

Abstract
Reaction phenotyping studies to identify specific enzymes involved in the metabolism of drug candidates are increasingly important in drug discovery efforts. Experimental approaches used for CYP reaction phenotyping include incubations with cDNA expressed CYP enzyme systems and incubations containing specific CYP enzyme inhibitors. Since both types of experiments present specific advantages as well as known drawbacks, these studies are generally viewed as complementary approaches. Although glucuronidation pathways are also known to present potential drug–drug interaction issues as well as challenges related to their polymorphic expression, reaction phenotyping approaches for glucuronidation are generally limited to cDNA expressed systems due to lack of availability of specific UGT inhibitors. This article presents a limited review of current approaches to reaction phenotyping studies used within the pharmaceutical industry.
Keywords: cytochrome P450, metabolism, reaction phenotyping, UDP-glucuronosyltransferase
INTRODUCTION
Several of the cytochrome P450 (CYP) enzymes involved in drug metabolism are known to exhibit polymorphic expression in human populations (most notably CYP2D6, CYP2C9 and CYP2C19), contributing to variable exposure levels for drugs metabolized by those enzymes (1–5). In addition, induction of metabolism may increase the clearance of therapeutic agents leading to sub-therapeutic exposures and lack of pharmacological effect. Alternatively, inhibition of metabolism may reduce clearance and lead to supra-therapeutic exposures resulting in undesired side-effects or toxicity. Such drug–drug interactions (DDI) in which one agent alters the exposure of a concomitantly administered drug through effects on CYP mediated metabolism are well documented (6,7). While such interactions generally tend to be viewed as potential liabilities, it has also been recognized that these interactions may present opportunities as well: witness the now-widespread practice of including a potent CYP3A4 inhibitor in the anti-HIV drug cocktails in order to increase exposures to the concomitant drugs, the so-called “ritonavir boost” (8,9).
In addition to CYP mediated metabolism, several other enzymes, the most significant of which are the flavin-containing monooxygenases (FMO), can mediate the oxidative metabolism of various drugs and xenobiotics. The metabolites generated by FMO enzymes are frequently the same as those generated by CYP enzymes; hence there is the need to clearly understand the underlying enzymology and biotransformation mechanisms. FMO1 has the broadest substrate acceptance, typically metabolizing large lipophilic compounds such as imipramine or orphenadrine, typically used as marker substrates (10).
In contrast to FMO1, FMO3 has the ability to selectively metabolize small amine-containing compounds such as trimethyl-amine (TMA), which is a useful probe substrate. Inclusion of FMO3 in a reaction phenotyping assessment is currently viewed as being of greater importance due to the causative relationship between a defective FMO3 gene (P153L mutation) and a condition known as Trimethylaminuria (fish-odor syndrome). This genetic disorder results from an inability to metabolize pungent smelling TMA via FMO3 to the odorless TMA N-oxide (11).
Less extensively studied, but potentially just as significant are polymorphisms and DDI involving other drug metabolism routes, such as UDP-glucuronosyltransferase (UGT) mediated glucuronidation pathways (12–14). In addition to the variability and DDI concerns outlined above for CYP enzymes, an additional concern with UGT is the potential for inhibition of UGT1A1, the isoform responsible for conjugation of bilirubin, since inhibition of that pathway could lead to reduction of bilirubin clearance, resulting in hyperbilirubinemia.
Whether viewed as a potential liability or a potential tool for exposure enhancement, prediction of possible metabolism-related DDI requires an understanding of the specific enzymes involved in metabolism of the drug in question. Furthermore, growing efforts to tailor therapies to specific genotypes, coupled with expanding knowledge of phenotypic variations in drug metabolizing enzymes combine to make understanding the molecular basis of metabolism critical information for evaluating potential drug candidates.
To address these issues, a number of approaches have been devised to perform such “reaction phenotyping” studies (15–18). Since reaction phenotyping studies, in particular those involving CYP enzymes, have been extensively reviewed by others, we propose to limit the discussion below to a more practical discussion of current screening methods used within the pharmaceutical industry.
EXPRESSED ENZYME STUDIES
Perhaps the most common approach to reaction phenotyping is the use of cDNA expressed enzyme systems to evaluate metabolism of lead compounds. This approach relies on transfection of a non-metabolizing cell line with cDNA coding for a specific drug metabolizing enzyme (CYP, UGT, etc). Incubations of the lead compound with a battery of cell lines, each expressing a different enzyme are conducted. The rate of disappearance of parent or the rate of appearance of the metabolite(s), if known, is then monitored by appropriate analytical tools, frequently LC/MS/MS. By comparing the rates of metabolism across cell lines, one can then identify which enzymes are capable of metabolizing the lead compound.
CYP Oxidation
For oxidative studies, the test compound is incubated with a panel of individually-expressed recombinant human CYP enzymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 in a typical panel) expressed in baculovirus-infected insect cell membranes. The incubation mixture typically contains the test compound at a final concentration of 1 μM or 10 μM, expressed CYP enzyme (20 nM), phosphate buffer (100 mM, pH 7.4), magnesium chloride (6.7 mM) and NADPH (1 mM) in a total volume of 1 ml. The reaction, conducted in triplicate, is initiated by the addition of NADPH followed by incubation at 37°C. Aliquots (0.1 ml) are taken at 0, 5, 10, 20, 30 and 60 min, and the reaction quenched by the addition of three volumes of acetonitrile. The samples are centrifuged at 10,000×g at room temperature for 2 min to pellet precipitated proteins. Supernatants are then transferred to clean vials and stored at −20°C until analysis. Incubations without NADPH and/or incubations with insect microsomes without human CYP cDNA are used as negative controls.
If no more than 20% of the parent is metabolized and the curve obtained by plotting the concentration of parent remaining (Ct) vs time (t) appears linear (Fig. 1), the initial rate of metabolism by recombinant human enzymes can be estimated by applying linear regression fitting of the following equation:
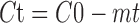 |
1 |
where C0 is the initial concentration of the test compound (1 μM or 10 μM under the above conditions) and m is the initial rate of metabolism.
Fig. 1.

Concentration vs time curve for disappearance of parent from initial concentrations of 10 and 1 μM. Linear regression fit was used to estimate initial slopes from first three data points (no more than 20% metabolism). Dividing slope by initial concentration yields an estimate of the rate constant (0.011 and 0.014 min−1 for 10 and 1 μM starting concentrations, respectively)
The turnover rate constant, k (min−1) can then be estimated from the initial rate according to the following equation:
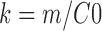 |
2 |
Alternatively, if more than 20% of the parent is metabolized and the curve obtained by plotting Ctvst curve can be reliably fit with an exponential curve (Fig. 2), the turnover rate constant (k) can be determined directly by non-linear regression fitting of a first-order equation to the Ctvst curve:
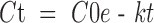 |
3 |
Ideally, pilot studies should be conducted to establish the concentration range for the test compound to ensure that substrate concentrations are in the linear range (well below the Km of the targeted enzyme). In practice, however, the evaluations are more commonly conducted in a screening format with two or three initial starting concentrations of test compound. Turnover rate constants are estimated as described above and if the estimates appear to be in reasonable agreement, the concentrations are assumed to be sufficiently below the Km to be non-saturating.
Fig. 2.

Concentration vs time curve for disappearance of parent from initial concentrations of 10 μM and 1 μM. Nonlinear regression fitting was used to estimate the rate constant (0.012 and 0.015 min−1 for 10 and 1 μM starting concentrations, respectively)
Various methods for scaling the results to predict in vivo drug clearance have been described, but for screening purposes, the usual approach is to use the following equation to calculate intrinsic clearances (CLint) for each of the individually expressed enzymes based on the estimated turnover rate constants:
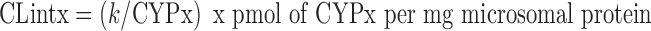 |
4 |
where CYPx is the concentration of recombinant CYP enzyme in the incubation (20 nM = 20 pmol/ml under the above conditions) (18–22). Estimated concentrations (pmol/mg microsomal protein) of the various CYP enzymes in human liver are listed in Table I below.
Table I.
| Enzyme | Microsomal protein (pmol/mg) |
|---|---|
| CYP1A2 | 45 to 52 |
| CYP2A6 | 36 to 68 |
| CYP2B6 | 11 to 39 |
| CYP2C8 | 24 to 64 |
| CYP2C9 | 73 to 96 |
| CYP2C19 | 14 to 19 |
| CYP2D6 | 8 to 10 |
| CYP2E1 | 49 to 61 |
| CYP3A4 | 108 to 111 |
The contribution of an individual CYP enzyme to the overall oxidative metabolism of a drug candidate can be estimated by Eq. 5 as follows (15).
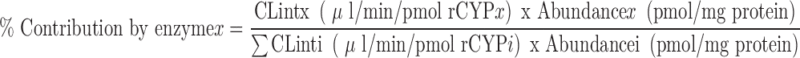 |
5 |
In the illustration shown in Table II, turnover of the drug candidate is observed in incubations with expressed CYP3A4 and CYP1A2, but not with any other enzymes.
Table 2.
Concentration (μM) of parent remaining after incubating with expressed CYP enzymes
| Time (min) | CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 | CYP2B6 | CYP2C8 |
|---|---|---|---|---|---|---|---|
| 0 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 5 | 1.00 | 1.00 | 1.00 | 1.00 | 0.97 | 1.00 | 1.00 |
| 10 | 1.00 | 1.00 | 1.00 | 1.00 | 0.94 | 1.00 | 1.00 |
| 20 | 1.00 | 1.00 | 1.00 | 1.00 | 0.88 | 1.00 | 1.00 |
| 30 | 1.00 | 1.00 | 1.00 | 1.00 | 0.82 | 1.00 | 1.00 |
| 60 | 0.99 | 1.00 | 1.00 | 0.68 | 1.00 | 1.00 |
Non-linear regression fitting was used to estimate the rate constants for metabolism by CYP3A4 and CYP1A2 (Fig. 3).
Fig. 3.

Concentration vs time curve for disappearance of parent from expressed CYP enzyme incubations with initial substrate concentrations of 1 μM. No turnover was observed in incubations containing CYP2B6, CYP2C9, CYP2C19 or CP2C8. Nonlinear regression fitting was used to estimate the rate constants for CYP1A2 (0.0002 min−1) and CYP3A4 (0.0065 min−1)
Assuming microsomal CYP450 content of 52 pmol of CYP1A2/mg microsomal protein and 111 pmol of CYP3A4/mg microsomal protein (Table I), the intrinsic clearance predictions with the above observations were predicted to be: 0.5 μl/min/mg microsomal protein for CYP1A2 and36 μl/min/kg for CYP3A4.
Applying Eq. 5, CYP3A4 is predicted to mediate >98% of the overall oxidative microsomal metabolism for this compound. Based on this finding, it is recommended that a DDI study to evaluate the effects of known CYP3A4 inhibitors on the human PK of this compound be conducted early in development (Fig. 4).
Fig. 4.

Flowchart for typical reaction phenotyping evaluation of a lead drug candidate. In all cases where results indicate a heightened risk of untoward effects, a decision to advance or terminate a compound must be made in the context of the risk–benefit balance and include consideration of anticipated in vivo drug concentrations at efficacious doses, nature of the untoward effect, alternatives to the lead compound and nature of the disease target
FMO Oxidation
For FMO oxidative studies, the test compound is incubated with individually expressed recombinant human FMO enzymes (FMO1 and FMO3) expressed in baculovirus-infected insect cell membranes. The incubation mixture typically contains the test compound at a final concentration of either 1 or 10 μM, expressed FMO enzyme (20–50 nM), phosphate buffer (100 mM, pH 7.4), magnesium chloride (6.7 mM) and NADPH (1 mM) in a total volume of 1 ml. The reaction, conducted in triplicate, is initiated by the addition of NADPH followed by incubation at 37°C. Aliquots (0.1 ml) are taken at 0, 5, 10, 20, 30 and 60 min, and the reaction quenched by the addition of three volumes of acetonitrile. The samples are centrifuged at 10,000×g at room temperature for 2 min to pellet precipitated proteins. Supernatants are then transferred to clean vials and stored at −20°C until analysis. Incubations without NADPH and/or incubations with insect microsomes without human CYP cDNA are used as negative controls.
Rates of metabolism by the FMO enzymes can be calculated from the observed loss of parent by methods directly analogous to those described above for CYP enzymes. However, since the relative abundances of the various FMO enzyme activities in human liver are not well established, it is not yet possible to use the method described above for microsomal CYP oxidation to estimate the relative contributions of the individual FMO enzymes to the overall metabolic rate. Nevertheless, in the absence of observed CYP mediated metabolism, FMO mediated clearance may provide useful information on the metabolic fate of the drug candidate. Furthermore, identification of a compound that is predominately metabolized by FMO3 can highlight a risk of Trimethylaminuria in populations with polymorphic expression of FMO3 (Fig. 4).
UGT Conjugation
For glucuronidation studies, the test compound is incubated with a panel of individually-expressed recombinant human UGT enzymes (typically UGT1A1, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, and UGT2B15) expressed in baculovirus-infected insect cell membranes. The incubation mixture typically contains the test compound at a final concentration of 5 μM, expressed UGT (0.1 mg protein/ml), Tris–HCl buffer (pH 7.5), magnesium chloride (10 mM), alamethicin (25 μg/ml) and UDPGA (2.5 mM). The mixture (without the UDPGA cofactor) is pre-incubated at 37°C for 5 min, after which the reaction is started by the addition of UDPGA.
Incubations are performed at 37°C. Aliquots (200 μl) are collected at 0, 10, 20, 30 and 60 min and quenched with two volumes of ice-cold acetonitrile. The samples are centrifuged at 10,000×g at room temperature for 2 min to pellet precipitated proteins. The supernatants are then transferred to clean vials and stored at −20°C until analysis. Incubations without UDPGA are used as negative controls.
Rates of glucuronidation by the various UGT isoforms can be calculated from the observed loss of parent by methods directly analogous to those described above for CYPs. However, since the relative abundances of the various UGT enzyme activities in human liver have not been well established, it is not yet possible to use the method described above for microsomal oxidation to estimate the relative contributions of the individual UGT enzymes to the overall glucuronidation rate, although alternative methods using multiple regression analyses of human liver microsomal incubations have been described (23–25). Instead, reaction phenotyping studies for glucuronidation are generally used as an alert for compounds that may be metabolized by UGT1A1 and thus present a risk for potential competition with bilirubin glucuronidation, or to provide an overall alert for compounds that may be metabolized by only one isoform and, thus, present a higher risk for drug–drug interactions than a compound that may have more balanced elimination by multiple pathways. More detailed studies to estimate Vmax and Km may be conducted to provide additional estimates of the risk level, but these types of evaluations are not generally considered a routine reaction phenotyping screen (Fig. 4).
Expressed Enzyme Study Caveats
The expressed enzyme approach is straightforward, relatively simple, and is commonly used. However, there are a number of drawbacks to this approach.
The various enzymes involved in drug metabolism are not expressed to the same level in normal drug-metabolizing cells. For example, the average level of CYP3A4 in microsomes prepared from human liver is reported to be around 111 pmol/mg protein whereas levels of CYP2D6 are reported to be around 8 pmol/mg protein. As a result, rates of metabolism in expressed systems must be normalized for expression levels as described above. However, significant variability in CYP enzyme levels has been observed in human liver microsomal preparations from different donors (23,26). Consequently, the CYP enzyme activity of a “typical” human liver donor may not provide an adequate indication of the range of activities likely to be encountered clinically.
Different enzymes will likely exhibit different kinetic behaviors (Km and Vmax values). As a result, a compound that is a substrate for both CYP3A4 and CYP2C9, for example, may exhibit a more rapid turnover with CYP2C9 than with CYP3A4 at low concentrations if the CYP2C9 Km is lower than the CYP3A4 Km. If, however, the Vmax for CYP2C9 is lower than the Vmax for CYP3A4, then high (saturating) drug concentrations would exhibit more rapid turnover with CYP3A4 than with CYP2C9 leading to the opposite conclusion. Although full kinetic evaluations of discovery compounds are not practical in a screening environment, it behooves the discovery scientist to remain aware of these potential issues.
As discussed above, extrapolation of glucuronidation rates to predict the relative contributions of the various UGT’s to overall turnover is challenging due to limited information regarding the relative activities of the various UGTs in human liver. In addition, there appears to be significant inter-individual variability in UGT enzyme activities (27,28). Furthermore, UGT activity in extrahepatic tissues, such as small intestine, may contribute to the overall metabolism of drugs in humans (28,29).
Since the UGT active site is on the interior of the microsomal vesicles, most glucuronidation screening preparations include a detergent or pore-forming antibiotic, such as alamethicin, to disrupt the membrane barrier and enhance access of both substrate and cofactor to the UGT active site (30–33). Detergents have been shown to alter intrinsic UGT activity and, while alamethicin appears to have less of an impact on intrinsic enzyme activity than detergent-based enhancers, there is still some concern that in vitro glucuronidation rates determined in the presence of activity enhancers may not reflect in vivo activity (34,35). In addition, since UGT substrates must be delivered to the luminal side of the endoplasmic reticulum (ER) to access the active site of UGT, uptake into the ER may be a limiting factor for in vivo glucuronidation. Unfortunately, screening methods to model uptake into the ER are not yet available. As a consequence of these considerations, the extrapolation of in vitro UGT-mediated metabolic rates to in vivo clearance predictions is generally believed to be less robust than the corresponding extrapolation of in vitro CYP-mediated metabolic rates.
SELECTIVE INHIBITION STUDIES
A second approach to reaction phenotyping involves conducting incubations in hepatocytes, microsomes, or some other in vitro preparation using normal tissues as the enzyme source, but including selective chemical or immuno-inhibitors of specific enzymatic pathways. Again, by performing a battery of incubations with various inhibitors, and comparing the relative rates of metabolism, one can identify which inhibitor reduces the overall metabolism to the greatest extent and thereby uncover the metabolic pathway that contributes the most to the clearance of a compound.
CYP Inhibitors
Since this approach generally uses a “normal” tissue as the source of the in vitro preparation, the relative expression levels of the various enzymes reflect the normal in vivo expression levels. As a result, this approach is generally favored for experiments intended to deconvolute the contributions from various enzymes when multiple pathways are involved in metabolism of a compound.
For oxidative studies, the test compound is incubated with human liver microsomes. The incubation mixture typically contains the test compound at a concentration of 3 μM, microsomal protein (1 mg/ml), phosphate buffer (100 mM, pH 7.4), magnesium chloride (6.7 mM), NADPH (1 mM) and a specific CYP inhibitor. A list of typical inhibitors and the final incubation concentrations used are summarized in Table III.
Table III.
Inhibitors Used for Microsomal Reaction Phenotyping Incubations
| CYP | Inhibitor | Stock Solution | Final concentration (μM) |
|---|---|---|---|
| CYP1A1/2 | Furafylline (38,39,40) | 5 mM in acetonitrile | 20 |
| CYP2C9 | Sulfaphenazole (40,41) | 5 mM in acetonitrile | 20 |
| CYP2C19 | 3-Benzylnirvanol (42,43) | 1.25 mM in acetonitrile | 5 |
| CYP2D6 | Quinidine(40,44,45) | 1.25 mM in acetonitrile | 5 |
| CYP3A4 | Ketoconazole(40) | 250 μM acetonitrile | 1 |
| Troleandomycin (44,46,47) | 12.5 mM in acetonitrile | 100 |
Final concentration of organic solvent in the incubation mixture is 0.4% (v/v) except for troleandomycin (0.8%).
For incubations involving time-dependent CYP inhibitors, CYP1A1/2 (furafylline) and CYP3A4 (troleandomycin), incubation mixtures containing microsomes, inhibitor and NADPH are preincubated for 15 min and the reaction is initiated by adding the test compound. For all other CYP inhibitors, the inhibitor is added concurrently with the test compound for a 5-min preincubation and the reaction is initiated by adding NADPH. Incubations are conducted in triplicate at 37°C. Aliquots of samples (0.1 ml) are taken at 0, 5, 10, 20, 30 and 60 min, and the reaction quenched by the addition of three volumes of acetonitrile. The samples are centrifuged at 10,000×g at room temperature for 2 min to pellet precipitated proteins. The supernatants are then transferred to clean vials and stored at −20°C until analysis. Incubations with acetonitrile but without inhibitor are used as positive controls.
The extent of inhibition is determined by comparing the extent of metabolism observed at 60 min in the presence of inhibitors with the extent of metabolism observed in comparable incubations without inhibitors.
An example is shown below in Table IV for results of a typical microsomal study performed with specific CYP inhibitors.
Table 4.
Percent inhibition of metabolism of a drug candidate in human liver microsomes by chemical inhibitors of CYPs
| Inhibitor | Furafylline (CYP1A1/2) | Sulfaphenazole(CYP2C9) | Benzylnirvanol(CYP2C19) | Quinidine (CYP2D6) | Troleandomycin (CYP3A4) |
|---|---|---|---|---|---|
| Inhibition (%) | 30 | 11 | 4 | 13 | 72 |
The data in Table IV indicate that CYP3A4 and CYP1A1/2 are the primary enzymes responsible for metabolism of this compound. In a subsequent confirmatory study, incubations conducted in the presence of both furafylline and troleandomycin exhibited no detectable metabolism of the substrate. Had metabolism been observed in the presence of both inhibitors, further investigation would have been needed to investigate additional routes of metabolism.
FMO Inhibitors
Although selective substrates of FMO enzymes are currently available, selective inhibitors of FMO have yet to be identified. Consequently, studies analogous to those described above for CYP enzymes cannot be conducted at present. However, it is possible to thermally inactivate FMO enzymes by pre-incubating microsomes for 2 min at 50°C in the absence of NADPH. Incubations can then proceed with addition of NADPH and incubation at 37°C as described above. By comparing the percent of parent compound remaining in incubations with and without the 50°C inactivation pretreatment, it is possible to discern the FMO contribution to overall metabolism.
UGT Inhibitors
Currently available inhibitors (and substrates) of UGT enzymes do not have the needed specificity to allow studies analogous to those described above for CYP enzymes.
Immuno-inhibitors
Although not widely used, monoclonal antibody (MAb) inhibitors of CYP enzymes can provide clear advantages over the more commonly used chemical inhibitors. These advantages include excellent specificity (>90%) and individual MAbs to all of the major CYP enzymes involved in drug metabolism. Gelboin et al. (36) have reviewed the use of MAbs in depth. Metabolic incubations involving MAbs are generally conducted as follows: 0.57 to 30 μl (1.2–1,000 μg) of ascites protein containing MAb are preincubated with 30 to 50 pmol of expressed CYP enzyme or 150 to 300 pmol of CYP in human liver microsomes in 0.5 ml of buffer (100 mM Tris, pH 7.5) at 37°C for 5 min (37). Incubations typically contain the test compound at a concentration of 1 to 10 μM. Control incubations contain a MAb against hen egg white lysozyme to assess non-specific reactions
Aliquots of samples (0.1 ml) are taken at 0, 5, 10, 20, 30 and 60 min, and the reaction quenched by the addition of three volumes of acetonitrile. The samples are centrifuged at 10,000×g at room temperature for 2 min to pellet precipitated proteins. The supernatants are then transferred to clean vials and stored at −20°C until analysis.
Percent inhibition is calculated based on activity in the presence of the specific MAb relative to the activity with the control MAb.
Caveats for Selective Inhibition Studies
As with the expressed enzyme approach, the interplay between enzyme kinetics and drug concentration can provide misleading conclusions unless studies are conducted at multiple concentrations.
For compounds exhibiting low turnover, the use of specific inhibitors presents an additional challenge since it may be very difficult to detect a decrease in an already very low metabolic rate. In such cases, monitoring for appearance of metabolite(s) is an option if the metabolic products are known.
In addition, various CYP enzymes, most notably CYP2D6, exhibit polymorphic expression in the general population. As a result, the relative levels of expression of the various CYP enzymes in any single donor may not reflect the “average” human liver expression levels. Generally this is dealt with by combining microsomes from multiple human donors to generate a pool to represent the “average” human. Even with this approach, full characterization of the expression levels of the various CYP enzymes in the pooled microsomal preparation is needed to ensure appropriate interpretation of the results.
GENERAL CONSIDERATIONS
Although all of the approaches described above can be utilized with analytical methods to assess either disappearance of parent or appearance of metabolites, the general trend in drug discovery is to monitor disappearance of parent. This is due to the fact that the metabolic fate of discovery compounds is usually incompletely understood. In addition, even in cases where the metabolism has been relatively well characterized, metabolite standards are often not readily available. As a consequence, discovery compounds with low overall metabolic turnover present a special challenge for reaction phenotyping since the investigator is attempting to discern changes in a low overall rate of metabolism. For those compounds, it may be necessary to first elucidate the metabolic products formed and then prepare standards of those metabolites in order to conduct more rigorous reaction phenotyping studies by monitoring their rate of formation. In such cases, the investigator’s ability to detect metabolism is limited by the sensitivity of the analytical assay to detect the metabolite. Unfortunately, this approach is highly resource-intensive and usually is limited to those compounds already selected for advancement.
CONCLUSIONS
Reaction phenotyping assays have become an integral part of drug discovery screening as tools to minimize drug–drug interaction potential and to avoid complications due to polymorphic expression of drug metabolizing enzymes. cDNA expressed enzyme systems are the most widely used approach and are available for both CYP and FMO enzymes as well as UGT and other enzymes. The complementary approach of using specific inhibitors is also widely used for CYP oxidation, but the dearth of specific UGT and FMO inhibitors has impeded the application of this approach to glucuronidation, FMO mediated oxidation and other metabolic pathways.
Since full kinetic characterization is generally not practical in drug discovery screening, assumptions regarding relative Km and Vmax of the enzymes involved, as well as variations in enzyme activities across tissues and individuals must be considered for interpretation of screening results. While reaction phenotyping results for CYP oxidation are generally viewed as moderately robust, significant opportunities remain for additional improvements, especially for UGT-mediated metabolic pathways.
References
- 1.Rodrigues A. D., Rushmore T. H. Cytochrome P450 pharmacogenetics in drug development: in vitro studies and clinical consequences. Curr. Drug Metab. 2002;3:289–309. doi: 10.2174/1389200023337522. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura K., Goto F., Ray W. A., McAllister C. B., Jacqz E., Wilkinson G. R., Branch R. A. Interethnic differences in genetic polymorphism of debrisoquin and mephenytoin hydroxylation between Japanese and Caucasian populations. Clin. Pharmacol. Ther. 1985;38:402–408. doi: 10.1038/clpt.1985.194. [DOI] [PubMed] [Google Scholar]
- 3.Kimura M., Ieiri I., Mamiya K., Urae A., Higuchi S. Genetic polymorphism of cytochrome P450s, CYP2C19, and CYP2C9 in a Japanese population. Ther. Drug Monit. 1998;20:243–247. doi: 10.1097/00007691-199806000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Jose R., Chandrasekaran A. The pharmacogenetics of CYP2C9 and CYP2C19: ethnic variation and clinical significance. Curr. Clin. Pharm. 2007;2(1):93–109. doi: 10.2174/157488407779422302. [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Martin E., Martinez C., Ladero J. M., Agundez J. A. G. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol. Diagn. Ther. 2006;10:29–40. doi: 10.1007/BF03256440. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka E. Clinically important pharmacokinetic drug–drug interactions: role of cytochrome P450 enzymes. J. Clin. Pharm. Ther. 1998;23:403–416. doi: 10.1046/j.1365-2710.1998.00086.x. [DOI] [PubMed] [Google Scholar]
- 7.Dresser G. K., Spence J. D., Bailey D. G. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin. Pharm. 2000;38:41–57. doi: 10.2165/00003088-200038010-00003. [DOI] [PubMed] [Google Scholar]
- 8.Hsu A., Granneman G. R., Bertz J. Ritonavir: Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin. Pharm. 1998;35:275–291. doi: 10.2165/00003088-199835040-00002. [DOI] [PubMed] [Google Scholar]
- 9.Hsu A., Granneman G. R., Cao G., Carothers L., Japour A., El-Shourbagy T., Dennis S., Berg J., Erdman K., Leonard J. M., Sun E. Pharmacokinetic Interaction between ritonavir and indinavir in healthy volunteers. Antimicrob. Agents Chemother. 1998;42:2784–2791. doi: 10.1128/aac.42.11.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koukouritaki S. B., Simpson P., Yeung C. K., Rettie A. E., Hines R. N. Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr. Res. 2002;51:236–243. doi: 10.1203/00006450-200202000-00018. [DOI] [PubMed] [Google Scholar]
- 11.Larsen-Su S., Williams D. E. Dietary indole-3-carbinol inhibits FMO activity and the expression of flavin-containing monooxygenase form 1 in rat liver and intestine. Drug Metab. Dispos. 1996;24:927–931. [PubMed] [Google Scholar]
- 12.Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003;3:136–158. doi: 10.1038/sj.tpj.6500171. [DOI] [PubMed] [Google Scholar]
- 13.Levesque E., Delage R., Benoit-Biancamano M.-O., Caron P., Bernard O., Couture F., Guillemette C. The impact of UGT1A8, UGT1A9, and UGT2B7 genetic polymorphisms on the pharmacokinetic profile of mycophenolic acid after a single oral dose in healthy volunteers. Clin. Pharm. Ther. 2007;81:392–400. doi: 10.1038/sj.clpt.6100073. [DOI] [PubMed] [Google Scholar]
- 14.Kiang T. K. L., Ensom M. H. H., Chang T. K. H. UDP-glucuronosyltransferases and clinical drug–drug interactions. Pharmacol. Ther. 2005;106:97–132. doi: 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Rodrigues A. D. Integrated cytochrome P450 reaction phenotyping: attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochem. Pharmacol. 1999;57:465–480. doi: 10.1016/S0006-2952(98)00268-8. [DOI] [PubMed] [Google Scholar]
- 16.Parkinson A. An overview of current cytochrome P450 technology for assessing the safety and efficacy of new materials. Toxicol. Pathol. 1996;24:48–57. doi: 10.1177/019262339602400107. [DOI] [PubMed] [Google Scholar]
- 17.Rodrigues A. D. Use of in vitro human metabolism studies in drug development. An industrial perspective. Biochem. Pharmacol. 1994;48:2147–2156. doi: 10.1016/0006-2952(94)00312-2. [DOI] [PubMed] [Google Scholar]
- 18.Emoto C., Murase S., Iwasaki K. Approach to the prediction of the contribution of major cytochrome P450 enzymes to drug metabolism in the early drug-discovery stage. Xenobiotica. 2006;36:671–683. doi: 10.1080/00498250600709778. [DOI] [PubMed] [Google Scholar]
- 19.Houston J. B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharm. 1994;47:1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 20.Iwatsubo T., Hirota N., Ooie T., Suzuki H., Simada N., Chiba K., Ishizaki T., Green G. E., Tyson C. A., Sugiyama Y. Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol. Ther. 1997;73:147–171. doi: 10.1016/S0163-7258(96)00184-2. [DOI] [PubMed] [Google Scholar]
- 21.Obach R. S., Baxter J. G., Liston T. E., Silber B. M., Jones B. C., MacIntyre R., Rance D. J., Wastal P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997;283:46–58. [PubMed] [Google Scholar]
- 22.Proctor N. J., Tucker G. T., Rostami-Hodjegan A. Predicting drug clearance from recombinantly expressed CYPs: intersystem extrapolation factors. Xenobiotica. 2004;34:151–178. doi: 10.1080/00498250310001646353. [DOI] [PubMed] [Google Scholar]
- 23.Ikushiro S.-I., Yoshikazu E., Yoshihisa K., Yamada S., Sakaki T. Monospecific antipeptide antibodies against human hepatic UDP-glucuronosyltransferase 1A subfamily (UGT1A) isoforms. Drug Metab. Pharmacokinet. 2006;21:70–74. doi: 10.2133/dmpk.21.70. [DOI] [PubMed] [Google Scholar]
- 24.Lin J. H., Wong B. K. Complexities of glucuronidation affecting in vitro–in vivo extrapolation. Curr. Drug Metab. 2002;3:623–646. doi: 10.2174/1389200023336992. [DOI] [PubMed] [Google Scholar]
- 25.Court M. H., Duan S. X., Von Moltke L. L., Greenblatt D. J., Patten C. J., Miners J. O., MacKenzie P. I. Interindividual variability in acetaminophen glucuronidation by human liver microsomes: identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J. Pharmacol. Exp. Ther. 2001;299:998–1006. [PubMed] [Google Scholar]
- 26.Yeo K. R., Rostami-Hodjegan A., Tucker G. T. Abundance of cytochromes P450 in human liver: a meta-analysis. Br. J. Clin. Pharmacol. 2004;57:687–688. [Google Scholar]
- 27.Krishnaswamy S., Duan S. X., Von Moltke L. L., Greenblatt D. J., Court M. H. Validation of serotonin (5-hydrosytryptamine) as an in vitro substrate probe for human UDP-glucuronosyltransferase (UGT) 1A6. Drug Metab. Disp. 2003;31:133–139. doi: 10.1124/dmd.31.1.133. [DOI] [PubMed] [Google Scholar]
- 28.Fisher M. B., Vandenbranden M., Findlay K., Burchell B., Thummel K. E., Hall S. D., Wrighton S. A. Tissue distribution and interindividual variation in human UDP-glucuronosyltransferase activity: relationship between UGT1A1 promoter genotype and variability in a liver bank. Pharmacogenetics. 2000;10:727–739. doi: 10.1097/00008571-200011000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Strassburg C. P., Kneip S., Topp J., Obermayer-Straub P., Barutt A., Turkey R. H., Manns M. P. Polymorphic gene regulation and interindividual variation of UDP-glucuronosyltransferase activity in human small intestine. J. Biol. Chem. 2000;46a:36164–36171. doi: 10.1074/jbc.M002180200. [DOI] [PubMed] [Google Scholar]
- 30.Fulceri R., Banhegyi G., Gamberucci A., Giunti R., Mandl J., Benedetti A. Evidence for the intraluminal positioning of p-nitrol UDP-glucuronosyltransferase activity in rat liver microsomes. Arch. Biochem. Biophys. 1994;309:43–46. doi: 10.1006/abbi.1994.1081. [DOI] [PubMed] [Google Scholar]
- 31.Fisher M. B., Campanale K., Ackermann B. L., Banderbranden M., Wrighton S. A. In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab. Disp. 2000;25:560–566. [PubMed] [Google Scholar]
- 32.Little J. M., Lehman P. A., Nowell S., Samokyszyn V., Radominska A. Glucuronidation of all-trans-retinoic acid and 5,6-epoxy-all-trans-retinoic acid. Drug Metab. Disp. 1997;25:5–11. [PubMed] [Google Scholar]
- 33.He K., Ludtke S. J., Heller W. T., Huang H. W. Mechanism of alamethicin insertion into lipid bilayers. Biophys. J. 1996;71:2669–2679. doi: 10.1016/S0006-3495(96)79458-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lett E., Kriszt W., de Sandro V., Ducrotoy G., Richert L. Optimal detergent activation of rat liver microsomal UDP-glucuronosyltransferases toward morphine and 1-naphthol: contribution to induction and latency studies. Biochem. Pharmacol. 1992;43:1649–1653. doi: 10.1016/0006-2952(92)90225-8. [DOI] [PubMed] [Google Scholar]
- 35.Trapnell C. B., Klecker R. W., Jamis-Dow C., Colling J. M. Glucuronidation of 3¢-azido-3¢-deoxythymidine (zidovudine) by human liver microsomes—relevance to clinical pharmacokinetic interactions with atovaquone, fluconazole, methadone and valproic acid. Antimicrob. Agents Chemother. 1998;42:1592–1596. doi: 10.1128/aac.42.7.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gelboin H. V., Krausz K. Monoclonal Antibodies and Multifunctional Cytochrome P450: Drug Metabolism as Paradigm. J. Clin. Pharmacol. 2006;46:353–372. doi: 10.1177/0091270005285200. [DOI] [PubMed] [Google Scholar]
- 37.Krausz K. W., Goldfarb I., Buters J. T., Yang T. J., Gonzalez F. J., Gelboin H. V. Monoclonal antibodies specific and inhibitory to human cytochromes P450 2C8, 2C9, and 2C19. Drug Metab. Dispos. 2001;29:1410–1423. [PubMed] [Google Scholar]
- 38.Sesardic D, Boobis AR, Murray BP, Murray S, Segura J, de la Torre R, Davies DS. Furafylline is a potent and selective inhibitor of cytochrome P4501A2 in man. Br. J. Clin. Pharm. 1990;29:651–663. doi: 10.1111/j.1365-2125.1990.tb03686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke SE, Ayrton AD, Chenery RJ. Characterization of the inhibition of P4501A2 by furafylline. Xenobiotica. 1994;24:517–526. doi: 10.3109/00498259409043254. [DOI] [PubMed] [Google Scholar]
- 40.Bourrie M, Meunier V, Berger Y, Fabre G. Cytochrome P450 isoform inhibitors as a tool for the investigation of metabolic reactions catalyzed by human liver microsomes. J. Pharmacol. Exp. Ther. 1996;277:321–332. [PubMed] [Google Scholar]
- 41.Mancy A, Dijols S, Poli S, Guengerich FP, Mansuy D. Interaction of sulfaphenazole derivatives with human liver cytochromes P4502C: Molecular origin of the specific inhibitory effects of sulfaphenazole on CYP2C9 and consequences for the substrate binding site topology of CYP2C9. Biochemistry. 1996;35:16205–16212. doi: 10.1021/bi961950t. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki H, Kneller MB, Haining RL, Trager WF, Rettie AE. (+)-N-3-benzyl-nirvanol and (−)-N-3-benzylphenobarbital: New potent and selective in vitro inhibitors of CYP2C19. Drug Metab. Dispos. 2002;30:235–239. doi: 10.1124/dmd.30.3.235. [DOI] [PubMed] [Google Scholar]
- 43.Walsky RL, Obach RS. Verification of the selectivity of (+)N-3-benzylnirvanol as a CYP2C19 inhibitor. Drug Metab. Dispos. 2003;31:343. doi: 10.1124/dmd.31.3.343. [DOI] [PubMed] [Google Scholar]
- 44.Newton DJ, Wang RW, Lu AY. Cytochrome P450 inhibitors. Evaluation of specificities in the in vitro metabolism of therapeutic agents by human liver microsomes. Drug Metab. Dispos. 1995;23:154–158. [PubMed] [Google Scholar]
- 45.Rodrigues AD, Roberts EM. The in vitro interaction of dexmedetomidine with human liver microsomal cytochrome P4502D6 (CYP2D6) Drug Metab. Dispos. 1997;25:651–655. [PubMed] [Google Scholar]
- 46.Yamazaki H, Shimada T. Comparative studies of in vitro inhibition of cytochrome P450 3A4-dependent testosterone 6beta-hydroxylation by roxithromycin and its metabolites, troleandomycin, and erythromycin. Drug Metab. Dispos. 1998;26:1053–1057. [PubMed] [Google Scholar]
- 47.Marre F, de Sousa G, Orloff AM, Rahmani R. In vitro interaction between cyclosporin A and macrolide antibiotics. Br. J. Clin. Pharm. 1993;35:447–448. doi: 10.1111/j.1365-2125.1993.tb04166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]