf (accessed 8/13/07).
- 13.L. Z. Benet. Why highly variable drugs are safer. Advisory Committee for Pharmaceutical Sciences Meeting Transcript, October 6, 2006. http://www.fda.gov/ohrms/dockets/ac/06/transcripts/2006–421t2–01.pdf (accessed 8/12/07).