

Abstract
Caveolin-1, the signature protein of endothelial cell caveolae, has many important functions in vascular cells. Caveolae are thought to be the transcellular pathway by which plasma proteins cross normal capillary endothelium, but, unexpectedly, cav-1−/− mice, which lack caveolae, have increased permeability to plasma albumin. The acute increase in vascular permeability induced by agents such as vascular endothelial growth factor (VEGF)-A occurs through venules, not capillaries, and particularly through the vesiculo-vacuolar organelle (VVO), a unique structure composed of numerous interconnecting vesicles and vacuoles that together span the venular endothelium from lumen to ablumen. Furthermore, the hyperpermeable blood vessels found in pathological angiogenesis, mother vessels, are derived from venules. The present experiments made use of cav-1−/− mice to investigate the relationship between caveolae and VVOs and the roles of caveolin-1 in VVO structure in the acute vascular hyperpermeability induced by VEGF-A and in pathological angiogenesis and associated chronic vascular hyperpermeability. We found that VVOs expressed caveolin-1 variably but, in contrast to caveolae, were present in normal numbers and with apparently unaltered structure in cav-1−/− mice. Nonetheless, VEGF-A-induced hyperpermeability was strikingly reduced in cav-1−/− mice, as was pathological angiogenesis and associated chronic vascular hyperpermeability, whether induced by VEGF-A164 or by a tumor. Thus, caveolin-1 is not necessary for VVO structure but may have important roles in regulating VVO function in acute vascular hyperpermeability and angiogenesis.
Caveolae (also referred to as plasmalemmal vesicles) were described by Palade and Bruns in capillary endothelial cells as 50- to 100-nm diameter smooth membrane-bound vesicles.1,2 Palade and Bruns proposed that caveolae shuttled across capillary endothelium from lumen to ablumen, carrying with them “cargoes” of plasma and in this manner provided the small amounts of plasma proteins that are required for maintaining tissue health. Later work demonstrated that caveolae could also form short chains of two to three linked vesicles that spanned the short distance across the capillary endothelium.3 Together these studies implied that, whether shuttling or interconnected into short chains, capillary caveolae were de facto the elusive “large pores” that physiologists had postulated to account for plasma protein extravasation.4,5
Since their initial discovery, much has been learned about caveolae and their signature protein, caveolin.6,7,8,9,10 Caveolin is thought to be necessary for caveolae formation and overexpression of caveolin can induce caveolae in cells that normally lack them.11 Caveolin exists in three isoforms.12,13,14 The first two isoforms, cav-1 and cav-2, are highly expressed in vascular endothelium, pericytes and smooth muscle, among other cell types, whereas cav-3 is confined to muscle.15 Caveolae and caveolin have many functions besides plasma protein transport, including regulation of cholesterol homeostasis and sorting of signaling molecules such as endothelial nitric oxide synthase, heterotrimeric G proteins, and nonreceptor tyrosine kinases.7,9,14,16,17
Cav-1−/− mice have contributed much to our understanding of caveolin and caveolae. Cav-1−/− mice are viable and fertile but lack caveolae and exhibit various types of vascular dysfunction, including impaired nitric oxide and Ca2+ signaling.12,18,19,20,21,22 However, there is controversy on some other points, such as whether tumor growth and angiogenesis are altered in cav-1−/− mice, and, if so, in what direction and by what mechanism.22,23,24,25,26,27,28,29 Recently, cav-1−/− mice have been found to be systemically hyperpermeable to plasma albumin25,30,31; this finding was unexpected in that caveolae have been thought to be necessary for transporting plasma proteins across capillary endothelium under basal conditions.1,2,3
However, vascular permeability is not of a single type.32 In contrast to the normal, low level basal vascular permeability (BVP) of normal tissues, two distinctly different types of increased vascular permeability are found in pathological conditions.32 Vascular permeabilizing factors, such as vascular endothelial growth factor (VEGF)-A, histamine, and others, induce acute vascular hyperpermeability (AVH), a characteristic feature of acute inflammation. Chronic vascular hyperpermeability (CVH), on the other hand, is found in the pathological angiogenesis induced by tumors, healing wounds, and chronic inflammatory diseases; as its name implies, CVH persists for long periods of time—days to weeks and sometimes indefinitely. AVH and CVH differ from BVP not only in terms of the much greater amounts of plasma that extravasate but also with respect to the microvessels that leak. BVP takes place in capillaries.2,3,4 In contrast, AVH takes place primarily in postcapillary venules33,34,35,36,37 and is thought to involve an organelle, the vesiculo-vacuolar organelle (VVO), that is uniquely present in venular endothelial cells. VVOs are grapelike clusters of hundreds of uncoated, trilaminar unit membrane-bound, interconnecting vesicles and vacuoles that extend across the relatively tall cytoplasm of venular endothelium from lumen to ablumen. The relationship of VVOs to caveolae is uncertain.36,37,38,39,40 Unlike caveolae, which are of relatively uniform size, the vesicles and vacuoles that comprise VVOs vary widely in size from caveolae-sized vesicles to those with a cross-sectional areas more than 10-fold greater.37 They attach to each other and to the endothelial plasma membrane by stomata that are normally closed by thin diaphragms. In this respect, VVO stomata and diaphragms closely resemble the analogous structures by which caveolae attach to each other and to the luminal and abluminal plasma membranes of capillary endothelium.36,37,38,41 On exposure to acute permeabilizing agents such as VEGF or histamine, the diaphragms interconnecting VVO vesicles and vacuoles open to provide a trans-endothelial cell pathway for plasma extravasation.36,37,42 Others have reported leakage through a paracellular route, independent of VVOs.43,44 In CVH, yet another type of blood vessel, the “mother” vessel, accounts for the bulk of vascular hyperpermeability.45 Mother vessels are greatly enlarged, thin-walled, pericyte-poor sinusoids that derive from pre-existing normal venules after longer exposures to VEGF and other angiogenic stimuli.46 VVOs participate in mother vessel formation and associated CVH.45,47
The experiments reported here made use of cav-1−/− mice to investigate the relationship between caveolae and VVOs and the role of caveolin-1 in VVO structure, in AVH and CVH, and in pathological angiogenesis. We report here that some, but not all, VVO vesicles and vacuoles express caveolin-1. Nonetheless, VVOs are present in normal numbers and with unaltered structure in cav-1−/− mice. Further, we find that AVH and CVH are strikingly reduced in cav-1−/− mice. Angiogenesis is also reduced in cav-1−/− mice, whether induced by an adenoviral vector expressing VEGF-A164 (Ad-VEGF-A164) or by a tumor, the B16 melanoma.
Materials and Methods
Animals, Adenoviral Vector, and Tumors
Four- to 6-week-old female wild-type C57BL/6 and caveolin 1 knockout (cav-1−/−) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Angiogenesis was induced in flank skin either with (Ad-VEGF-A164)46,47,48 or with the B16 melanoma.49 All studies were performed under protocols approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Tissue Processing for Light Microscopic Immunohistochemistry
Animals were sacrificed by CO2 narcosis. Flank skin was fixed in 4% paraformaldehyde and processed either for frozen or paraffin sections and immunohistochemical analysis, as described previously.50 Two different rabbit polyclonal antibodies, one directed against N-terminal amino acids 1 to 97 (BD Biosciences, San Jose, CA) and the other against N-terminal amino acids 1 to 20 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), were used to identify caveolin-1. Mean vascular density was calculated by counting CD31-positive structures with lumens in the five most highly vascularized fields at ×400 magnification. Statistical analysis was performed using an unpaired t-test.
Electron Microscopy
Tissues were fixed and processed for electron microscopy as described previously.51 Morphometric analysis was performed on randomly selected electron micrographs of wild-type and cav-1−/− flank skin venules and capillaries for quantification of VVOs, vesicles, and caveolae.35,37 Data were analyzed with the Kruskal-Wallis nonparametric analysis of variance test and with Dunn’s multiple comparisons test.
Electron Microscopic Immuno-Nanogold Cytochemistry
Immuno-nanogold cytochemistry was performed as described previously.50 Tissues were fixed for 4 hours at room temperature in 4% paraformaldehyde-0.02 mol/L PBS, pH 7.4, and were washed in 0.02 mol/L PBS, pH 7.4. before immersion in 30% sucrose in 0.02 mol/L PBS, pH 7.4, overnight at 4°C. Tissues were embedded in OCT compound (Miles, Elkhart, IN), snap-frozen, and stored in liquid nitrogen. The same two rabbit polyclonal antibodies used for light microscopic immunohistochemistry were used. Ten-micrometer frozen sections were cut, collected on precleaned glass slides, air-dried for 20 minutes, and processed as described previously,50 except for differences in the antibodies used. After immunocytochemistry, sections were stained either with uranyl acetate or lead citrate or left unstained and examined in a Philips CM10 transmission electron microscope. Four control experiments were performed to ensure the specificity of immuno-nanogold staining: (i) replace the primary antibody with an irrelevant rabbit IgG; (ii) omit the primary antibody; (iii) omit the secondary antibody; and (iv) omit the HQ silver enhancement step.
Miles Assay and Quantification of Vascular Volume and Permeability
The Miles assay was performed as described previously.45 Mice were anesthetized with Avertin and injected i.v. with 0.1 ml of 0.5% Evans blue dye, and their flanks were injected intradermally with recombinant VEGF-A (National Cancer Institute, Bethesda, MD) or with an equivalent volume (50 μl) of HBSS. Mice were sacrificed 30 minutes later, and a sample of heart blood was collected. Skin test sites and platelet-poor plasma were extracted in formamide at 56°C for 48 hours. The amount of extracted Evans blue dye was determined by measuring absorbance at 620 nm and calculated against a standard curve. Data are presented as microliters of plasma.45,48
To quantify angiogenesis and vascular permeability at angiogenic sites, we made use of a double tracer method.48 Angiogenesis was induced by injecting 5 × 108 plaque-forming units of Ad-VEGF-A164 into flank skin. Four days later, at the height of the angiogenic response, mice were injected i.v. with 0.1 ml of 0.5% Evans blue dye, followed 25 minutes later by an i.v. injection of 10 μCi of 125I-bovine serum albumin. Mice were sacrificed 5 minutes after the second injection. Heart blood was collected as above and angiogenic sites were subjected to gamma counting. Evans blue dye was extracted in formamide as above, and vascular volume and vascular leakage were calculated as described.45,48
To compare vascular permeability in tissues of wild-type and cav-1−/− mice, 125I-bovine serum albumin was injected i.v. as above. An aliquot of blood (100 μl) was taken by retro-orbital puncture into heparin (10 μl) within 30 seconds thereafter for determining the initial injected dose of tracer. After 1 hour, mice were sacrificed, and tissues were excised, weighed, and subjected to gamma counting. Radioactivity was normalized to tissue weight and calculated as percentage of injected dose of tracer per gram of tissue ± SD.
Results
Caveolin-1 Expression in Vascular Endothelium of cav-1−/− and Wild-Type C57BL/6 Mice
Immunohistochemistry was performed on flank skin with two different antibodies that were directed against different N-terminal peptides of caveolin-1. Both antibodies gave identical results, demonstrating strong labeling of both capillary and venular endothelium in wild-type mice and an absence of detectable vascular staining in cav-1−/− mice (Figure 1, A and B); in contrast, vascular CD31 staining was equivalent in wild-type and cav-1−/− mice (Figure 1, C and D).
Figure 1.

Immunostaining for caveolin-1 in flank skin of wild-type (A) and cav-1−/− (B) mice with the N1–20 anti-caveolin antibody. Numerous blood vessels are labeled in wild-type skin, but staining in cav-1−/− skin is entirely negative. Similar results (not shown) were observed with the N1–97 antibody. CD31 antibodies showed equivalent vascular staining in wild-type (C) and cav-1−/− (D) mice. v identifies some of the unlabeled blood vessels in (B). Magnification bars = 50 μm.
To localize caveolin-1 within endothelial cells we performed electron microscopic immuno-nanogold cytochemistry with these same antibodies. As expected, caveolae were numerous in the capillary endothelium of wild-type mice (Figure 2, A and B; Table 1), and nearly all labeled strongly with anti-caveolin-1 antibodies (Figure 2C). In contrast, few caveolae were found in the capillary endothelium of cav-1−/− mice; total caveolae-sized (50 to 100 nm) vesicles in the cytoplasm were reduced by nearly 80% and caveolae attached to the luminal or abluminal plasma membranes by nearly 90% (Figure 2, D–F; Table 1). In addition, the few remaining caveolae in cav-1−/− capillary endothelium were negative for caveolin-1 (Figure 2F).
Figure 2.

Electron micrographs of flank skin capillaries of wild-type (A–C) and cav-1−/− mice (D–F). Boxes in (A) and (D) indicate portions enlarged in (B) and (E), respectively. Numerous 50- to 100-nm vesicles (caveolae), some indicated by arrows, are present in the cytoplasm and attached to both luminal and abluminal plasma membranes of wild-type capillary endothelial cells (A and B), whereas caveolae are largely, although not entirely, absent from capillaries of cav-1−/− mice (D and E). Immuno-nanogold staining for caveolin-1 demonstrates strong labeling of caveolae in wild-type capillaries (C) and absence of labeling in their cav-1−/− counterparts (F). L, lumen; asterisk, multivesicular body. Magnification bars = 1 μm.
Table 1.
Caveolae and VVOs in Capillary and Venular Endothelium of Wild-Type and Cav-1−/− Mice
| Blood vessel types | No. of vessels studied | Total vessel luminal perimeter evaluated (μm) | Total vessel abluminal perimeter evaluated (μm) | Total vessel area evaluated (μm2) | No. of VVOs/μm | No. of vesicles or vacuoles per VVO | No. of individual caveolae or vesicles/μm2 cytoplasm | No. of caveolae or vesicles facing on lumen or ablumen/μm |
|---|---|---|---|---|---|---|---|---|
| Capillaries +/+ | 33 | 198.3 | 205.8 | 79.9 | 10.82 ± 0.46 | 3.4 ± 0.12 | ||
| Capillaries −/− | 25 | 186.0 | 192.5 | 86.4 | 2.33 ± 0.16* | 0.36 ± 0.02* | ||
| Venules +/+ | 21 | 140.6 | 143.3 | 114.1 | 1.08 ± 0.08 | 10.25 ± 0.63 | 1.5 ± 0.25 | 0.97 ± 0.09 |
| Venules −/− | 17 | 152.9 | 155.7 | 131.6 | 0.96 ± 0.08 | 12.36 ± 1.30 | 0.60 ± 0.10† | 0.21 ± 0.04* |
P < 0.0001, as determined by Kruskal-Wallis ANOVA and Dunn’s multiple comparison test.
P < 0.005.
Turning to venules, we found that in wild-type mice nearly all caveolae-sized (50 to 100 nm) vesicles that were in contact with or closely apposed to the endothelial cell luminal or abluminal plasma membranes labeled strongly for caveolin-1 (Figure 3, A–E). However, only one third to one half of the larger, intracytoplasmic VVO vesicles and vacuoles were labeled, and these were generally labeled less intensely, with fewer nanogold particles per vesicle or vacuole. We could not quantify labeling more precisely because nanogold particles frequently obscured underlying vesicles and vacuoles. As with capillaries, venule immuno-nanogold labeling for caveolin was entirely negative in cav-1−/− mice (Figure 4C).
Figure 3.

Electron microscopic immuno-nanogold labeling of wild-type venular endothelium with antibodies against caveolin-1. A and B: Larger VVO vesicles and vacuoles labeled variably, some being strongly positive, whereas others are unlabeled. In contrast, 50- to 100-nm vesicles facing the luminal and abluminal surfaces, corresponding to the location of caveolae, labeled consistently. C and D: Enlargements of boxed areas in A, as indicated. E: Lateral borders of adjoining endothelial cells are closely apposed (open arrowheads) and display a normal adherens junction (arrow). Magnification bars = 250 nm.
Figure 4.

A and B: Electron micrographs illustrating structurally normal VVOs in venular endothelium of cav-1−/− mice. C: Electron microscopic section of cav-1−/− venular endothelium shows lack of immuno-nanogold labeling with anti-caveolin antibody. L, lumen. In B, arrow indicates the normally closed specialized junction, and, below, normally apposed interendothelial cell interface. Magnification bars = 1 μm.
VVOs and Isolated Vesicles in Cav-1−/− and Wild-Type Mice
We next performed electron microscopy to evaluate the frequency of VVOs and caveolae in the venular endothelium of cav-1−/− mice. As described previously,37 VVOs were arbitrarily defined as clusters of three or more (generally many more) interconnecting, smooth membrane-coated vesicles and vacuoles, typically of widely varying size. Single uncoated, caveolae-sized (50 to 100 nm) vesicles or two such interconnected vesicles were separately classified; many of these were attached to the luminal or abluminal plasma membrane and thus are probably caveolae. We found that VVO frequency and vesicle/vacuole composition in cav-1−/− mice did not differ from that of their wild-type counterparts (Figure 4; Table 1). However, the numbers of single or double caveolae-sized uncoated vesicles were strikingly reduced in the venular endothelium of cav-1−/− venular endothelium; total numbers of vesicles in the cytoplasm were reduced by 60% in cav-1−/− mice and those attached to the luminal or abluminal plasma membrane were reduced by nearly 80% (Table 1). In both capillary and venular endothelial cells, lateral plasma membranes contained normal-appearing adherens junctions and interendothelial cell clefts that were indistinguishable between wild-type and cav-1−/− mice (Figures 2, A and D, 3E, and 4B).
AVH in Cav-1−/− and Wild-Type Mice
Several groups25,30,31 have reported that macromolecules such as albumin are cleared more rapidly from plasma in cav-1−/− than in control mice. Our own data indicate that plasma albumin accumulated to a significantly greater extent in skeletal muscle and mesentery of cav-1−/− mice than in that of their wild-type counterparts; however, in flank skin, a tissue not previously reported on, there was no difference (Supplemental Figure S1, see http://ajp.amjpathol.org). To evaluate AVH in cav-1−/− mice, we performed the Miles assay. As shown in Figure 5, permeability after intradermal injection of HBSS did not differ significantly between wild-type and cav-1−/− mice; however, AVH induced by VEGF-A was greatly reduced in cav-1−/− compared with that in wild-type mice.
Figure 5.
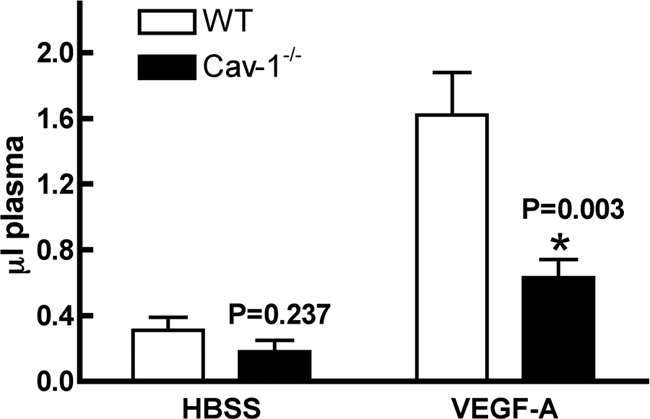
Miles assay performed in flank skin of wild-type (WT) and cav-1−/− mice. Evans blue dye was injected just before intradermal injection of HBSS or VEGF-A, and skin sites were harvested 30 minutes later. Dye was extracted and microliters of accumulated plasma (mean ± SEM) was calculated (see Materials and Methods). Data are from 10 wild-type and 10 cav-1−/− mice.
Angiogenesis and Associated CVH Induced by Ad-VEGF-A164 in Cav-1−/− and Wild-Type Mice
To define the role of caveolin-1 in VEGF-A-induced angiogenesis, we made use of a standard in vivo model in which an adenoviral vector expressing VEGF-A164 (Ad-VEGF-A164) is injected into flank skin.46,47,48 In wild-type C57BL/6 mice, Ad-VEGF-A164 induced the expected, strong angiogenic response with formation of numerous mother vessels and extensive edema (Figure 6, A and B); in contrast, angiogenesis, mother vessel formation and edema were greatly reduced in cav-1−/− mice (Figure 6, C and D). These findings were confirmed by assessing microvascular density (Figure 6E). As an additional means of quantifying angiogenesis and vascular permeability, we made use of a recently developed, double tracer technique that quantifies both intravascular plasma volume (a measure of angiogenesis) and, separately, the amount of extravasated plasma, both expressed as microliters of plasma.45,48 As shown in Figure 6F, the volumes of both intravascular plasma and extravasated plasma were greatly reduced in cav-1−/− mice.
Figure 6.

Angiogenic response 4 days after injection of Ad-VEGF-A164 into flank skin of wild-type (WT) (A and B) and cav-1−/− mice (C and D). A and C: Macroscopic appearance of angiogenic sites 30 minutes after i.v. injection of Evans blue dye. Note the striking difference in overall angiogenic response and bluing intensity between wild-type and cav-1−/− mice. B and D: Histological analysis shows striking angiogenesis with mother vessel formation and edema in wild-type versus cav-1−/− reactions; differences in skin thickness reflect differences in tissue edema. E: Microvascular density (mean ± SEM) as determined in wild-type (N = 7) and cav-1−/− (N = 6) angiogenic sites. *P < 0.0001. F: Intravascular and extravasated volumes (microliters) of plasma (mean ± SEM) in skin sites of wild-type (N = 8) versus cav-1−/− (N = 8) mice injected 4 days previously with Ad-VEGF-A164. See Materials and Methods and references45,48 for calculations. Magnification bars = 100 μm.
Tumor Growth and Angiogenesis in Cav-1−/− and Wild-Type Mice
To determine whether the lack of caveolin-1 affected tumor growth and associated angiogenesis, we injected B16 melanoma cells into the subcutaneous space of syngeneic wild-type C57BL/6 and cav-1−/− mice. Tumors grew to large sizes in wild-type mice, but growth in cav-1−/− mice was greatly retarded and was accompanied by diminished angiogenesis and mother vessel generation (Figure 7, A–D).
Figure 7.

Differential growth and vascularity of B16 melanomas in the subcutaneous space of wild-type (WT) and cav-1−/− mice. A: Appearance of typical tumors (of 16 studied) growing in wild-type and cav-1−/− mice. B: Tumor weights (mean ± SEM). C and D: Vascularity of tumors in wild-type and cav-1−/− mice. T, tumor; asterisks indicate some mother vessels of varying size in tumors growing in wild-type mice and are greatly reduced in cav-1−/− mice. Magnification bars = 100 μm.
Discussion
The data presented here demonstrate that the frequency of caveolae was greatly diminished, although not entirely eliminated, in skin capillary and venular endothelial cells of cav-1−/− mice (Table 1). If caveolae are defined as single or doublet vesicles of 50 to 100 nm diameter that were free in cytoplasm, they were reduced by almost 80% in capillary endothelium in cav-1−/− mice compared with wild-type mice; if they are instead defined as 50- to 100-nm diameter vesicles attached to the luminal or abluminal plasma membrane, they were reduced by ∼90% in cav-1−/− mice (Table 1). Using the same criteria, 50- to 100-nm diameter vesicles were reduced by ∼60 and ∼80%, respectively, in the venular endothelium of cav-1−/− mice. The calculated frequency of residual caveolae in venular endothelium of cav-1−/− mice may be an overestimate in that some of the vesicles counted as caveolae were probably attached to vesicles or vacuoles in other section planes and therefore were parts of VVOs and not in fact caveolae.
In wild-type mice, nearly all capillary caveolae (Figure 2C) and 50- to 100-nm diameter vesicles (presumably caveolae) in venular endothelium (Figure 3) labeled strongly with antibodies against caveolin-1. On the other hand, only one third to one half of the larger VVO vesicles and vacuoles in venular endothelium were so labeled, and these generally labeled with less intensity than smaller, caveolae-sized vesicles (Figure 3). Nonetheless, VVOs were found in equal numbers and with indistinguishable vesicle and vacuole composition in the venular endothelium of cav-1−/− and wild-type mice (Table 1). No caveolin-1 staining was observed in either the capillaries or venules of cav-1−/− mice (Figures 1, 2F, and 4C).
These findings are of interest for several reasons. First, they demonstrate that, although caveolin-1 is important for caveolae formation, it is not absolutely essential in that small numbers of caveolae, ∼10 to 20% of normal, persisted in the capillaries of cav-1−/− mice (Figure 2, D–F; Table 1). Second, caveolin-1 is not essential for the formation of VVOs in venules (Figure 4; Table 1). These findings speak to the possible relationship of VVOs to caveolae. Some years ago we reported that the areas and volumes of VVO vesicles and vacuoles did not fall on a continuum but rather were multiples of a small, 50- to 100-nm diameter vesicle, ie, a unit vesicle the size of caveolae.37 On this basis, we proposed that VVOs were formed from caveolae, variable numbers of which, we suggested, fused together to form dimers and multimers, up to vacuoles as much as 10 times the size of unit caveolae. In view of our new data, this conclusion seems less likely. Caveolae, ie, single or doublet 50- to 100-nm vesicles, were greatly reduced in venular endothelium, whereas VVOs were not diminished in either number or composition. It would seem, therefore, that VVOs are composed of a population of uncoated vesicles and vacuoles that is distinct from that of caveolae, although they contain variable amounts of caveolin.
The pathways by which plasma proteins cross vascular endothelium have been the subject of debate for many decades (reviewed in5). It had come to be accepted that, at least in the case of BVP, plasma proteins cross normal capillary endothelium by way of shuttling or linked caveolae.1,2,3 However, it has now been reported by three groups25,30,31 that macromolecules of several types are cleared more rapidly from plasma in cav-1−/− than in control mice. To account for this finding, Schubert et al31 reported a shortening of “tight” junctions in the lung capillary endothelium of cav-1−/− mice and on that basis suggested that increased paracellular permeability accounted for the increased basal permeability they observed. On the other hand, Rosengren et al30 found no difference in adherens junctions in hyperpermeable abdominal wall capillaries of cav-1−/− mice, and we noted no morphological differences in either adherens junctions or in the length of interendothelial cell interfaces in skin capillaries. However, the vascular beds of different tissues may behave differently. The ultrastructural studies performed by Schubert et al,31 Rosengren et al,30 and our group examined three different tissues, lung, peritoneal wall, and flank skin, respectively. In addition, although Schubert et al31 found increased accumulation of extravasated albumin in many tissues in cav-1−/− mice, in at least one tissue (aorta) such accumulation was reduced. Furthermore, we found increased albumin accumulation in skeletal muscle and mesentery of cav-1−/− mice, but no significant difference in such accumulation in skin (Supplemental Figure S1, see http://ajp.amjpathol.org), a tissue not previously reported on. Thus, although overall BVP to albumin may be increased in cav-1−/− mice, there are distinct differences of permeability in different tissues and perhaps in the pathways by which albumin and other plasma proteins extravasate.
Using the Miles assay, we found that AVH in skin was strikingly decreased in cav-1−/− compared with that in wild-type mice (Figure 5). This difference in AVH could be tissue specific because, as already noted, BVP in skin did not differ between control and cav-1−/− mice. However, another explanation is that different types of microvessels participate in BVP and AVH.32 BVP takes place in capillaries, whereas AVH involves postcapillary venules. In AVH, as in BVP, there is disagreement as to whether plasma proteins extravasate by transcellular or paracellular routes. Majno et al43 and subsequently others44 proposed that the leakage induced by vascular permeabilizing agents resulted from a pulling apart of adjacent venular endothelial cells. On the other hand, several groups, including ours, have reported a transcellular route for plasma protein extravasation in AVH.36,37,42,52,53 We found that tracer proteins such as ferritin and horseradish peroxidase extravasate through VVOs after exposure to VEGF, histamine, and other vascular permeabilizing agents.36,37,39,40,42 Therefore, the finding that AVH is strikingly impaired in cav-1−/− mice suggests that, although caveolin-1 is not essential for VVO structure, it may be very important for VVO function, even though only one third to one half of large VVO vesicles and vacuoles expressed caveolin-1 as determined by immuno-nanogold labeling (Figure 3).
Bauer et al23 reported that AVH is decreased in transgenic mice that selectively overexpress caveolin-1 in vascular endothelium, whereas we found AVH to be strikingly reduced in cav-1−/− mice (Figure 5). This paradox can be resolved if it is assumed that just the right amount of caveolin-1 is necessary for proper regulation of VEGF-A-induced permeability and that both lower and higher levels of caveolin-1 impair that response. In accord with this suggestion, others have called attention to the importance of both the levels and the appropriate intracellular distribution of caveolin-1 in endothelial nitric oxide synthase signaling; endothelial nitric oxide synthase has an important role in vascular permeability, and this function was found to be perturbed both in cav-1−/− mice and in caveolin-1-overexpressing mice.9,23,24,29,54,55
Our data also indicate that caveolin-1 has an important role in pathological angiogenesis and associated CVH. VEGF-A, other growth factors, and tumors initiate angiogenesis by inducing pre-existing microvessels, primarily venules, to enlarge to form a characteristic type of hyperpermeable, pericyte-poor sinusoid, the “mother” vessel.46,47,56,57,58 VVOs contribute to this process by donating their membranes to the rapidly expanding plasma membrane that is required as venules enlarge to form mother vessels. Further, residual, short chains of VVOs provide an important pathway for mother vessel hyperpermeability.45 Mother vessels formed poorly in cav-1−/− mice in response to either Ad-VEGF-A164 or B16 melanomas (Figures 6 and 7). Because the increased permeability of angiogenesis occurs primarily through mother vessels, it is not surprising that CVH was also reduced. These data provide further evidence that, although structurally intact in cav-1−/− mice, VVOs function poorly in the absence of caveolin-1. To a lesser extent, Ad-VEGF-A164 also induces capillaries to form mother vessels in wild-type mice, although these are smaller than the ones induced from venules; it is possible, therefore, that the lack of caveolin-1 and caveolae in capillary endothelium may also have contributed to the failure of mother vessel formation in cav-1−/− mice.
There remains controversy as to the role of caveolin-1 in pathological angiogenesis and associated chronic vascular hyperpermeability. Different reports indicate that angiogenesis and permeability are increased, decreased, or unaltered in cav-1−/− mice. Consistent with our findings (Figures 6 and 7), Griffoni et al26 reported that knockdown of caveolin-1 by antisense oligonucleotides impaired angiogenesis, both in vitro in a fibrin gel assay and in vivo on chicken chorio-allantoic membranes. In addition, Jasmin et al59 found that ischemic cerebral injury generated an impaired angiogenic response in cav-1−/− mice, and Woodman et al22 reported decreased angiogenesis induced by basic fibroblast growth factor in Matrigel plugs, and, as reported here (Figure 7), decreased B16 tumor growth and associated angiogenesis. On the other hand, Bauer et al23 found that angiogenesis induced by Ad-VEGF164 and by tissue ischemia was significantly impaired in transgenic mice in which caveolin-1 was overexpressed. As noted above with respect to AVH, this finding could be explained if a “just right” level of caveolin-1 is required for an optimal response to VEGF-A and that higher or lower levels impair angiogenesis. However, there are also data in the literature that are contrary to our findings and to those of the authors cited above. Thus, Lin et al27 reported that the Lewis lung carcinoma induced increased angiogenesis and associated vascular permeability when grown in cav-1−/− mice. Finally, there are data indicating that tumor-associated vascular permeability is unchanged in cav-1−/− mice. Thus, DeWever et al25 found that permeability to 125I-albumin, although increased in normal tissues of cav-1−/− mice, was not statistically different in B16 melanomas grown in cav-1−/− versus wild-type mice. In addition, in contrast to our findings and those of Woodman et al,22 these B16 tumors grew larger and generated increased numbers of new blood vessels in cav-1−/− mice. Further work will be required to reconcile these disparate findings. Differences in tumors studied and in techniques used to evaluate angiogenesis and permeability, failure to distinguish among the several different types of vascular permeability (BVP, AVH, and CVH), and the study of different vascular beds may have contributed to the differences observed.
Supplementary Material
Footnotes
Address reprint requests to Harold F. Dvorak, M.D., Department of Pathology, Beth Israel Deaconess Medical Center, 330 Brookline Ave., RN227C, Boston, MA 02215. E-mail: hdvorak@bidmc.harvard.edu.
Supported in part by U.S. Public Health Service grants HL-64402 and P01 CA92644 and by a contract from the National Foundation for Cancer Research.
S.H.-C. and D.F. contributed equally to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Palade GE, Bruns RR. Structural modulations of plasmalemmal vesicles. J Cell Biol. 1968;37:633–649. doi: 10.1083/jcb.37.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RR, Palade GE. Studies on blood capillaries. II. Transport of ferritin molecules across the wall of muscle capillaries. J Cell Biol. 1968;37:277–299. doi: 10.1083/jcb.37.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simionescu N. Cellular aspects of transcapillary exchange. Physiol Rev. 1983;63:1536–1579. doi: 10.1152/physrev.1983.63.4.1536. [DOI] [PubMed] [Google Scholar]
- Rippe B, Haraldsson B. Transport of macromolecules across microvascular walls: the two-pore theory. Physiol Rev. 1994;74:163–219. doi: 10.1152/physrev.1994.74.1.163. [DOI] [PubMed] [Google Scholar]
- Dvorak A. Electron microscopic-facilitated understanding of endothelial cell biology. Aird W, editor. New York: Cambridge University Press,; Contributions Established during the 1950s and 1960s. 2007:pp 643–656. [Google Scholar]
- Carver LA, Schnitzer JE. Caveolae: mining little caves for new cancer targets. Nat Rev Cancer. 2003;3:571–581. doi: 10.1038/nrc1146. [DOI] [PubMed] [Google Scholar]
- Frank PG, Pavlides S, Cheung MW, Daumer K, Lisanti MP. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am J Physiol Cell Physiol. 2008;295:C242–C248. doi: 10.1152/ajpcell.00185.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol. 2003;23:1161–1168. doi: 10.1161/01.ATV.0000070546.16946.3A. [DOI] [PubMed] [Google Scholar]
- Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004;94:1408–1417. doi: 10.1161/01.RES.0000129178.56294.17. [DOI] [PubMed] [Google Scholar]
- Navarro A, Anand-Apte B, Parat MO. A role for caveolae in cell migration. FASEB J. 2004;18:1801–1811. doi: 10.1096/fj.04-2516rev. [DOI] [PubMed] [Google Scholar]
- Vallejo J, Hardin CD. Expression of caveolin-1 in lymphocytes induces caveolae formation and recruitment of phosphofructokinase to the plasma membrane. FASEB J. 2005;19:586–587. doi: 10.1096/fj.04-2380fje. [DOI] [PubMed] [Google Scholar]
- Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, Shirani J, Razani B, Tang B, Jelicks LA, Factor SM, Weiss LM, Tanowitz HB, Lisanti MP. Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol. 2002;160:2207–2217. doi: 10.1016/S0002-9440(10)61168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- Williams TM, Lisanti MP. The Caveolin genes: from cell biology to medicine. Ann Med. 2004;36:584–595. doi: 10.1080/07853890410018899. [DOI] [PubMed] [Google Scholar]
- Tang Z, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–2261. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- Frank PG, Lisanti MP. Caveolin-1 and caveolae in atherosclerosis: differential roles in fatty streak formation and neointimal hyperplasia. Curr Opin Lipidol. 2004;15:523–529. doi: 10.1097/00041433-200410000-00005. [DOI] [PubMed] [Google Scholar]
- Frank PG, Pavlides S, Lisanti MP. Caveolae and transcytosis in endothelial cells: role in atherosclerosis. Cell Tissue Res. 2009;335:41–47. doi: 10.1007/s00441-008-0659-8. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Medina FA, de Almeida CJ, Dew E, Li J, Bonuccelli G, Williams TM, Cohen AW, Pestell RG, Frank PG, Tanowitz HB, Lisanti MP. Caveolin-1-deficient mice show defects in innate immunity and inflammatory immune response during Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74:6665–6674. doi: 10.1128/IAI.00949-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Schubert W, Sotgia F, Cohen AW, Capozza F, Bonuccelli G, Bruno C, Minetti C, Bonilla E, Dimauro S, Lisanti MP. Caveolin-1−/−- and caveolin-2−/−-deficient mice both display numerous skeletal muscle abnormalities, with tubular aggregate formation. Am J Pathol. 2007;170:316–333. doi: 10.2353/ajpath.2007.060687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman SE, Ashton AW, Schubert W, Lee H, Williams TM, Medina FA, Wyckoff JB, Combs TP, Lisanti MP. Caveolin-1 knockout mice show an impaired angiogenic response to exogenous stimuli. Am J Pathol. 2003;162:2059–2068. doi: 10.1016/S0002-9440(10)64337-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer PM, Yu J, Chen Y, Hickey R, Bernatchez PN, Looft-Wilson R, Huang Y, Giordano F, Stan RV, Sessa WC. Endothelial-specific expression of caveolin-1 impairs microvascular permeability and angiogenesis. Proc Natl Acad Sci USA. 2005;102:204–209. doi: 10.1073/pnas.0406092102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouet A, DeWever J, Martinive P, Havaux X, Bouzin C, Sonveaux P, Feron O. Antitumor effects of in vivo caveolin gene delivery are associated with the inhibition of the proangiogenic and vasodilatory effects of nitric oxide. FASEB J. 2005;19:602–604. doi: 10.1096/fj.04-2682fje. [DOI] [PubMed] [Google Scholar]
- DeWever J, Frerart F, Bouzin C, Baudelet C, Ansiaux R, Sonveaux P, Gallez B, Dessy C, Feron O. Caveolin-1 is critical for the maturation of tumor blood vessels through the regulation of both endothelial tube formation and mural cell recruitment. Am J Pathol. 2007;171:1619–1628. doi: 10.2353/ajpath.2007.060968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffoni C, Spisni E, Santi S, Riccio M, Guarnieri T, Tomasi V. Knockdown of caveolin-1 by antisense oligonucleotides impairs angiogenesis in vitro and in vivo. Biochem Biophys Res Commun. 2000;276:756–761. doi: 10.1006/bbrc.2000.3484. [DOI] [PubMed] [Google Scholar]
- Lin MI, Yu J, Murata T, Sessa WC. Caveolin-1-deficient mice have increased tumor microvascular permeability, angiogenesis, and growth. Cancer Res. 2007;67:2849–2856. doi: 10.1158/0008-5472.CAN-06-4082. [DOI] [PubMed] [Google Scholar]
- Liu J, Razani B, Tang S, Terman BI, Ware JA, Lisanti MP. Angiogenesis activators and inhibitors differentially regulate caveolin-1 expression and caveolae formation in vascular endothelial cells. Angiogenesis inhibitors block vascular endothelial growth factor-induced down-regulation of caveolin-1. J Biol Chem. 1999;274:15781–15785. doi: 10.1074/jbc.274.22.15781. [DOI] [PubMed] [Google Scholar]
- Sonveaux P, Martinive P, DeWever J, Batova Z, Daneau G, Pelat M, Ghisdal P, Gregoire V, Dessy C, Balligand JL, Feron O. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ Res. 2004;95:154–161. doi: 10.1161/01.RES.0000136344.27825.72. [DOI] [PubMed] [Google Scholar]
- Rosengren BI, Rippe A, Rippe C, Venturoli D, Sward K, Rippe B. Transvascular protein transport in mice lacking endothelial caveolae. Am J Physiol Heart Circ Physiol. 2006;291:H1371–H1377. doi: 10.1152/ajpheart.01364.2005. [DOI] [PubMed] [Google Scholar]
- Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP. Microvascular hyperpermeability in caveolin-1 (−/−) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, l-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem. 2002;277:40091–40098. doi: 10.1074/jbc.M205948200. [DOI] [PubMed] [Google Scholar]
- Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol. 1961;11:571–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Nagy J, Brekken R, Pettersson A, Manseau E, Pyne K, Mulligan R, Thorpe P, Dvorak H, Dvorak A. Ultrastructural localization of the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) receptor-2 (FLK-1, KDR) in normal mouse kidney and in the hyperpermeable vessels induced by VPF/VEGF-expressing tumors and adenoviral vectors. J Histochem Cytochem. 2000:545–556. doi: 10.1177/002215540004800412. [DOI] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Dvorak AM, Dvorak HF. Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc Res. 2000;59:24–37. doi: 10.1006/mvre.1999.2207. [DOI] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med. 1996;183:1981–1986. doi: 10.1084/jem.183.5.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Pyne K, Hammel I, Dvorak HF, Dvorak AM. Pathways of macromolecular extravasation across microvascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation. 1999;6:23–44. [PubMed] [Google Scholar]
- Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF. The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol. 1996;59:100–115. [PubMed] [Google Scholar]
- Dvorak AM, Feng D. The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem. 2001;49:419–432. doi: 10.1177/002215540104900401. [DOI] [PubMed] [Google Scholar]
- Kohn S, Nagy JA, Dvorak HF, Dvorak AM. Pathways of macromolecular tracer transport across venules and small veins. Structural basis for the hyperpermeability of tumor blood vessels. Lab Invest. 1992;67:596–607. [PubMed] [Google Scholar]
- Stan RV. Endothelial stomatal and fenestral diaphragms in normal vessels and angiogenesis. J Cell Mol Med. 2007;11:621–643. doi: 10.1111/j.1582-4934.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Hipp J, Pyne K, Dvorak HF, Dvorak AM. Reinterpretation of endothelial cell gaps induced by vasoactive mediators in guinea-pig, mouse and rat: many are transcellular pores. J Physiol. 1997;504(Pt 3):747–761. doi: 10.1111/j.1469-7793.1997.747bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majno G, Shea SM, Leventhal M. Endothelial contraction induced by histamine-type mediators: an electron microscopic study. J Cell Biol. 1969;42:647–672. doi: 10.1083/jcb.42.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Hirata A, Thurston G, Fujiwara T, Neal CR, Michel CC, McDonald DM. Endothelial gaps: time course of formation and closure in inflamed venules of rats. Am J Physiol. 1997;272:L155–170. doi: 10.1152/ajplung.1997.272.1.L155. [DOI] [PubMed] [Google Scholar]
- Nagy JA, Feng D, Vasile E, Wong WH, Shih SC, Dvorak AM, Dvorak HF. Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab Invest. 2006;86:767–780. doi: 10.1038/labinvest.3700436. [DOI] [PubMed] [Google Scholar]
- Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, Carter R, Krieger JE, Manseau EJ, Harvey VS, Eckelhoefer IA, Feng D, Dvorak AM, Mulligan RC, Dvorak HF. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251–275. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- Nagy JA, Shih SC, Wong WH, Dvorak AM, Dvorak HF. The adenoviral vector angiogenesis/lymphangiogenesis assay. Methods Enzymol. 2008;444:43–64. doi: 10.1016/S0076-6879(08)02803-6. [DOI] [PubMed] [Google Scholar]
- Zeng H, Qin L, Zhao D, Tan X, Manseau EJ, Van Hoang M, Senger DR, Brown LF, Nagy JA, Dvorak HF. Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J Exp Med. 2006;203:719–729. doi: 10.1084/jem.20051523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Ultrastructural localization of platelet endothelial cell adhesion molecule (PECAM-1. CD31) in vascular endothelium. J Histochem Cytochem. 2004;52:87–101. doi: 10.1177/002215540405200109. [DOI] [PubMed] [Google Scholar]
- Dvorak A. Procedural guide to specimen handling for the ultrastructural pathology service laboratory. J Electron Microsc Tech. 1987;6:255–301. [Google Scholar]
- Feng Y, Venema VJ, Venema RC, Tsai N, Behzadian MA, Caldwell RB. VEGF-induced permeability increase is mediated by caveolae. Invest Ophthalmol Vis Sci. 1999;40:157–167. [PubMed] [Google Scholar]
- Michel CC, Neal CR. Openings through endothelial cells associated with increased microvascular permeability. Microcirculation. 1999;6:45–54. [PubMed] [Google Scholar]
- Feron O, Balligand JL. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc Res. 2006;69:788–797. doi: 10.1016/j.cardiores.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- Björndahl MA, Cao R, Burton JB, Brakenhielm E, Religa P, Galter D, Wu L, Cao Y. Vascular endothelial growth factor-a promotes peritumoral lymphangiogenesis and lymphatic metastasis. Cancer Res. 2005;65:9261–9268. doi: 10.1158/0008-5472.CAN-04-2345. [DOI] [PubMed] [Google Scholar]
- Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi JH, Claesson-Welsh L, Alitalo K. Vascular endothelial growth factor C induces angiogenesis in vivo. Proc Natl Acad Sci USA. 1998;95:14389–14394. doi: 10.1073/pnas.95.24.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen LJ, Cao R, Hedlund EM, Wang Z, Zhao X, Wetterskog D, Funa K, Brakenhielm E, Cao Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest. 2007;117:2766–2777. doi: 10.1172/JCI32479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin JF, Malhotra S, Singh Dhallu M, Mercier I, Rosenbaum DM, Lisanti MP. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res. 2007;100:721–729. doi: 10.1161/01.RES.0000260180.42709.29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.