

Abstract
In an effort to develop novel and more potent therapies to treat tuberculosis, a new class of chemical agents, nitrofuranylamides, is being developed. The present study examines biopharmaceutic properties and preclinical pharmacokinetics of nitrofuranylamides at early stages of drug discovery to accelerate the optimization of leads into development candidates. The first tested compound, Lee 562, had high anti-tuberculosis activity in vitro, but exhibited poor metabolic stability resulting in a high systemic clearance, a short elimination half-life and low oral bioavailability in vivo in rats. Thus, two follow-up compounds were designed and tested that included structural modifications for increased metabolic stability. Both compounds showed improved metabolic stability compared to Lee 562, with Lee 878 being much more stable than Lee 952. As a consequence, the oral bioavailability of Lee 878 reached ~27% compared to 16% for the other two compounds. This observation prompted us to select compounds based on metabolic stability screening and a new set of nine compounds with high in vitro activity were tested for metabolic stability. The most stable compound in the assay, Lee 1106 was selected for further pharmacokinetic evaluation in rats. Surprisingly, Lee 1106 exhibited poor oral bioavailability, 4.6%. Biopharmaceutic evaluation of the compound showed that the compound has poor aqueous solubility and a high clogP. Based on these results, a screening paradigm was developed for optimization of the nitrofuranylamide lead compounds in a timely and cost-effective manner that might also be applicable to other classes of anti-infective drugs.
Key words: anti-infectives, lead optimization, metabolic stability, pharmacokinetics, preclinical drug development, tuberculosis
INTRODUCTION
Mycobacterium tuberculosis (M.tb) is the causative agent of tuberculosis (TB) in humans. As indicated by World Health Organization statistics, one-third of the world population is currently infected with TB, and it is anticipated that close to 10% of these infected individuals develop active TB at some point in their lifetime. TB is the second leading cause of infectious disease mortality in the world with approximately 2 to 3 million deaths per year (1). Association of TB with HIV has proven to be fatal; a quarter of a million TB deaths are HIV infection associated (2). Current pharmacotherapeutic regimens require treatment of TB with multiple drugs, including isoniazid, rifampicin, ethambutol and pyrazinamide for a period of six months. Treatment of drug resistant TB is more difficult requiring the usage of second-line agents that are associated with increased toxicity (3). Adherence to prolonged treatment regimens required to kill persistent or latent bacteria and thus clear the infection is often poor, leading to treatment failures and development of resistance to therapy. Recently, the emergence of extensively drug resistant tuberculosis (XDR-TB) (4) has raised concerns that we may lose control over the spread of TB. Though currently there are intensive efforts to develop new TB therapies, it is worth reflecting that no new major classes of drugs have been approved for the treatment of TB since the introduction of the rifamycins into the market in 1971 (5). Thus, the availability of more potent antibiotics that could effectively reduce the duration of therapy against M.tb is highly desirable.
Besides good in vivo activity, an ideal TB drug should have minimal side effects, high oral bioavailability, and a favorable pharmacokinetic profile, i.e., linear pharmacokinetics and a long terminal half-life that would allow once-a-day dosing with predictable and reproducible systemic exposure, and compatibility with existing HIV medications for joint therapy. In an effort to develop new and more potent therapies to treat TB, a novel class of anti-infectives with high in vitro activity against M.tb, nitrofuranylamides, has recently been characterized by our group (6–9).
The main objective of the current work is to gain an understanding of the preclinical pharmacokinetics of the nitrofuranylamide series of compounds, and to improve the pharmacokinetic properties of these compounds via structural modification. Because optimization of pharmacokinetic properties is frequently a rate limiting step in the preclinical development of anti-tuberculosis agents (10–12), the pharmacokinetically-guided lead optimization used in this publication may also be useful for screening other compounds in early drug discovery projects.
MATERIALS AND METHODS
Chemicals and Reagents
All tested nitrofuranylamides were synthesized in the laboratory of Dr. R.E. Lee at the University of Tennessee Health Science Center, Memphis, TN, including Lee 562 (5-nitro-furan-2-carboxylic acid [6-(4-benzyl-piperazin-1-yl)-pyridin-3-ylmethyl]-amide), Lee 878 (1-benzyl-4-{4-[(5R)-3-(5-nitro-2-furyl)-4, 5-dihydroisoxazol-5-yl] phenyl} piperazine), Lee 952 (N-methyl-N-[4-(2-morpholin-4-ylethoxy) benzyl]-5-nitro-2-furamide), and Lee 1106 (1-{4-[3-(5-nitro-2-furyl)-4, 5-dihydroisoxazol-5-yl] phenyl}-4-[4-(trifluoromethoxy) phenoxy] piperidine) (Fig. 1). Details on the synthesis of the test compounds are described elsewhere (8,9,13). Acetonitrile, HPLC grade water, acetic acid and ammonium acetate were purchased from Fisher Scientific (Pittsburgh, PA). Drug free rat plasma was purchased from Aleken Biologicals (Nash, TX).
Fig. 1.

Chemical structures of Lee 562, Lee 878, Lee 952 and Lee 1106. The boxes and circles depict important structural differences between the first generation compound Lee 562, the second generation compounds Lee 878 and Lee952, and the third generation compound Lee 1106
Pharmacokinetic Studies in Rats
Catheterized male Sprague-Dawley rats (jugular vein alone for oral study and jugular vein and femoral vein for intravenous study) from Harlan Bioscience (Indianapolis, IN), weighing approximately 250g, were kept on a 12h light/dark cycle with food and water available ad libitum. Groups of rats (n = 4–5) received either an intravenous (IV) or oral dose of a test compound at a dose level of 10mg/kg or 100mg/kg, respectively. For oral administrations, the animals were fasted overnight and until 4h after administration of test compound. Serial blood samples (~250μL) were collected predose and at predetermined time points postdose. Plasma was separated immediately by centrifugation (3,000×g for 10min at 4°C) and stored at −80°C until analysis. Feces and urine specimens were collected for Lee 562 and Lee 1106 for a period of 24h after administration. The study protocol was approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center.
Microsomal Incubations
Microsomal metabolic stability of the compounds was assessed in pooled rat liver microsomal preparations (Cellzdirect, Austin, TX) by monitoring disappearance of the parent compound over an incubation period of 90min. The percentage of intact parent compound in the samples was estimated by comparing analyte concentrations before and after incubation using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay. Reactions were started by adding 25μL of microsomal protein solution (10mg/ml) to 25μL of test compound (50μM) and 200μL of NADPH regenerating solution (1.3mM NADP+, 3.3mM glucose-6-phosphate, 3.3mM MgCl2 and 1unit/mL glucose-6-phosphate dehydrogenase in pH7.4 phosphate buffer solution). The mixture was incubated at 37°C and samples were taken at 0, 15, 30, 45, 60 and 90min. The reaction was terminated by the addition of three volumes of ice-cold acetonitrile containing an internal standard, Lee 563. Controls were treated in a similar manner using heat-denatured microsomes. Samples were centrifuged at 3,000×g for 10min at 4°C and the supernatants were removed for analysis. An aliquot (10μL) was analyzed by LC-MS/MS.
Solubility and Lipophilicity
Aqueous solubility of the compounds was estimated at two different pH values using a miniaturized shake-flask method (14). Approximately 1mg of the test compound was shaken with 500μL of either pH6.0 or pH7.4 buffer in a glass vial at 400rpm and room temperature for 24h. The resulting mixture was centrifuged and the concentration in the supernatant was determined by LC-MS/MS. The octanol-water partition coefficient (clogP) of the compounds as measure of their lipophilicity was calculated using ChemDraw Ultra version 7.0 (CambridgeSoft Corporation, Cambridge, MA).
Protein Binding
Protein binding of the compounds was determined using equilibrium dialysis. Biologically relevant concentrations of test compound were prepared (low and high) in rat plasma. 200μL of the plasma sample was placed in the central chamber and 350μL of blank isotonic phosphate buffer, pH7.4 in the peripheral chamber of a dialysis device (MW cutoff, 6,000–8,000, RED® device, Pierce Biotechnology Inc, Rockford, IL). The chambers were covered with a seal and incubated at 37°C for 4–6h at a rocking speed of 100rpm. At the end of incubation, the volumes of plasma and recipient buffer were measured to identify and account for volume shift, if any. Aliquots of plasma and buffer were used to determine the drug concentration using an LC-MS/MS assay. The free fraction of the drug was calculated as ratio of the concentrations in the buffer and in plasma.
Plasma Concentration Measurements by LC-MS/MS
A calibration curve ranging from 1–500μg/L was constructed for each test compound by spiking the test compound into 50μL of blank rat plasma. All test compounds were found to be stable in acetonitrile/5mM ammonium acetate buffer (50:50, v/v). A structural analogue of the test compounds, Lee 563 (ethyl 4-(5-{[(5-nitro-2-furoyl) amino] methyl} pyridin-2-yl) piperazine-1-carboxylate), was added as internal standard to all calibration standards and all plasma specimens. Plasma proteins were precipitated by the addition of three volumes of acetonitrile. The samples were centrifuged at 3,000×g for 10min at 4°C and the supernatants were removed for analysis.
Chromatographic separations were carried out using a Shimadzu liquid chromatograph (Shimadzu Corporation, USA) consisting of two pumps, online degasser, system controller and a CTC Leap auto sampler (Leap Technologies, Carrboro, NC). A gradient of acetonitrile and 10mM ammonium acetate was used at a flow rate of 0.3mL/min. A Phenomenex® Luna, 3μ C18 (2), 50 × 2mm column (Phenomenex, Torrance, CA) protected with a guard column was used for the separation. Ten microliters of sample was injected onto the column and the eluate was led directly into an API 3000 triple-quadrupole mass spectrometer (Applied Biosystems ABI/MDS-Sciex, Foster City, CA) equipped with an electrospray ion source. The quadrupoles were operated in the positive ion mode. The resulting multiple reaction monitoring chromatograms were used for quantification using Analyst software version 1.4.1 (Applied Biosystems ABI/MDS-Sciex, Foster City, CA).
Pharmacokinetic Data Analysis
Plasma concentration-time data were analyzed by non-compartmental analysis using WinNonlin 5.0.1 (Pharsight Corporation, Mountain View, CA). The area under the plasma concentration–time curve from time 0 to infinity (AUCinf) was calculated by the trapezoidal rule with extrapolation to time infinity. The terminal half-life (t1/2) was calculated as 0.693/λz, where λz is the terminal phase rate constant. The plasma clearance (CL) was calculated using the equation 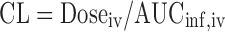 , where Doseiv and AUCinf, iv are the IV dose and corresponding area under the plasma concentration-time curve from time 0 to infinity, respectively. Volume of distribution based on terminal phase (Vz) was calculated using
, where Doseiv and AUCinf, iv are the IV dose and corresponding area under the plasma concentration-time curve from time 0 to infinity, respectively. Volume of distribution based on terminal phase (Vz) was calculated using 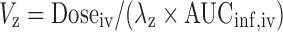 . The peak plasma concentration (Cmax) and the time when it occurred (tmax) in the oral dose group were obtained by visual inspection of the plasma concentration–time curves. Clearance after oral dose (CL/F) was calculated as
. The peak plasma concentration (Cmax) and the time when it occurred (tmax) in the oral dose group were obtained by visual inspection of the plasma concentration–time curves. Clearance after oral dose (CL/F) was calculated as 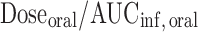 . Volume of distribution after oral dose (Vz/F) was calculated as
. Volume of distribution after oral dose (Vz/F) was calculated as 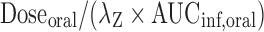 . Oral bioavailability (F) was calculated using
. Oral bioavailability (F) was calculated using 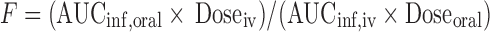 , where Doseoral, Doseiv, AUCinf, iv, and AUCinf, oral are the oral and IV dose and the corresponding areas under the plasma concentration-time curves from time 0 to infinity, respectively.
, where Doseoral, Doseiv, AUCinf, iv, and AUCinf, oral are the oral and IV dose and the corresponding areas under the plasma concentration-time curves from time 0 to infinity, respectively.
RESULTS
Pharmacokinetics of Lee 562
The first evaluated lead compound was Lee 562 (Fig. 1). The plasma concentration-time profiles of Lee 562 after single IV and oral administration are shown in panels a and b of Fig. 2, the derived pharmacokinetic parameters in Table I. Lee 562 appeared rapidly in plasma after oral administration with peak plasma levels (Cmax) observed in less than 15min after administration. There was a rapid decline in the plasma levels followed by a smaller second peak about 6h after the dose administration. The second peak in the profile after oral administration indicates the possibility of enterohepatic recirculation of the compound or erratic absorption. The compound was distributed extensively in the body with a volume of distribution of 28.9L/kg. A high clearance of 12.9L h−1 kg−1 indicates rapid elimination. The amount of compound excreted unchanged into urine or feces was less than 0.1% of the dose by both routes of administration suggesting that the high clearance is the result of extensive metabolism. The high clearance resulted in a short terminal half-life of 1.3hr after IV and 3.7h after oral administration, and an oral bioavailability of only 15.9%. To investigate the extent of metabolism, Lee 562 was incubated in rat liver microsomal preparations. The microsomal metabolism of Lee 562 was rapid and extensive, with nearly complete loss (>99.5%) of the parent compound within 90min of incubation.
Fig. 2.

Measured plasma concentration-time profiles (mean ± SD) after intravenous (10 mg/kg; a, c, e, g) or oral (100 mg/kg) (b, d, f, h) administration of nitrofuranylamides to rats (n = 5–6 per group): Lee 562 (a and b), Lee 878 (c and d), Lee 952 (e and f), Lee 1106 (g and h)
Table I.
Pharmacokinetic Parameters (Mean ± SD) of Lee 562, Lee 878, Lee 952 and Lee 1106
| Route | Compound | t (h) | AUCinf (μg h/L) | Volume of Distribution L/kg | Clearance (L h−1 kg−1) | % Dose Excreted | Bioavailability (%) | |
|---|---|---|---|---|---|---|---|---|
| Urine | Feces | |||||||
| IV | Lee 562 | 1.30 ± 0.89 | 956 ± 442 | 28.9 ± 29.4 | 12.9 ± 7.6 | 0.001 ± 0.0003 | 0.001 ± 0.001 | |
| Lee 878 | 2.63 ± 0.35 | 19,091 ± 1,724 | 2.00 ± 0.36 | 0.527 ± 0.05 | ND | ND | ||
| Lee 952 | 7.34 ± 2.27 | 1,846 ± 82.2 | 58.0 ± 20.1 | 5.42 ± 0.24 | ND | ND | ||
| Lee 1106 | 10.3 ± 1.41 | 22,423 ± 4,096 | 6.72 ± 1.10 | 0.456 ± 0.07 | 0.035 ± 0.01 | 0.14 ± 0.06 | ||
| Oral | Lee 562 | 3.69 ± 0.86 | 1,519 ± 458 | 386 ± 187 | 71.6 ± 23.2 | 0.009 ± 0.02 | 0.023 ± 0.02 | 15.9 ± 4.8 |
| Lee 878 | ND | 52,324 ± 14,834b | ND | 2.07 ± 0.71c | ND | ND | 27.4 ± 7.8c | |
| Lee 952 | 3.39 ± 1.16a | 2,936 ± 1,119 | 192 ± 127 | 37.7 ± 12.5 | ND | ND | 15.9 ± 6.1 | |
| Lee 1106 | 9.24 ± 1.71 | 10,230 ± 9,101 | 231 ± 174 | 16.0 ± 10.3 | 0.025 ± 0.02 | 11.8 ± 7.09 | 4.6 ± 4.1 | |
ND Not determined
aNot reliable determined due to limited log-linear phase
bAUC0–48 h
cAUC0–48 h was used for calculation of clearance and oral bioavailability
Microsomal Metabolic Stability of Second Generation Compounds Lee 878 and Lee 952
Metabolic stability of a compound is investigated to determine metabolic half-life and intrinsic clearance which can be major determinants for in vivo drug efficacy, and to aid in structural optimization of lead compounds in drug discovery settings (15). Analysis of the structure of Lee 562 led us to attribute its metabolic instability in part to the amide bond of the compound. Thus, two follow-up second generation compounds, Lee 878 and Lee 952, were tested that included structural modifications to improve metabolic stability. For Lee 878, the amide bond was replaced by an isoxazole ring; for Lee 952, the amide hydrogen was substituted with a methyl group (Fig. 1). The microsomal metabolism of Lee 952 was rapid and extensive, with >95% loss of the parent compound after 90min of incubation (4.3% remaining). The metabolic half-life appears to be less than 15min. The desired cutoff for metabolic stability in most preclinical programs is more than 30% of parent remaining stable after 90min of incubation (16). In contrast, Lee 878 exhibited acceptable metabolic stability under this criterion with 31.4% of the parent compound remaining stable at the end of incubation.
Pharmacokinetics of Second Generation Compounds Lee 878 and Lee 952
The plasma concentration-time profiles and pharmacokinetic parameters of Lee 878 and Lee 952 after single IV and oral administration to rats are shown in panels c–f of Fig. 2 and Table I, respectively. Oral absorption of Lee 878 was slow with peak plasma concentration (Cmax) occurring about 6h after administration. In contrast, oral absorption of Lee 952 was rapid with peaks observed within 15min of administration. Lee 952 distributed more extensively in the body than Lee 878 with a distribution volume of 58L/kg compared to 2.0L/kg. As expected from the microsomal metabolic stability studies, the mean plasma clearance was with 5.4L h−1 kg−1 compared to 0.53L h−1 kg−1 much larger for Lee 952 compared to Lee 878. Consequently, oral bioavailability of Lee 878 was found to be 27.4%, while Lee 952 exhibited poor oral bioavailability with only 15.9%. Since the terminal phase of the concentration-time profile was not captured after oral administration of Lee 878, AUC0–48h was used to determine oral bioavailability. Despite its substantially lower clearance, Lee 878 exhibited a much shorter terminal half-life of 2.6h after IV administration compared to Lee 952 with 7.3h. This can be explained with Lee 878’s substantially smaller distribution volume compared to Lee 952, which indicates less drug distribution to peripheral tissues and thus easier access of drug compound to elimination processes.
Metabolic Stability of Third Generation Compounds
Since the microsomal metabolic stability was found to be correlated with the oral bioavailability of Lee 562, Lee 878 and Lee 952, a new set of nine compounds within the nitrofuranylamide series was screened for metabolic stability. Chemical structures of the compounds and microsomal metabolic stability data are shown in Fig. 3. Lee 1106 exhibited superior metabolic stability with more than 70% of the parent compound remaining stable after 90min of incubation. Thus, Lee 1106 was selected for further pharmacokinetic evaluation in rats. The structural modifications of Lee 562 that lead to the other three compounds, Lee 878, Lee 952 and Lee 1106, are shown in Fig. 1.
Fig. 3.

Chemical structures of third generation nitrofuranylamides. The values provided in parenthesis indicate the metabolic stability in an in vitro microsomal preparation assay (percent parent compound remaining stable after 90 min of incubation)
Pharmacokinetics of the Third Generation Compound Lee 1106
The plasma concentration-time profiles of Lee 1106 after single IV and oral administration are shown in panels g and h of Fig. 2, the corresponding pharmacokinetic parameters in Table I. Absorption of Lee 1106 was slow, with peak plasma concentration (Cmax) observed 9h after administration. Oral bioavailability of the compound was 4.6%. Lee 1106 showed a distribution volume of 6.7L/kg. As expected from the metabolic stability data, the plasma clearance of the compound was low, 0.46L h−1 kg−1. The unchanged compound excreted in urine was less than 1% of the dose by both routes of administration, whereas about 0.1% and 12% of the dose was excreted unchanged in feces after IV and oral administration, respectively. The low plasma clearance of Lee 1106 resulted in a longer terminal half-life than observed in the other tested nitrofuranylamides, 10.3h after the IV and 9.24h after the oral administration.
Solubility Screening
Aqueous solubility is one of the most important properties for a compound intended to be developed as an orally active molecule. The solubility of the investigated compounds is presented in the Table II. A compound is conventionally classified as highly soluble when the largest dose of the compound is soluble in less than 250mL water over a pH range from 1.0 to 7.5 (17). Though there is no strict cutoff value for solubility available, solubility above 50μg/mL is arbitrarily considered acceptable for compound progression in most preclinical development programs (10). It is assumed that this level of solubility would never pose a problem in limiting bioavailability. Lee 562, Lee 878 and Lee 1106 did not pass this solubility requirement, but the solubility for Lee 952 clearly exceeds the cutoff value. Predicted octanol-water partition coefficient, clogP values for Lee 562, Lee 878, Lee 952 and Lee 1106 are shown in Table II. A graph showing the relationship between the clogP and the aqueous solubility of all the tested compounds is presented in Fig. 4a.
Table II.
Aqueous Solubility and Plasma Protein Binding of Lee 562, Lee 878, Lee 952 and Lee 1106
| Compound | Molecular Weight | MIC90 (μg/mL) | clogP | Solubility (mg/L) | % Protein Binding | |
|---|---|---|---|---|---|---|
| pH 6.0 | pH 7.4 | |||||
| Lee 562 | 421.4 | 0.0062 | 2.41 | 18.5 | 4.43 | 96.43 |
| Lee 878 | 432.5 | 0.00005 | 4.29 | 3.34 | 0.24 | 99.92 |
| Lee 952 | 389.4 | 0.09 | 1.91 | 1975 | 759 | 44.53 |
| Lee 1106 | 517.5 | 0.025 | 6.41 | BLQ | 2.67 | 99.99 |
MIC 90 Minimum inhibitory concentration to inhibit the growth of 90% of organisms determined against M. tuberculosis H37Rv using microbroth dilution in Middlebrook 7H9
BLQ Below the limit of quantification
Fig. 4.

Correlation between a clogP and aqueous solubility, b clogP and plasma protein binding in the investigated nitrofuranylamides. The plasma protein binding of the compounds Lee 992 and Lee 1053 could not be determined due to instability in plasma
Plasma Protein Binding
The compounds Lee 562, Lee 878 and Lee 1106 are extensively bound to plasma proteins with 96.4%, 99.9% and >99.9% binding, respectively (Table II). In contrast, Lee 952 was only 44.5% bound to plasma proteins. A graph showing relationship between clogP and the protein binding of all the tested compounds is presented in Fig. 4b.
DISCUSSION
The first tested lead compound, Lee 562, exhibited a high systemic clearance, short terminal half-life, and low oral bioavailability of 15.9%. These observations were further supported by the poor metabolic stability of Lee 562, which was attributed to its amide linkage and the benzyl piperazine side chain in the structure. Thus, two second generation follow-up compounds were tested that included structural modifications for increased metabolic stability. For Lee 878, the amide bond was replaced by an isoxazole ring; for Lee 952, the amide hydrogen was substituted with a methyl group and the benzyl piperazine was replaced with a solubilizing morpholino group. Both compounds showed improved metabolic stability in the rat microsome assay compared to Lee 562, with Lee 878 being much more stable than Lee 952. As expected, this in vitro observation translated into an increased in vivo stability (lower clearance) of Lee 878 compared to Lee 562 and Lee 952, with a 20- and 10-fold higher systemic exposure, respectively. As a consequence, oral bioavailability of Lee 878 reached ~27% compared to 16% for Lee 952 and Lee 562.
The low oral bioavailability of Lee 562 and Lee 952 was predictable, considering that the mean plasma clearance after IV administration was 12.9L h−1 kg−1 and 5.4L h−1 kg−1 respectively. Based on their extensive metabolism, their plasma clearance and a typical hepatic blood flow of 3.3L h−1 kg−1 in rats [18], the theoretical hepatic extraction ratio (ER) for these compounds could be estimated as 3.9 and 1.6 for Lee 562 and Lee 952, respectively. As the maximum physiologically possible hepatic ER is 1, i.e. complete removal and/or metabolic conversion of all drug presented to the liver, the calculated ERs for Lee 562 and Lee 952 exceeding 1 suggest the presence of extrahepatic metabolism and/or sequestration of drug into blood cells. Overall, the low oral bioavailability of these compounds could be attributed to high first-pass metabolism. For Lee 562, poor aqueous solubility apart from the first-pass inactivation could also be a contributing factor to its low oral bioavailability.
Similarly, assuming that all clearance of Lee 878 is hepatic, the hepatic ER for Lee 878 could be estimated as 0.16. Considering first-pass inactivation alone, this would suggest that the maximum oral bioavailability for Lee 878 could theoretically reach 84% [(1 − ER)×100] as opposed to the observed bioavailability of 27.4%. Possible reasons for this difference in bioavailability could be poor aqueous solubility, transporter-mediated efflux in the gastrointestinal membrane, and/or degradation/metabolism in the gastrointestinal tract.
The elimination half-life of Lee 878 was only 2.6h, mainly due to the surprisingly small volume of distribution of this compound. Considering the high clogP of 4.3 for Lee 878 and the corresponding low aqueous solubility, the limited volume of distribution of Lee 878 is most likely explained by its excessive binding (99.92%) to plasma proteins. A high plasma protein binding reduces the free, pharmacologically active concentrations of a drug and limits its tissue distribution. This might be the major reason why Lee 878 was found to have low in vivo efficacy in a mouse model of M.tb infection despite favorable systemic exposure, high metabolic stability and high in vitro activity (9).
Since the microsomal metabolic stability was found to be correlated with the oral bioavailability of the first and second generation compounds we tested, a new set of nine third generation compounds belonging to the same series was screened for microsomal metabolic stability to guide selection of a stable compound for pharmacokinetic evaluation.
As Lee 1106 exhibited superior metabolic stability, its pharmacokinetic properties were evaluated in vivo. The compound exhibited favorable pharmacokinetic properties such as a low systemic clearance and a long terminal half-life. However, oral bioavailability of Lee 1106 is poor despite a mean plasma clearance after IV administration of only 0.46L h−1 kg−1. Assuming that all clearance for Lee 1106 is mediated by the liver, the hepatic ER for Lee 1106 would be calculated as 0.14 and the maximum oral bioavailability based on first-pass inactivation alone would be 86%. However, Lee 1106 exhibited a very low oral bioavailability of 4.6%. Poor aqueous solubility of the compound could explain to some extent the observed low oral bioavailability of the compound, which might be overcome by optimizing the dosage form using solubilizing excipients (19). Further, possible degradation/metabolism and/or transporter-mediated efflux of the compound in the gastrointestinal tract could also contribute to this observation. In addition, poor aqueous solubility, high plasma protein binding and high clogP of Lee 1106 suggest hydrophobicity of the molecule. Lee 1106 has a molecular weight exceeding 500Da (517.5Da) and a clogP exceeding 5 (6.41), thereby clearly deviating from the Lipinski’s rule of five, a scoring function used to predict orally active molecules (20).
A closer look at the structures of the compounds suggests that Lee 878 and Lee 1106 have a larger number of aromatic rings imparting more hydrophobicity to the molecule which in turn decreases their aqueous solubility. Lee 952 has fewer aromatic rings in its structure and exhibits high aqueous solubility. As shown in Fig. 4a, aqueous solubility in our set of compounds was inversely correlated with clogP as measure of hydrophobicity (p = 0.004, Pearson correlation coefficient: −0.76). Similarly, protein binding was consistently above 96% with higher clogP values in all the tested compounds (Fig. 4b).
Based on these observations, aqueous solubility and metabolic stability are the most important determinants of oral bioavailability of the tested nitrofuranylamide compounds. Thus, an early evaluation of aqueous solubility and protein binding coupled with metabolic stability could be useful for predicting the oral bioavailability of novel nitrofuranylamide compounds. Accordingly, a screening paradigm was proposed for lead optimization of compounds of the nitrofuranylamide class based on our experiences in evaluating first and second generation compounds (Fig. 5). As a first step in this process, compounds will be designed for synthesis using in silico approaches such as quantitative structure activity relationships (QSAR) to predict antimicrobial efficacy. In the next step, a variety of in silico predictions (quantitative structure-property relationships) such as aqueous solubility, metabolic stability, protein binding and clogP will be used for rank-ordering the compounds for synthesis. The most promising compounds will then be synthesized and tested for in vitro activity against M.tb. Potent compounds resulting from the in vitro activity screen will be studied for aqueous solubility and in vitro pharmacokinetic properties such as metabolic stability and protein binding. The compounds with higher stability, higher aqueous solubility and lower protein binding will be selected for pharmacokinetic and bioavailability evaluation in rats. The compounds that exhibit high oral bioavailability and long elimination half-life in rats will be progressed into more expensive and time-consuming in vivo evaluations of activity in animal models such as mouse models of M.tb infection. The information obtained at each step will be used for updating the design of compounds for synthesis and the filtering used at each step will allow to concentrate development resources on a smaller number of more promising compounds which makes the lead optimization process more efficient.
Fig. 5.

Screening paradigm for lead optimization of anti-TB compounds
CONCLUSION
In summary, we determined the pharmacokinetic properties of key compounds in the nitrofuranylamide series and developed a screening paradigm that could be useful for the further optimization of leads in the group of anti-infectives against M.tb. This paradigm may also be useful for lead optimization of other series of anti-infectives.
Acknowledgements
This study was supported by research grant 1R01 AI 2415-01 by the National Institutes of Health.
References
- 1.Global Alliance for TB Drug Development. www.tballiance.org (2007).
- 2.World Health Organization. Fact sheet on Tuberculosis. Geneva: WHO (March, 2006).
- 3.Blumberg H. M., Burman W. J., E Chaisson R., et al. American thoracic society/centers for disease control and prevention/infectious diseases society of America: treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 2003;167(4):603–662. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 4.Center for Disease Control and Prevention. Morbidity and mortality weekly report. Atlanta: CDC (Apr 2006).
- 5.Burman W. J., Jones B. E. Treatment of HIV-related tuberculosis in the era of effective antiretroviral therapy. Am. J. Respir. Crit. Care Med. 2001;164(1):7–12. doi: 10.1164/ajrccm.164.1.2101133. [DOI] [PubMed] [Google Scholar]
- 6.Tangallapally R. P., Yendapally R., Lee R. E., et al. Synthesis and evaluation of nitrofuranylamides as novel antituberculosis agents. J. Med. Chem. 2004;47(21):5276–5283. doi: 10.1021/jm049972y. [DOI] [PubMed] [Google Scholar]
- 7.Tangallapally R. P., Yendapally R., Lee R. E., Lenaerts A. J., Lee R. E. Synthesis and evaluation of cyclic secondary amine substituted phenyl and benzyl nitrofuranyl amides as novel antituberculosis agents. J. Med. Chem. 2005;48(26):8261–8269. doi: 10.1021/jm050765n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tangallapally R. P., Lee R. E., Lenaerts A. J., Lee R. E. Synthesis of new and potent analogues of anti-tuberculosis agent 5-nitro-furan-2-carboxylic acid 4-(4-benzyl-piperazin-1-yl)-benzylamide with improved bioavailability. Bioorg. Med. Chem. Lett. 2006;16(10):2584–2589. doi: 10.1016/j.bmcl.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 9.Tangallapally R. P., Sun D., Rakesh R. , Budha N., Lee R. E., Lenaerts A. J., Meibohm B., Lee R. E. Discovery of novel isoxazolines as anti-tuberculosis agents. Bioorg. Med. Chem. Lett. 2007;17(23):6638–6642. doi: 10.1016/j.bmcl.2007.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caldwell G. W., Ritchie D. M., Masucci J. A., Hageman W., Yan Z. The new pre-preclinical paradigm: compound optimization in early and late phase drug discovery. Curr. Top Med. Chem. 2001;1(5):353–66. doi: 10.2174/1568026013394949. [DOI] [PubMed] [Google Scholar]
- 11.Laughon B. E. New tuberculosis drugs in development. Curr. Top. Med. Chem. 2007;7(5):463–473. doi: 10.2174/156802607780059736. [DOI] [PubMed] [Google Scholar]
- 12.Eddershaw P. J., Beresford A. P., Bayliss M. K. ADME/PK as part of a rational approach to drug discovery. Drug Discov. Today. 2000;5(9):409–414. doi: 10.1016/S1359-6446(00)01540-3. [DOI] [PubMed] [Google Scholar]
- 13.Tangallapally R. P., Yendapally R., Daniels A. J., Lee R. E., Lee R. E. Nitrofurans as novel anti-tuberculosis agents: identification, development and evaluation. Curr. Top. Med. Chem. 2007;7(5):509–526. doi: 10.2174/156802607780059772. [DOI] [PubMed] [Google Scholar]
- 14.Glomme A., Marz J., Dressman J. B. Comparison of a miniaturized shake-flask solubility method with automated potentiometric acid/base titrations and calculated solubilities. J. Pharm. Sci. 2005;94(1):1–16. doi: 10.1002/jps.20212. [DOI] [PubMed] [Google Scholar]
- 15.Thompson T. N. Optimization of metabolic stability as a goal of modern drug design. Med. Res. Rev. 2001;21(5):412–449. doi: 10.1002/med.1017. [DOI] [PubMed] [Google Scholar]
- 16.R. E. White. High-throughput screening in drug metabolism and pharmacokinetic support of drug discovery. Annu. Rev. Pharmacol. Toxicol40:133–157 (2000). [DOI] [PubMed]
- 17.Amidon G. L., Lennernas H., Shah V. P., Crison J. R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995;12(3):413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 18.Davies B., Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993;10(7):1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 19.Strickley R. G. Solubilizing excipients in oral and injectable formulations. Pharm. Res. 2004;21(2):201–30. doi: 10.1023/B:PHAM.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 20.Lipinski C. A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44(1):235–49. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]