

Abstract
Little is known about the roles of beta-arrestins in the regulation of brain CB1 cannabinoid receptors. The present study investigated the role of beta-arrestin2 in cannabinoid behavioral effects using beta-arrestin2-/- mice and their wild-type counterparts. A variety of cannabinoid ligands from different chemical classes that exhibit a variety of efficacies for activation of CB1 receptors were investigated, including Δ9-tetrahydrocannabinol (THC), CP55940, methanandamide, JWH-073 and O-1812. THC produced both greater antinociception and greater decreases in body temperature in beta-arrestin2-/- compared to beta-arrestin2+/+ mice. However, there were no significant differences in either assay for the other CB1 agonists. Antagonist radioligand binding indicated no difference in the density of cannabinoid CB1 receptors in the cerebellum, cortex or hippocampus of beta-arrestin2+/+ and -/- mice. These data demonstrate that beta-arrestin2 may regulate cannabinoid CB1 receptor sensitivity in an agonist-specific manner.
Keywords: beta-arrestin2, knock-out, cannabinoid, CB1 receptor, CP55940, JWH-073, O-1812, Δ9-tetrahydrocannabinol, SR141716A, antinociception, mouse
INTRODUCTION
Cannabinoid CB1 receptors appear to mediate the majority of the CNS actions of cannabinoid compounds (Breivogel and Childers 1998). CB1 are G-protein coupled receptors (GPCRs) and couple primarily to the Gαi and/or Gαo subtypes (Childers and Breivogel 1998; Prather et al. 2000). Cannabinoid agonists acting through CB1 receptors elicit catalepsy, analgesia, decreases in spontaneous activity and body temperature (Adams and Martin 1996), and disruption of memory (Heyser et al. 1993).
Four subtypes of arrestins and seven subtypes of G-protein coupled Receptor Kinases (GRKs) have been cloned. Arrestin1 and 4 and GRK1 and 7 are visual subtypes and regulate photoreceptor molecules (rhodopsin). Arrestin2 and 3 (also known respectively as beta-arrestin1 and 2) and GRK2, 3, 5, and 6 are distributed widely, and are believed to regulate a variety of GPCRs (Luttrell and Lefkowitz 2002). There is some evidence that each subtype of GRK phosphorylates a subset of GPCRs, and that each subtype of arrestin binds a subset of GPCRs. Alternatively, it may be that each of the two beta-arrestins binds the same GPCRs but exerts different effects upon the receptor. The role of specific subtypes of arrestin and/or GRK has been assessed for only a few GPCRs. Some evidence for which subtypes of GRK and arrestin modulate cannabinoid receptors was provided by co-transfection of CB1, beta-arrestin2 and GRK3 into oocytes. These studies indicated that beta-arrestin2 and GRK3 could induce desensitization of CB1 (Jin et al. 1999). A second study involving cannabinoid receptors indicated that expression of dominant-negative forms of GRK2 and beta-arrestin inhibited the desensitization of the response to cannabinoid ligands in cultured hippocampal neurons (Kouznetsova et al. 2002).
Much of the work to match specific GPCRs with beta-arrestin and GRK subtypes has been accomplished in transfected cell lines, where it has been noted that over-expression of these proteins may lead to non-selective or irrelevant interactions (Gainetdinov et al. 2000). Thus, the activity of specific subtypes in the regulation of specific GPCRs remains largely undetermined. Mouse brain naturally expresses each of these proteins, so comparison of wild-type and transgenic beta-arrestin2 knock-out mice represents a strategy that provides results that may be more physiologically-relevant. Previous studies have shown that beta-arrestin2-/-mice were more sensitive to the acute effects of the mu opioid agonist morphine (Bohn et al. 1999), and exhibited attenuated tolerance to chronic morphine (Bohn et al. 2002). Interestingly, a morphine treatment that failed to produce significant tolerance still resulted in morphine dependence in these animals (Bohn et al. 2000a), but morphine did not show enhanced side effects (Raehal et al. 2005). Other investigators have found that GRK5-/- mice exhibited supersensitivity and impaired densensitization to muscarinic agonists (Gainetdinov et al. 1999).
The current study compared the acute effects of several cannabinoid receptor agonists in beta-arrestin2+/+ and -/- mice to examine the effect of beta-arrestin2 deletion on the efficacy of cannabinoids in whole mice. The classification of each compound in this study as a full or partial agonist is based on its ability to activate G-proteins via CB1 receptors in brain membranes as determined by [35S]GTPγS binding. The ED50 values reported here are previously-published reports of in vivo activity for the latency to tail withdrawal from a hot water bath (tail flick test of antinociception) and depression of rectal temperature in mice. Since the values for the two assays were quite similar, the average of the reported values for each compound is given below. CP55940 is a widely-used full agonist (Breivogel et al. 2001), and exhibits ED50 values around 0.1 mg/kg (Fan et al. 1994). Δ9-tetrahydrocannabinoid (THC) is the primary physiologically-active cannabinoid agonist found in Cannabis sativa. In contrast to CP55940, THC is a weak partial agonist (Sim et al. 1996; Burkey et al. 1997; Breivogel et al. 1998) and exhibits ED50 values around 1 mg/kg (Fan et al. 1994; Wiley et al. 1998). Methanandamide is a metabolically-stable analog of the endogenous cannabinoid, anandamide, that exhibits high efficacy and is classified as a full agonist or high efficacy partial agonist (Breivogel et al. 1998; Breivogel and Childers 2000). Methanandamide has reported ED50 values around 20 mg/kg (Adams et al. 1995). O-1812 is another analog of the endogenous cannabinoid, anandamide, which exhibits efficacy for G-protein activation similar to THC (Breivogel et al. 2001) with ED50 values around 0.03 mg/kg (Di Marzo et al. 2001). JWH-073 is an aminoalkylindole cannabinoid agonist with ED50 values around 2 mg/kg (Wiley et al. 1998). The efficacy of each agonist for activation of G-proteins in brain membranes is confirmed in the present study.
METHODS
SUBJECTS
C57BL/6 beta-arrestin2 knock-out and littermate wild-type mice were generously supplied by the laboratory of Dr. Robert Lefkowitz of Duke University (Durham, NC) and were bred in the animal facility of Campbell University School of Pharmacy. At Campbell University, homozygous wild-type were bred with homozygous wild-type and homozygous knock-outs were bred with homozygous knock-outs, obviating genotyping of the offspring. It has been reported that results from similar types of experiments using the same strain of beta-arrestin2-/- mice bred in this manner did not differ from those obtained in mice bred from heterozygous breeders (Raehal et al. 2005). All experiments were conducted in accordance with NIH guidelines for the care and use of laboratory animals and with an approved animal protocol from the Campbell University Animal Care and Use Committee.
IN VIVO ASSAYS
Drugs or vehicle (1:1:18 emulphor:ethanol:ddH2O) were administered by intraperitoneal (i.p.) injection at 0.01 ml/g of body weight. Due to the agonists, O-1812 and JWH-073 being available in limited supply and the knock-out mice being similarly limited in number, concentrations of each agonist were selected based on the ED50 values in the tail immersion and rectal temperature tests. Each drug was administered at a dose 10-26 times higher than the previously-determined ED50 values (see Table 1). These higher doses were chosen because in the previous studies the drugs were administered i.v., while they were given i.p. in the current study. Additionally, the previous studies used a 10 s cut-off time for the tail immersion test, while the current study utilized a 30 s cut-off to give a greater range of possible values and greater sensitivity to detect differences among groups of animals. O-1812 was used in the current study at a higher dose compared to its previously reported ED50, since an initial test assay using 0.1 mg/kg produced very little effect.
Table 1.
Comparison of previously-reported ED50 values and doses of agonists used.
| Drug | previous ED50 (mg/kg) | current dose (mg/kg) | ~ratio |
|---|---|---|---|
| CP55940 | 0.14, 0.05 1 | 0.3, 1 | 3, 10 |
| THC | 0.85, 0.78 4 | 10 | 12 |
| methanandamide | 20, 24 2 | 50, 250 | 2.3, 11 |
| O-1812 | 0.014, 0.050 3 | 2.5 | 78 |
| JWH-073 | 0.42, 1.1 4 | 20 | 26 |
The “previous ED50” values are those reported for the latency to tail withdrawal test and hypothermia in mice, respectively, though some were converted from other units (e.g. μmol/kg) to mg/kg. “Current dose” is that used in the present study and the “~ratio” is the current dose divided by the average of the previous ED50 values.
Latency to tail withdrawal as a measurement of antinociceptive activity
This method was similar to that previously described (Winters et al. 1988). In initial experiments, mice were held and the tip of the tail was immersed in a water bath with the temperature set to 50°C, 53°C or 56°C. When the tail was flicked from the water or after 30 s (cut-off time) elapsed, the time in the water was recorded and the mouse returned to its cage. In subsequent studies, latency was measured once prior to (basal latency), and at several times following (test latency), an i.p. injection of drug or vehicle. For this test and the hot plate test (see below), data were expressed as a percentage of the Maximum Possible Effect (%MPE) = (test latency − basal latency) / (cut-off time − basal latency) * 100% for each animal (Dewey et al. 1970). Mean and SEM for latency to tail withdrawal (in seconds) and %MPE were calculated for each group of 5-9 mice.
Hot plate measurement of antinociceptive activity
Mice were placed under a large beaker on a digitally-controlled Thermolyne Mirak (Dubuque, IA) hot plate with the temperature set to 56°C once prior to and at several times following an i.p. injection of drug or vehicle. When the mouse licked a rear paw, jumped, or the 60-s cut-off time elapsed, the mouse was removed immediately from the plate and the time on the plate was recorded. %MPE was calculated as above.
Rectal Temperature
Rectal temperature was determined to a tenth of a degree by a thermistor probe attached to a Traceable Digital Thermometer from Control Company (Friendswood, TX), lubricated with glycerin and inserted 2 cm. Temperature was taken prior to and at various times after i.p. injection of drug or vehicle. For each animal, the change in rectal temperature is determined as final minus pre-drug temperature and the data expressed as mean change in rectal temperature for each group.
AGONIST-STIMULATED [35S]GTPγS AND [3H]SR141716A RECEPTOR BINDING
Preparation of Brain Membranes
Brain regions were dissected from whole brains on ice and homogenized in cold membrane buffer (50 mM Tris-HCl pH 7.4, 3 mM MgCl2, 0.2 mM EGTA) and then centrifuged at 48,000 × g for 10 min at 4°C. Pellets were re-suspended in membrane buffer by homogenization, and then centrifuged again at 48,000 × g for 10 min at 4°C. Pellets from the second centrifugation were homogenized in membrane buffer, divided into cryovials and stored at −80°C until use.
[35S]GTPγS Binding to Brain Membranes
On the day of assay membranes were thawed, resuspended in membrane buffer containing 100 mM NaCl using a Polytron and preincubated for 10 min at 30°C in 0.004 units/ml adenosine deaminase (240 units/mg protein, Sigma Chemical Co.) to remove endogenous adenosine. Membranes were assayed for protein content (Bradford 1976) before addition to assay tubes. Assay tubes contained membrane homogenate with 10-20 μg of protein, membrane buffer with 100 mM NaCl, 0.1 nM [35S]GTPγS, 30 μM GDP, 1 mg/ml (w/v) bovine serum albumin (BSA) and various concentration of each agonist. Assays were incubated for 1 hr, and non-specific binding was determined in the presence of 30 μM unlabelled GTPγS. Each assay was terminated by filtration under vacuum through Whatman GF/B glass fiber filters, followed by three washes with cold Tris-HCl buffer, pH 7.4. Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency for [35S] after overnight extraction of the filters in 3.5 ml ScintiSafe Econo 1 scintillation fluid (Fisher Scientific).
[3H]SR141716A Receptor Binding to Brain Membranes
On the day of assay membranes were thawed, resuspended in assay buffer with 100 mM NaCl using a Polytron and assayed for protein content (Bradford 1976). Membrane homogenate containing 16 μg of membrane protein was added to assay tubes containing assay buffer, 1 mg/ml (w/v) bovine serum albumin (BSA) and [3H]SR141716A. Assays were incubated for 1 hr; non-specific binding was determined in the presence of 5 μM unlabelled SR141716A. Each assay was terminated by filtration under vacuum through Whatman GF/B glass fiber filters that had been soaked in cold Tris-HCl buffer, pH 7.4 with 5 mg/ml BSA, followed by seven washes with cold Tris-HCl buffer, pH 7.4 with 0.5 mg/ml BSA. Bound radioactivity was determined by liquid scintillation spectrophotometry at ~42% efficiency for [3H] after overnight extraction of the filters in 3.5 ml ScintiSafe Econo 1 scintillation fluid (Fisher Scientific).
Drugs and chemicals
SR141716A, [3H]SR141716A (16.9 Ci/mmol) and Δ9-tetrahydrocannabinol (THC) were provided by the NIDA Drug Supply Program/Research Triangle Institute (Research Triangle Park, NC). CP55940 and R-(+)-methanandamide were obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA). JWH-073 was provided by Dr. John Huffman of Clemson University and O-1812 was provided by Dr. Bill Martin of Virginia Commonwealth University and Raj Razdan of Organix, Inc. (Woburn, MA). [35S]guanosine-5’-O-(3-thiotriphosphate) ([35S]GTPγS) (1250 Ci/mmol) was purchased from Perkin Elmer (Boston, MA). Emulphor was generously donated by Dr. Allyn Howlett and Dr. Somnath Mukhopadhyay of North Carolina Central University. All other reagent grade or tissue culture grade chemicals and enzymes were obtained from Sigma Chemical Co. (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA).
DATA ANALYSIS
Significant effects of drugs were determined by 2-way ANOVA comparing the effect of each drug versus vehicle at each time after injection (time versus treatment) at p < 0.05 using Prism (GraphPad software, San Diego, California). Significant differences between the effects of each drug treatment in beta-arrestin2+/+ and -/-mice were determined by 2-way ANOVA (time versus genotype) at p < 0.05, followed by Bonferroni’s post-test (at p < 0.05) using Prism, to determine at which time points beta-arrestin2+/+ and -/- mice exhibited differences in response. In the text of the Results section, p values are those for 2-way ANOVAs. In the figures and legends, p values for differences between the genotypes at specific time points are from Bonferroni’s post-test, unless otherwise specified. All data are mean ± SEM of experiments performed with 5-9 mice per treatment group.
Specific binding of [35S]GTPγS was determined by subtracting the amount of binding obtained in the presence of 30 μM unlabeled GTPγS (non-specific) from values obtained in its absence. Net agonist-stimulated [35S]GTPγS binding values were calculated by subtracting basal binding values (obtained in the absence of agonist) from agonist-stimulated values (obtained in the presence of agonist) and percent stimulation was determined by dividing net values by the respective basal binding values. Data were fit to single-site concentration-effect curve models using Prism to obtain Emax and EC50 values. Specific binding of [3H]SR141716A was determined by subtracting the amount of binding obtained in the presence of 5 μM unlabeled SR141716A (non-specific) from values obtained with the same concentration of [3H]SR141716A in its absence. Kd and Bmax values for SR141716A were obtained by fitting specific binding values to a single-site binding model using Prism, and then averaged to obtain the reported data. All binding data presented are mean ± SEM from experiments performed in triplicate in tissue from at least three different mice. Significant differences in the amount of receptor binding between beta-arrestin2+/+ and -/- mouse brain membranes was determined by Student’s t-test at p < 0.05 comparing Bmax, Kd or total specific binding values (where complete saturation analysis was not performed).
RESULTS
Behavioral Effects of Cannabinoids in Beta-Arrestin2+/+ and -/- Mice
Mice were tested for latency to tail withdrawal from a water bath set to different temperatures (Figure 1). Beta-arrestin2-/- mice exhibited greater latencies to withdraw (2-way ANOVA for temperature versus genotype, p < 0.020) than beta-arrestin2+/+ mice (7.0 ± 1.4 s vs. 14.9 ± 3.0 s), at the lowest temperature tested of 50°C (p < 0.001 by Bonferroni’s post-test). This difference was not present at the higher temperatures of 53°C and 56°C, where the latencies for both genotypes were approximately 2.5 s and 1.5 s, respectively. Therefore, all experiments in which the effects of cannabinoid drugs were tested were performed using a water bath temperature of 53°C to achieve a basal latency around 2.5s, and to avoid the complication of differences in basal latency across genotypes.
FIGURE 1.
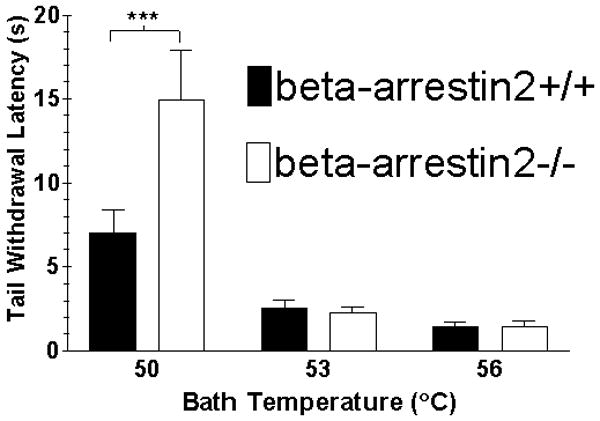
Effect of water bath temperature on basal tail withdrawal latencies. The tips of the tails of beta-arrestin2+/+ and -/- mice were immersed in a water bath set to 50, 53 or 56°C and the time it took each animal to remove its tail from the water was measured. Values represent mean ± SEM; n = 10 mice in each group. 2-way ANOVA temperature versus genotype, p = 0.02; ***p < 0.001 by Bonferroni’s post-test.
The vehicle used to dissolve each agonist for whole animal assays, 1:1:18 ethanol: emulphor: water, was evaluated in the tail withdrawal and rectal temperature assays using the same protocol as were used for each agonist (Figure 2, right panels). Results for the tail withdrawal test indicated that vehicle did not produce significant effects in either genotype compared to pre-injection responses (1-way ANOVA, p > 0.10), and there were no differences between the genotypes as assessed by 2-way ANOVA for genotype versus time (p = 0.87). Vehicle did produce small decreases in rectal temperature compared to pre-injection values in beta-arrestin2+/+ mice from 60 - 360 minutes (1-way ANOVA, p < 0.001 followed by Dunnett’s test) and beta-arrestin2-/- mice from 60 - 240 minutes (p < 0.001) post-injection. However, there were no differences between the genotypes in the rectal temperature response to vehicle (2-way ANOVA, p = 0.52). Since there was a small effect of vehicle on rectal temperature in these mice, all effects of drugs in these assays were compared to the response of the same genotype to vehicle by 2-way ANOVA for time versus treatment (drug or vehicle).
FIGURE 2.
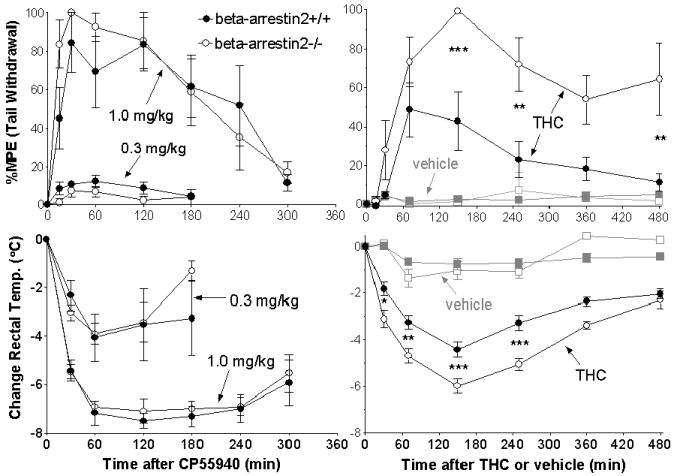
Effects of CP55940 and THC in beta-arrestin2+/+ and -/- mice. Mice were assayed before and at different times after i.p. injection of either 1.0 or 0.3 mg/kg CP55940 (left) or 10 mg/kg THC or vehicle (right) for latency to tail withdrawal (above) and rectal temperature (below). Change in rectal temperature and %Maximum Possible Effect (%MPE) were calculated as described under Methods. Data shown are mean ± SEM with n = 5 for each genotype for CP55940; n = 6-9 for THC, and n = 6 for for vehicle. *p < 0.05, ** p < 0.01, ***p < 0.001, at the times marked, for beta-arrestin2+/+ versus -/- by Bonferroni’s post-test.
Tests of the response to the full agonist, CP55940, were conducted at 1.0 and 0.3 mg/kg (Figure 2, left panels). At 1.0 mg/kg, CP55940 increased the latency to tail withdrawal to the cut-off time of 30 s in some mice from both groups at 30 minutes after injection, and the antinociceptive effects lasted for at least 4 hours (Figure 2, top left panel). At the same time as the tail withdrawal test, rectal temperature measurements in the same mice showed mean decreases in body temperature of around 7°C that lasted between 1 and 4 hours after injection (Figure 2, bottom left panel). At 0.3 mg/kg, the effects of CP55940 were lesser in magnitude; 8-13% of the Maximum Possible Effect (MPE) in the tail withdrawal test (Figure 2, top left panel) and a 4°C drop in body temperature (Figure 2, bottom left panel). It was observed that CP55940 at both doses affected antinociception and decreased the body temperature of the mice, determined by 2-way ANOVA at p < 0.05 for time versus treatment comparing CP55940 to vehicle (the effects of vehicle are shown in Figure 2, right panels). On the other hand, there were no differences between the beta-arrestin2+/+ and -/- mice in the magnitude of the effects at any time at either dose (2-way ANOVA p > 0.05 for each assay comparing time versus genotype).
In experiments with THC (Figure 2, right top panel), 10 mg/kg produced significantly greater effects than vehicle in the tail withdrawal test between 1 and 2.5 hours in beta-arrestin2+/+ mice (p < 0.001). In beta-arrestin2-/- mice the effect was greater than vehicle beginning at 1 hour and continuing for at least 8 hours post-injection (p < 0.001). Both beta-arrestin2+/+ and -/- mice experienced greater rectal temperature decreases following THC than vehicle beginning at 30 minutes and continuing for at least 8 hours. While the +/+ mice had a maximum decrease of 4.4°C, the -/- mice had a decrease of 6.0°C at the same time point (Figure 2, right bottom panel). In contrast to the effects of CP55940, the differences between beta-arrestin2+/+ and -/- mice were determined to be statistically significant by 2-way ANOVA (p < 0.05 and 0.02), for antinociception and temperature depression, respectively. The time points at which the two genotypes exhibited significant differences were determined by Bonferroni’s posttest (shown in Figure 2).
One additional full agonist (methanandamide) and two additional partial agonists (O-1812 and JWH-073) for the cannabinoid receptor were also assayed. The identification of the relative efficacies of all five compounds was determined previously (see Introduction), and confirmed by our laboratory using agonist-stimulated [35S]GTPγS binding in wild-type mouse cerebellar membranes (see Table 2 and below for results).
Table 2.
Agonist concentration-effect curve Emax, relative efficacy and EC50 values.
| Drug | Emax (%stimulation) | Rel. Efficacy | logEC50 | EC50 (nM) |
|---|---|---|---|---|
| CP55940 | 134 ± 23 | 1.00 | -7.7 ± 0.1 | 20 |
| THC | 54 ± 10 | 0.40 | -6.6 ± 0.1 | 260 |
| methanandamide | 136 ± 24 | 1.02 | -5.7 ± 0.6 | 1900 |
| O-1812 | 72 ± 12 | 0.54 | -6.2 ± 0.4 | 610 |
| JWH-073 | 53 ± 9 | 0.40 | -6.3 ± 0.1 | 490 |
Each agonist was incubated at various concentrations with wild-type mouse cerebellar membranes to determine relative efficacies for activation of G-protein via CB1 receptors. Concentration-effect curves were constructed and analyzed for Emax and logEC50 values using Prism. Values shown are Mean ± SEM for 3-4 determinations for each agonist. Relative efficacy (Rel. Efficacy) values were calculated by dividing mean Emax values by the mean Emax value for CP55940; EC50 values in nM units were calculated by taking the inverse log of each mean logEC50 value and converting from molar to nM by multiplying by 109.
In the in vivo assays, 50 mg/kg methanandamide produced only 10% MPE in the tail withdrawal assay (Figure 3, left top panel) and a rectal temperature decrease of only 2°C (Figure 3, left bottom panel) in both beta-arrestin2+/+ and -/- mice. Drug effects were determined to be statistically significant compared to the effects of vehicle (p < 0.05 at each dose for each genotype in each assay), but did not differ between beta-arrestin2+/+ and -/- mice. Since the effects of 50 mg/kg methanandamide were minimal, a higher dose of 250 mg/kg was assayed. The higher dose produced 60-100% MPE in the tail withdrawal assay (Figure 3, left top panel) and a rectal temperature decreases of 6.5-8°C (Figure 3, left bottom panel), but there were still no significant differences between beta-arrestin2+/+ and -/- mice (p = 0.85 and p = 0.18, respectively). For this treatment, the difference in the mean responses of the wild-type and beta-arrestin2-/- mice did appear to be more different than the responses to the other treatments that were not significantly different. Closer inspection of the data from the individual mice reveals that one-third of the mice in the wild-type group showed almost no increase in latency to tail withdrawal and relatively small changes in temperature, while the other two-thirds of the wild-type mice showed responses to 250 mg/kg methanandamide that were greater in magnitude and were indistinguishable from the responses of the beta-arrestin2-/-mice. In assays with THC, the differences were clearly not attributable to a minority of mice failing to show a significant response to drug treatment as was seen with methanandamide. Two-thirds of the +/+ mice treated with THC had lower increases in latency to tail withdrawal and decreases in temperature than any of the -/- mice. The remaining one-third of +/+ mice had responses that were similar to those of the -/- mice, but at most time points, the +/+ mice were similar only to those -/- mice that showed the smallest responses.
FIGURE 3.
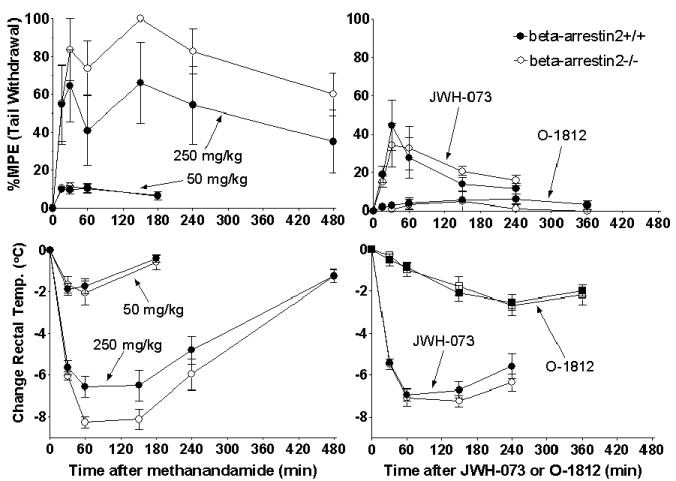
Effects of other cannabinoid agonists in beta-arrestin2+/+ and -/- mice. Mice were assayed before and at different times after i.p. injection of 50 and 250 mg/kg methanandamide (left) and 2.5 mg/kg O-1812 and 20 mg/kg JWH-015 (right) for latency to tail withdrawal (above) and rectal temperature (below). Data shown are mean ± SEM; n = 6 - 8 for each genotype for each drug.
O-1812 at 2.5 mg/kg produced no significant effects on latency to tail withdrawal (Figure 3, right top panel) in either beta-arrestin2+/+ (p = 0.66) or -/- mice (p = 0.66). However, O-1812 did produce significant decreases in rectal temperature (Figure 3, right bottom panel) from 2.5-6 hours in +/+ mice (p < 0.001) and from 4-6 hours in -/- mice (p < 0.001). O-1812 exerted maximal effects in both genotypes from 2.5-6 hours. JWH-073 at 20 mg/kg produced significant antinociception compared to vehicle in both beta-arrestin2+/+ (p < 0.02) and -/- (p < 0.01) mice from 30-60 min (Figure 3, right top panel). Rectal temperature (Figure 3, right bottom panel) was decreased greater than the effect of vehicle beginning at 30 min and remained so through 4 hours in both genotypes (p < 0.001). As with CP55940, the effects of methanandamide, O-1812 and JWH-073 were not different between beta-arrestin2+/+ and -/- mice.
To confirm that deletion of beta-arrestin2 selectively increases the efficacy of THC but not CP55940, a second assay of antinociception, the hot plate test, was performed using 1.0 mg/kg CP55940, 10 mg/kg THC or vehicle. At this temperature, there was no difference in pre-drug latencies between the beta-arrestin2+/+ (18.0 ± 3.0 s) and -/- (17.7 ± 3.6 s) mice. Vehicle did not produce any significant effects compared to pre-injection responses in either genotype (1-way ANOVA, p = 0.93 in beta-arrestin+/+ and p = 0.954 in -/- mice), and response to vehicle did not differ between +/+ and -/- mice (2-way ANOVA, p = 0.971). CP55940 produced 80-100% MPE in each genotype from the first time point of 15 min through the last at 2 hours post-injection, which was significantly greater (2-way ANOVA, p < 0.05) than the effect of vehicle at every time (Figure 4, left panel). As before, CP55940 did not exhibit significant differences between the genotypes (2-way ANOVA, p = 0.94). The effect of THC in beta-arrestin2+/+ mice did not exceed 25% MPE at any time and was not significantly greater (p = 0.82) than the effect of vehicle (Figure 4, right panel). However, in beta-arrestin2-/- mice THC produced significantly greater effects than vehicle (p < 0.02) from 60-120 minutes. As in the tail flick and rectal temperature assays above, the effect of THC in the hot plate test was significantly greater in beta-arrestin2-/- than in +/+ mice (2-way ANOVA: Interaction p = 0.53, genotype p < 0.01, time p < 0.05, Bonferroni’s posttest p < 0.05 at 90 minutes).
FIGURE 4.
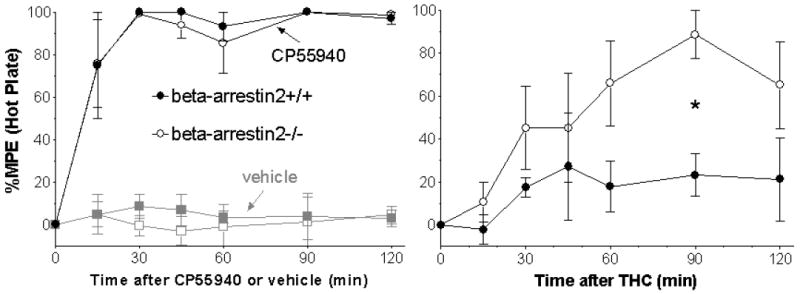
Effect of CP55940 and vehicle (left) and THC (right) in the hot plate test of antinociception. %Maximum Possible Effect (%MPE) were calculated as described in Methods. Data shown are mean ± SEM with n = 5 for each group. *p < 0.05 at 90 min for beta-arrestin2+/+ versus -/- by Bonferroni’s posttest.
CB1 Receptor Density in Brain Membrane Homogenates
Saturation analysis with the CB1 receptor-selective antagonist, [3H]SR141716A, was conducted in cerebellar membranes (Figure 5). Bmax values were 4.1 ± 0.3 pmol/mg versus 4.1 ± 0.4 pmol/mg and Kd values were 1.3 ± 0.1 versus 1.2 ± 0.1 nM, respectively. In cortex and hippocampus membranes, complete saturation analysis was not conducted, but the amount of specific binding obtained with 1.0 nM [3H]SR141716A was determined in beta-arrestin2+/+ and -/- mice (Figure 5, inset). In cortex membranes, [3H]SR141716A exhibited 1.6 ± 0.3 pmol/mg and 1.8 ± 0.2 pmol/mg of binding, respectively. In hippocampal membranes, [3H]SR141716A bound 1.7 ± 0.3 pmol/mg and 1.9 ± 0.2 pmol/mg, respectively. In cerebellar membranes analyzed in the same way (the amount of binding at 1.0 nM [3H]SR141716A) there was 2.5 ± 0.5 pmol/mg and 2.1 ± 0.4 pmol/mg of binding. None of the three brain areas investigated showed significant differences in the amount of CB1 receptor binding between beta-arrestin2+/+ and -/- mice (p > 0.05 by Student’s t-test for each brain region).
FIGURE 5.
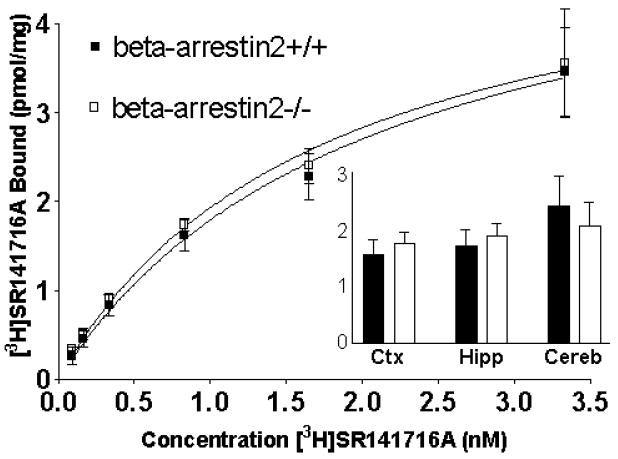
Cannabinoid CB1 receptor binding in brain membranes from beta-arrestin2+/+ and -/- mice. Cerebellar membranes were prepared and incubated with various concentrations of [3H]SR141716A. Relative CB1 receptor levels (inset) were also determined in membranes from cortex (Ctx), hippocampus (Hipp) and cerebellum (Cereb) by the amount of binding at a single [3H]SR141716A concentration (1 nM). Data shown are mean ± SEM for specific [3H]SR141716A binding (n = 3 or more). There were no differences between beta-arrestin2+/+ and -/-for any brain area (Student’s t-test, p > 0.05).
[35S]GTPγS Binding to Brain Membranes
Agonist-stimulated [35S]GTPγS binding to wild-type mouse cerebellar membranes was measured for each of the agonists used in the in vivo experiments in this study. This was done to confirm the relative agonist efficacy for each agonist to be able to classify each agonist as a full or partial agonist at the CB1 receptor. Results (Table 2) indicated that CP55940 and methanandamide were both full agonists, exhibiting Emax values of ~135% stimulation over basal [35S]GTPγS binding values. Though both full agonists, the potencies of CP55940 and methanandamide varied over a 100-fold range with logEC50 values of -7.7 and -5.7, respectively. The remaining agonists exhibited similar concentration-effect curves. THC, JWH-073 and O-1812 were all partial agonists and exhibited Emax values between 53% and 72% stimulation (between 40% and 52% of the values exhibited by the full agonists). JWH-073 and O-1812 had potencies similar to that of THC (log EC50 value -6.6) with logEC50 values of -6.3 and -6.2, respectively.
DISCUSSION
The effect of deletion of beta-arrestin2 on basal tail withdrawal latencies at lower water bath temperatures was nearly identical to that noted by Bohn et al. (2000), and indicates a role for beta-arrestin2 in the responsiveness of pain pathways in the central nervous system. Since these data were collected in the absence of a drug treatment, there is little evidence for the receptor system(s) or other biochemical mechanism involved in this effect. However, G-protein coupled receptors are implicated since they are the target of regulation by beta-arrestin2.
This is the first study to demonstrate the effects of cannabinoid receptor agonists in transgenic beta-arrestin2-/- mice, and the effects of only a few other substances in these mice have previously been published (Bohn et al. 1999, 2000a, 2002, 2003, 2004; Beaulieu et al. 2005; Bouxsein et al. 2005; Ferrari et al. 2005; Pi et al. 2005; Schmid et al. 2008). In the previous studies by Bohn et al. (1999, (2000), it was noted that morphine exhibited enhanced activity in beta-arrestin2-/- compared to beta-arrestin2+/+ mice. In the present study, CP55940 was assayed first at 1.0 mg/kg, and no differences between beta-arrestin2+/+ and -/- mice were observed. Since this dose produce nearly maximal activity in the antinociception assay, it was hypothesized that the reason for the lack of difference between the genotypes was due to a ceiling effect; it would not be possible to observe a greater effect in the beta-arrestin2-/- mice, since the effect in the +/+ mice was already near the maximum possible effect. Thus, a lower dose of CP55940 (0.3 mg/kg) was subsequently assayed to observe the effects at a less-than-maximally effective dose. The effects at this dose were also found to be unaffected by deletion of beta-arrestin2.
Next, the effects of the naturally-occurring cannabinoid agonist, THC, were found to be enhanced in the absence of beta-arrestin2, as seen previously for morphine (Bohn et al. 1999, 2000a,b,2003, 2004; Raehal et al. 2005). The doses of THC used produced maximal effects of approximately 50% MPE in the tail withdrawal assay, 25% MPE in the hot plate assay and a 4.4°C drop in body temperature in the beta-arrestin2+/+ mice and significantly greater effects in the -/- mice (100% MPE, 90% MPE and 6°, respectively).
Since CP55940 was known to be a full agonist and THC a partial agonist at CB1 receptors in brain membranes (Table 2) (Breivogel et al. 2001), it was hypothesized that beta-arrestin2 deletion affected sensitivity to cannabinoid receptor partial agonists, but not full agonists. To test this hypothesis, one other full agonist, methanandamide, and two other partial agonists with efficacy and potency similar to THC, JWH-073 and O-1812, were assayed. The relative efficacy and thus identification of each of these agonists as full or partial was confirmed using the in vitro [35S]GTPγS binding assay in the present study (see Table 2). The doses of each agonist used for the in vivo assays were chosen based on previously-published ED50 values for the same antinociception and hypothermia assays used in the present study. The effects of these agonists, CP55940 (Compton et al. 1993), THC (Wiley et al. 1998), methanandamide (Abadji et al. 1994), O-1812 (Di Marzo et al. 2001) and JWH-073 (Wiley et al. 1998) on body temperature and latency to tail withdrawal were similar to what has previously been observed in wild-type mice.
Though the levels of in vivo activity varied somewhat between the agonists, each agonist produced significant hypothermia, and all but one produced significant antinociception at the doses administered. If the activity of any of these agonists had been enhanced by deletion of beta-arrestin2, it is reasonable to expect that this effect would be observable at these doses. However, none of the data for any of the agonists (other than THC) showed a difference between beta-arrestin2+/+ and -/-mice.
The finding that the activity of THC was selectively increased by deletion of beta-arrestin2 paralleled the findings of Bohn, et al., (2004) where only the effects of morphine, or its derivative heroin, were enhanced in beta-arrestin2-/- mice. Interestingly, THC and morphine are partial agonist for G-protein activation at CB1 (Sim et al. 1996) or mu (Selley et al. 1997) receptors, respectively, and each is a plant-derived exogenous ligand for its respective receptor. However, it is clear from the present study that partial agonist activity alone is not responsible for this selectivity, since neither of the other two partial CB1 receptor agonists exhibited this effect.
To investigate whether the effect of deletion of beta-arrestin2 on basal latency to tail withdrawal at 50°C or the in vivo effects of THC might be based on differences in CB1 receptor density, the CB1-selective antagonist [3H]SR141716A was used to measure the total number of CB1 receptors in three different brain regions. Cerebellum, cortex and hippocampus represent three brain regions that express similarly high levels of CB1 receptors (Herkenham et al. 1990), and cortex and hippocampus express relatively high levels of beta-arrestin2, while cerebellum displays lower levels (Gurevich et al. 2002; 2004). There were no differences between beta-arrestin2+/+ and -/- mouse brain in cannabinoid CB1 receptor density in any brain region examined; analogous to what was found previously for the mu opioid receptor (Bohn et al. 1999). This is not surprising, since a difference in overall CB1 receptor density would likely affect the activity of all the agonists, or at least all of the partial agonists, and not just THC.
The main finding of this study is that in vivo effects of THC were selectively influenced by the absence of beta-arrestin2. However, the reason for the agonist-selective effects of beta-arrestin2 is clearly not due purely to the low efficacy of THC, since the differences between beta-arrestin2+/+ and -/- mice were not seen with either of the two other agonists with similarly low efficacy (O-1812 and JWH-073). A likely hypothesis for this selectivity is that THC is able to activate CB1 receptors in a manner that recruits only beta-arrestin2 to interact with it, while the other agonists are able to recruit either beta-arrestin1 or beta-arrestin2 to interact with CB1 receptors, analogous to what was observed for mu receptor agonists (Bohn et al. 2004). The fact that the activity of THC and morphine-based agonists were selectively affected by deletion of beta-arrestin2 suggests that agonist-selective involvement of beta-arrestin2 in the regulation of GPCRs may be a common phenomenon.
The biochemical mechanism by which even morphine exerts this agonist-specific effect has not been investigated. Some possibilities include recruitment of specific G-protein couple Receptor Kinases (GRKs) subtypes to phosphorylate GPCRs, induction of selective patterns of phosphorylation of GPCRs by GRKs (that affect selective binding of beta-arrestin subtypes), or selective coupling of beta-arrestins to GPCRs (even in the presence of identical phosphorylation).
The fact that the activity of THC was affected indicates an important role for beta-arrestin2 in the context of the recreational use/abuse of marijuana or the medicinal use of Marinol®, since THC is the main active component of each of these. Perhaps some future therapy will take advantage of the selective regulation of the activity of THC (and/or morphine) by modulating the activity of beta-arrestins.
Acknowledgments
This work was supported by the Campbell University School of Pharmacy and the NIDA Drug Supply Program. Beta-arrestin2+/+ and -/- breeder mice were generously provided by Dr. Robert J. Lefkowitz of Duke University Medical Center. Financial support (RKR) from NIDA grants DA-005488 and DA-008904 is gratefully acknowledged.
References
- Abadji V, Lin S, Taha G, Griffin G, Stevenson LA, Pertwee RG, Makriyannis A. (R)-methanandamide: a chiral novel anandamide possessing higher potency and metabolic stability. J Med Chem. 1994;37:1889–93. doi: 10.1021/jm00038a020. [DOI] [PubMed] [Google Scholar]
- Adams IB, Martin BR. Cannabis: pharmacology and toxicology in animals and humans. Addiction. 1996;91:1585–614. [PubMed] [Google Scholar]
- Adams IB, Ryan W, Singer M, Thomas BF, Compton D, Razdan RK, Martin BR. Evaluation of cannabinoid receptor binding and in vivo activities for anandamide analogs. J Pharmacol Exp Ther. 1995;273:1172–81. [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–73. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–8. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000a;408:720–3. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Xu F, Gainetdinov RR, Caron MG. Potentiated opioid analgesia in norepinephrine transporter knock-out mice. J Neurosci. 2000b;20:9040–5. doi: 10.1523/JNEUROSCI.20-24-09040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci. 2002;22:10494–500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Sotnikova TD, Medvedev IO, Lefkowitz RJ, Dykstra LA, Caron MG. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J Neurosci. 2003;23:10265–73. doi: 10.1523/JNEUROSCI.23-32-10265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–12. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Bouxsein ML, Pierroz DD, Glatt V, Goddard DS, Cavat F, Rizzoli R, Ferrari SL. Beta-Arrestin2 Regulates the Differential Response of Cortical and Trabecular Bone to Intermitent PTH in Female Mice. J Bone Miner Res. 2005;20:635–43. doi: 10.1359/JBMR.041204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR. The functional neuroanatomy of brain cannabinoid receptors. Neurobiol Dis. 1998;5:417–31. doi: 10.1006/nbdi.1998.0229. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR. Cannabinoid signal transduction in rat brain: comparison of cannabinoid agonists in receptor binding, G-protein activation and adenylyl cyclase inhibition. J Pharmacol Exp Ther. 2000;295:328–36. [PubMed] [Google Scholar]
- Breivogel CS, Selley DE, Childers SR. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPγS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J Biol Chem. 1998;273:16865–73. doi: 10.1074/jbc.273.27.16865. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Griffin G, Di Marzo V, Martin BR. Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol Pharmacol. 2001;60:155–63. [PubMed] [Google Scholar]
- Burkey TH, Quock RM, Consroe P, Roeske WR, Yamamura HI. Δ9-tetrahydrocannabinol is a partial agonist of cannabinoid receptors in mouse brain. Eur J Pharmacol. 1997;323:R3–R4. doi: 10.1016/s0014-2999(97)00146-5. [DOI] [PubMed] [Google Scholar]
- Childers SR, Breivogel CS. Cannabis and endogenous cannabinoid systems. Drug Alcohol Dep. 1998;51:173–87. doi: 10.1016/s0376-8716(98)00075-1. [DOI] [PubMed] [Google Scholar]
- Compton DR, Rice KC, DeCosta BR, Razdan RK, Melvin LS, Johnson MR, Martin BR. Cannabinoid structure-activity relationships: Correlation of receptor binding and in vivo activities. J Pharmacol Exp Ther. 1993;265:218–26. [PubMed] [Google Scholar]
- Dewey WL, Harris LSJ, Howes JF, Nuite JA. The effect of various neurohumoral modulators on the activity of morphine and the narcotic antagonists in the tail-flick and phenylquinone tests. J Pharmacol Exp Ther. 1970;175:435–42. [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, De Petrocellis L, Brandi I, Jefferson RG, Winckler RL, Davis JB, Dasse O, Mahadevan A, Razdan RK, Martin BR U-hwscsaBW-SKY-Bebbfcdac. Highly Selective CB1 Cannabinoid Receptor Ligands and Novel CB1/VR1 Vanilloid Receptor “Hybrid” Ligands. Biochem Biophys Res Commun. 2001;281:444–51. doi: 10.1006/bbrc.2001.4354. [DOI] [PubMed] [Google Scholar]
- Fan F, Compton DR, Ward S, Melvin L, Martin BR. Development of cross-tolerance between Δ9-tetrahydrocannabinol, CP 55,940 and WIN 55,212. J Pharmacol Exp Ther. 1994;271:1383–90. [PubMed] [Google Scholar]
- Ferrari SL, Pierroz DD, Glatt V, Goddard DS, Bianchi EN, Lin FT, Manen D, Bouxsein ML. Bone Response to Intermittent Parathyroid Hormone Is Altered in Mice Null for {beta}-Arrestin2. Endocrinol. 2005;146:1854–62. doi: 10.1210/en.2004-1282. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, Premont RT. Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron. 1999;24:1029–36. doi: 10.1016/s0896-6273(00)81048-x. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Caron MG, Lefkowitz RJ. Reply: receptor specificity of G-protein-coupled receptor kinases. Trends Pharmacol Sci. 2000;21:366–7. doi: 10.1016/s0165-6147(00)01538-8. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109:421–36. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 expression selectively increases during neural differentiation. J Neurochem. 2004;91:1404–16. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, De Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–6. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyser CJ, Hampson RE, Deadwyler SA. Effects of delta-9-tetrahydrocannabinol on delayed match to sample performance in rats: Alterations in short-term memory associated with changes in task specific firing of hippocampal cells. J Pharmacol Exp Ther. 1993;264:294–307. [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, Chavkin C, Mackie K. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–80. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouznetsova M, Kelley B, Shen M, Thayer SA. Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Mol Pharmacol. 2002;61:477–85. doi: 10.1124/mol.61.3.477. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G- protein-coupled receptor signals. J Cell Sci. 2002;115:455–65. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- Pi M, Oakley RH, Gesty-Palmer D, Cruickshank RD, Spurney RF, Luttrell LM, Quarles LD. Beta-Arrestin- and G Protein Receptor Kinase-Mediated Calcium-Sensing Receptor Desensitization. Mol Endocrinol. 2005;19:1078–87. doi: 10.1210/me.2004-0450. [DOI] [PubMed] [Google Scholar]
- Prather PL, Martin NA, Breivogel CS, Childers SR. Activation of cannabinoid receptors in rat brain by WIN 55212-2 produces coupling to multiple G protein α-subunits with different potencies. Mol Pharmacol. 2000;57:1000–10. [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on -arrestin-2 interactions in vivo. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selley DE, Sim LJ, Xiao R, Liu Q, Childers SR. Mu opioid receptor-stimulated [35S]GTPγS binding in rat thalamus and cultured cell lines: Signal transduction mechanisms underlying agonist efficacy. Mol Pharmacol. 1997;51:87–96. doi: 10.1124/mol.51.1.87. [DOI] [PubMed] [Google Scholar]
- Sim LJ, Hampson RE, Deadwyler SA, Childers SR. Effects of chronic treatment with Δ9-tetrahydrocannabinol on cannabinoid-stimulated [35S]GTPγS autoradiography in rat brain. J Neurosci. 1996;16:8057–66. doi: 10.1523/JNEUROSCI.16-24-08057.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley JL, Compton DR, Dai D, Lainton JAH, Phillips M, Huffman JW, Martin BR. Structure-activity relationships of indole- and pyrrole-derived cannabinoids. J Pharmacol Exp Ther. 1998;285:995–1004. [PubMed] [Google Scholar]
- Winters WD, Hance AJ, Cadd GG, Quam DD, Benthuysen JL. Ketamine-and morphine-induced analgesia and catalepsy. I. Tolerance, cross-tolerance, potentiation, residual morphine levels and naloxone action in the rat. J Pharmacol Exp Ther. 1988;244:51–7. [PubMed] [Google Scholar]