

Abstract
The potent new antiviral inhibitor GRL-98065 (1) of HIV-1 protease (PR) has been studied with PR variants containing the single mutations D30N, I50V, V82A and I84V that provide resistance to the major clinical inhibitors. Compound 1 had inhibition constants of 17-fold, 8-fold, 3-fold and 3-fold, respectively, for PRD30N, PRI50V, PRV82A and PRI84V relative to wild type PR. The chemically related darunavir had similar relative inhibition, except for PRD30N, where inhibitor 1 was approximately threefold less potent. The high resolution (1.11–1.60 Å) crystal structures of PR mutant complexes with inhibitor 1 showed small changes relative to the wild type enzyme. PRD30N and PRV82A showed compensating interactions with inhibitor 1 relative to those of PR, while reduced hydrophobic contacts were observed with PRI50V and PRI84V. Importantly, inhibitor 1 complexes showed fewer changes relative to wild type enzyme than reported for darunavir complexes. Therefore, inhibitor 1 is a valuable addition to the antiviral inhibitors with high potency against resistant strains of HIV.
Keywords: HIV-1, protease inhibitor, GRL-98065, crystal structure, enzyme kinetics, hydrogen bonds
INTRODUCTION
Highly active antiretroviral therapy (HAART) consisting of a combination of drugs that inhibit the HIV-1 protease (PR) or the reverse transcriptase has provided an effective treatment for AIDS 1–3. However, the success of long-term therapy is limited by the rapid emergence of drug resistance 4. Resistance to antiviral PR inhibitors arises by distinct mutations in the PR gene 5–7. Many of these mutations provide resistance by altering the amino acid residues forming the inhibitor binding site of the PR and thus, reducing the affinity for the inhibitor. Most of the current drugs show significantly reduced inhibition of PR with such resistant mutations. Therefore, new antiviral inhibitors are being developed 8. One successful approach is to design inhibitors that provide more interactions with the main chain atoms of PR 9. These interactions cannot easily be changed by mutation. Such new non-peptide inhibitors, like the recently FDA-approved darunavir (TMC-114), have already demonstrated success in clinical trials in treating infections with resistant virus.
PR mutations observed in resistant HIV include D30N, I50V, V82A and I84V, which individually provide high levels of resistance to the major clinical inhibitors. These four mutations alter residues forming the binding site for peptide substrates or inhibitors. Previously, we have analyzed the effect of darunavir on the structure and inhibition of PR with each of these mutations 10, 11. Darunavir showed lower affinity for PR with the D30N and I50V mutations. The mutation D30N, which removes the negative charge of the amino acid side chain, is selected frequently in clinical isolates on exposure to nelfinavir 12. PRD30N showed altered activity for different substrates compared to wild type PR 13. Crystal structures of PRD30N with different inhibitors have shown changes in the position of the side chain of residue 30 10, 14. Asp30 forms hydrogen bond interactions with the aniline group of darunavir. In the D30N mutant these direct interactions were replaced by a weaker water-mediated hydrogen bond interaction, in agreement with the 30-fold weaker inhibition of darunavir for PRD30N compared to wild type PR. Mutation I50V is very common on exposure to amprenavir 15, the drug that is chemically most related to darunavir. I50V also confers resistance to darunavir 16. Our structural analysis showed that PRI50V has reduced interactions with the aniline group of darunavir consistent with 9-fold worse inhibition. Mutations V82A and I84V are observed with all the clinical inhibitors 7. These mutations had smaller structural changes in the interactions with darunavir, and little change in the inhibition relative to wild type PR.
Compound 1 [(1R,5S,6R)-2,8-dioxabicyclo[3.3.0]oct-6-yl] N-[(2S,3R)-4-[(3,4-methylenedioxyphenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenyl-butan-2-yl] carbamate (Figure 1) is a potent new antiviral inhibitor derived from darunavir in which the aniline group has been replaced by a 1, 3-benzodioxole group 9, 17. This chemical change is expected to alter the affinity of the compound, especially for mutants PRD30N and PRI50V because the aniline group of darunavir interacts with Asp30 and Ile50. However, crystal structures show that many of the HIV PR interactions are similar for inhibitor 1 and darunavir 17. The antiviral data show that inhibitor 1 is highly potent against HIV-infected cells with IC50 values of 0.2–0.5 nM compared to 3 nM for darunavir, and has minimal cytotoxicity 9, 17. Here, the structures and in vitro inhibition data are reported for inhibitor 1 and PR variants with the single substitutions D30N, I50V, V82A and I84V. Inhibitor 1 and darunavir showed very similar in vitro inhibition of PR, PRV82A and PRI84V, and slightly worse inhibition of PRD30N and PRI50V relative to PR. Therefore, inhibitor 1 is likely to be as effective as darunavir in treating many resistant variants of HIV.
Figure 1. Structures of A) Inhibitor 1 (GRL-98065), B) Darunavir (TMC-114), and C) PR dimer indicating location of mutations.


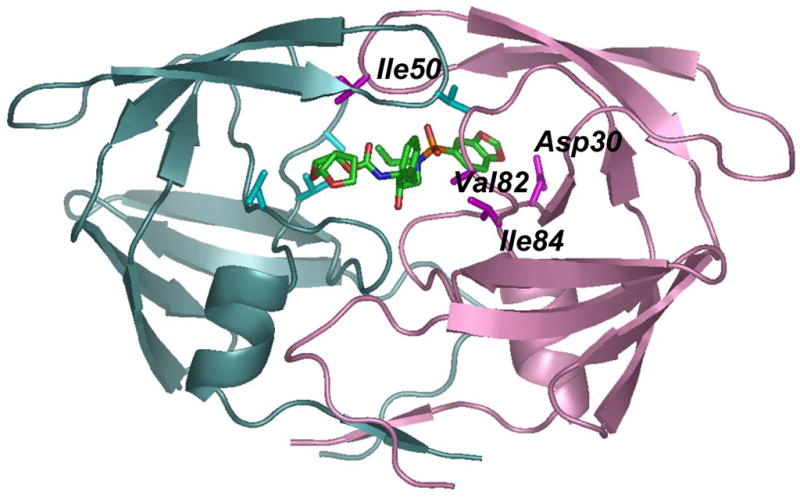
The backbone of one subunit is shown as pink ribbons with the sites of mutation (D30N, I50V, V82A and I84V) indicated by magenta side chains, while the other subunit is colored in light cyan with mutation sites in darker cyan. Only one subunit is labeled. Inhibitor 1 is colored by atom type.
RESULTS
Inhibition of HIV PR and the mutants PRV82A, PRI84V, PRD30N and PRI50V by inhibitor 1
The inhibition constants of inhibitor 1 against PR and the drug-resistant mutants PRD30N, PRI50V, PRV82A and PRI84V were measured using the fluorescence substrate RE(Edans)SQNY*PIVRK(Dabcyl)R 18, an analog of the MA-CA cleavage site in the Gag-Pol polyprotein. The kinetic parameters of these mutants for hydrolysis of the fluorescence substrate have been reported in 10. As shown in Table 1, wild type PR had an essentially identical inhibition constant of 0.3 nM for inhibitor 1 and darunavir. Moreover, inhibitor 1 and darunavir had similar relative inhibition of 1.6- to 3-fold for PRV82A and PRI84V relative to PR. PRD30N and PRI50V had higher Ki values of 17- and 8-fold relative to PR for inhibitor 1, compared with 6-fold and 4-fold for darunavir. Inhibitor 1 and darunavir showed the same order of relative inhibition of PR > PRV82A ~ PRI84V > PRI50V > PRD30N.
Table 1.
Inhibition data
| Protease | Ki (nM) (Relative) inhibitor 1 | Ki (nM) (Relative) darunavir |
|---|---|---|
| PR | 0.27 ± 0.06 (1) | 0.34 ± 0.08 (1) |
| PRD30N | 4.6 ± 0.7 (17) | 1.9 ± 0.3 (5.5) |
| PRI50V | 2.1 ± 0.4 (8) | 1.3 ± 0.3 (3.9) |
| PRV82A | 0.80 ± 0.06 (3) | 0.54 ± 0.04 (1.6) |
| PRI84V | 0.85 ± 0.15 (3.2) | 0.56 ± 0.10 (1.7) |
Description of the high-resolution crystal structures
The crystal structures of the drug-resistant mutants PRD30N, PRI50V, PRV82A and PRI84V have been determined in their complexes with the new antiviral inhibitor 1 at resolutions of 1.11–1.35 Å (Figure 1). The crystallographic statistics are summarized in Table 2. The wild type PR complex was described briefly in 17. Each crystallographic asymmetric unit contains a dimer with the residues numbered 1–99 and 1′–99′. Three structures were determined in the space group P21212 and the PRV82A complex was solved in the P212121 space group. The structures were refined to R-factors of 0.13–0.16 including solvent molecules, anisotropic B-factors and hydrogen atoms. These high-resolution crystal structures showed clear electron density for all the protease atoms, the inhibitor, and solvent molecules. Overall, the mutant structures were very similar to the wild type complex with RMS deviations of 0.21–0.22 Å for the main atoms of the structures in the same space group (P21212), and 0.63 Å for the PRV82A/inhibitor 1 complex in the space group P212121, due to the differences in lattice packing in the two space groups.
Table 2.
Data collection and refinement statistics
| PRD30N | PRI50V | PRV82A | PRI84V | |
|---|---|---|---|---|
| Space group | P21212 | P21212 | P212121 | P21212 |
| Unit cell dimensions: (Å) | ||||
| a | 58.42 | 58.29 | 50.88 | 58.42 |
| b | 86.23 | 86.23 | 58.15 | 86.18 |
| c | 46.01 | 46.01 | 62.11 | 45.99 |
| Resolution range (Å) | 50–1.20 | 50–1.28 | 50–1.11 | 50–1.35 |
| Unique reflections | 73,411 | 60,638 | 73,051 | 51,903 |
| Rmerge (%) overall (final shell) | 8.9 (36.8) | 7.7 (41.1) | 8.6 (23.0) | 5.1 (30.8) |
| I/σ(I) overall (final shell) | 14.7 (2.3) | 11.9 (2.2) | 18.2 (4.0) | 17.3 (4.7) |
| Completeness (%) overall(final shell) | 89.0 (51.0) | 89.5 (59.3) | 97.4 (80.3) | 98.5 (86.6) |
| Data range for refinement (Å) | 10–1.20 | 10–1.28 | 10–1.11 | 10–1.35 |
| R (%) | 16.1 | 15.8 | 13.0 | 13.3 |
| Rfree (%) | 19.5 | 20.2 | 16.3 | 17.5 |
| No. of solvent atoms (total occupancies) | 161.4 | 136.3 | 186.3 | 192.8 |
| RMS deviation from ideality | ||||
| Bonds (Å) | 0.013 | 0.012 | 0.015 | 0.012 |
| Angle distance (Å) | 0.034 | 0.031 | 0.033 | 0.032 |
| Average B-factors (Å2) | ||||
| Main-chain atoms | 15.4 | 15.1 | 7.9 | 14.7 |
| Side-chain atoms | 20.1 | 19.2 | 11.6 | 18.6 |
| Inhibitor | 11.8 | 12.3 | 7.2 | 11.3 |
| Solvent | 35.0 | 17.1 | 19.3 | 27.4 |
| Residual density (max/min) (eÅ−3) | 0.41/−0.33 | 0.39/−0.32 | 0.65/−0.43 | 0.41/−0.32 |
| Relative occupancy of inhibitor 1 | 0.6/0.4 | 0.6/0.4 | 1.0 | 0.6/0.4 |
The average B-factors were low for protein and inhibitor atoms. The higher resolution structures showed lower average B-factors. In particular, the PRV82A complex had the lowest B-factors for protein main-chain and side-chain atoms of 7.9 Å2 and 11.6 Å2, respectively, compared to the values of about 15 Å2 to 20 Å2 for the other structures. The inhibitor atoms also showed the lowest average B-factors for the PRV82A complex: 7.2 Å2 compared to 11.7 Å2 – 12.3 Å2 for the others. The average B-factors of the solvent molecules depend on the quality of the data as well as the number of solvent molecules included in refinement. The higher resolution data also provide better quality electron density maps in which more ordered solvent molecules are visible. The solvent atoms for the complexes with PRI50V and PRV82A showed lower average B-factors of 17.1 Å2 and 19.3 Å2, respectively, compared to 27.4 Å2 – 35.0 Å2 for the other structures.
Residues with alternate conformations
Alternate conformations were observed in all the crystal structures. Inhibitor 1 was bound in two conformations in all structures in the space group P21212 with relative occupancy of 0.6/0.4, although it had a single conformation in the PRV82A complex in space group P212121 (Figure 2). Alternate conformations were observed for the main chain atoms of residues 3–5 in PRV82A, as noted previously in complexes PRV82A/darunavir [PDB code 2IDW] 11 and PR/JE-2147 [PDB code 1KZK] 19. There were 23 residues with two alternate conformations modeled for the side chains in PR, 23 for mutant PRD30N, 29 for PRI50V, 22 for PRV82A and 25 for PRI84V. A similar pattern of the residues with alternate conformations was observed for the four structures (PR, PRI84V, PRD30N and PRI50V) in the same space group. Surface residues, especially charged residues with longer side-chains, such as arginine and lysine, showed more flexibility and were observed with more alternate conformations. However, several internal hydrophobic residues, such as Leu23, Ile33′, and Leu97/97′, had two alternate conformations. The side-chains of several residues located close to the inhibitor, Arg8, Asp30, Ile50, Pro81, Val82, and Ile84, showed alternate conformations in at least one subunit, as reported for darunavir complexes 11. Alternate conformations were observed at the sites of mutation in most structures, except for PRV82A, which had a single conformation for residues 30, 50, 82 and 84 in both subunits. The side chain of residue 30 had alternate conformations in the structures of PRD30N and PRI50V. The side chain of Val 82 had alternate conformations in both subunits of all structures, except for PRV82A. The side chain of Ile 84 had alternate conformations in the structure of PRI50V, while Ile/Val 84′ had alternate conformations in all structures, except for PRV82A. The main chain atoms of residues 50 and 50′ had alternate conformations in four structures, while the side chain of Ile 50′ had alternate conformations in the PRI84V structure.
Figure 2. Electron density map for inhibitor 1 in complex with PRV82A.

The omit Fo-Fc map, using Fc calculated without inhibitor 1, is contoured at 8.5σ level. Inhibitor 1 had a single conformation.
Solvent molecules in the crystals
The high quality of diffraction data for the structures allowed the modeling of many solvent molecules. The structures were modeled with more than 130 water molecules, ions and other small molecules from the crystallization solutions, including many with partial occupancy, depending on the shape of the electron density and the interactions with other molecules. Sodium, chloride and acetate ions were modeled in PR, PRI84V, PRD30N and PRI50V complexes, while phosphate, glycerol and DMSO molecules were fitted to density in the PRV82A structure. The sodium ion was identified for the first time in the PR and PRI84V complexes with darunavir 11 and illustrated in 10; although there was strong evidence in other published structures that some water molecules should be identified as Na+ (such as PDB code 1KZK, 1SDT, 1SDV). The sodium ion was recognized initially by the abnormally low B factor and positive peak when water was refined at that site, and then confirmed by the six octahedral oxygen donors at the mean distance of 2.42 Å. Valence calculations suggested that the atom was either a sodium or calcium ion 20, and only sodium was present in the crystallization conditions. The PR, PRD30N, PRI50V and PRI84V structures in space group P21212 each contain one sodium ion.
HIV-1 Protease interactions with inhibitor 1
The numerous protease-inhibitor interactions include strong hydrogen bonds, weaker C-H…O contacts, C-H… π contacts, and the weakest van der Waals contacts such as C-H…H-C, as described for darunavir complexes 21. Polar interactions are important for ligand binding to the PR. Ten direct hydrogen bonds, three water-mediated hydrogen bond interactions, and five C-H…O interactions with main chain atoms were observed between the major conformation of inhibitor 1 and the PR (Figure 3A). The inhibitor hydroxyl group formed hydrogen bonds with all four carboxylate oxygens of the catalytic Asp 25 and 25′ with distances of 2.7, 2.4, 3.2 and 2.8 Å, which is likely to mimic the interactions of the tetrahedral transition state of the proteolytic reaction. At one end of the inhibitor, the two oxygen atoms of the bis-THF group formed three hydrogen bonds with the main-chain amides of Asp 29′ and Asp 30′, and there is a C-H…O interaction with Gly 48′. At the other end of the inhibitor, one of the two oxygen atoms in the 1,3-benzodioxole group interacted with the main-chain amide and the side-chain carboxylate oxygen of Asp 30. The polar interaction with the carboxylate side chain of Asp 30 appeared to be similar to the proton-mediated interaction observed between the carboxylate side chains of Asp 30 and Glu at P2′ in peptide analogs 14. The other oxygen atom of the 1,3-benzodioxole group showed a water-mediated interaction with the amide of Gly 48 in the flap. The 1,3-benzodioxole group had a C-H…O interaction with Gly 48, C-H…π contacts with Ala 28, Val 32, and Ile 50′, and a water-mediated interaction with amide of Asp 29. The P1′ side chain had a C-H…O interaction with the carbonyl oxygen of Gly 27, while the nitrogen next to the P1 phenyl group of the inhibitor formed a hydrogen bond with the carbonyl oxygen of Gly 27′. The phenyl group has C-H…π contacts with Pro 81, Val 82 and Gly 49′. The flexible PR flaps were stabilized by the C-H…O interactions of Gly 48 and 48′ with the inhibitor. A water molecule (H2OA) linked the amides of Ile 50 and 50′ in the flaps with the carbonyl oxygen and one of the sulfonamide oxygen atoms of inhibitor 1. This water is conserved in all structures of PR with inhibitors/substrates 22, except for those urea-based inhibitors designed explicitly to substitute for this water 23–25. Equivalent hydrogen bond interactions were observed for the minor conformation of the inhibitor except for one water-mediated hydrogen bond. The water H2OB formed interactions connecting oxygen in the 1,3-benzodioxole group of inhibitor 1 with the amide of Gly 48′. This hydrogen bond was longer, and therefore, weaker for the major conformation of the inhibitor (3.5 Å) than for the minor one (3.0 Å). Also, the minor conformation of inhibitor 1 formed additional C-H…π contacts between the phenyl group and Leu 23′ and Ile 84′ compared to those of the major conformation.
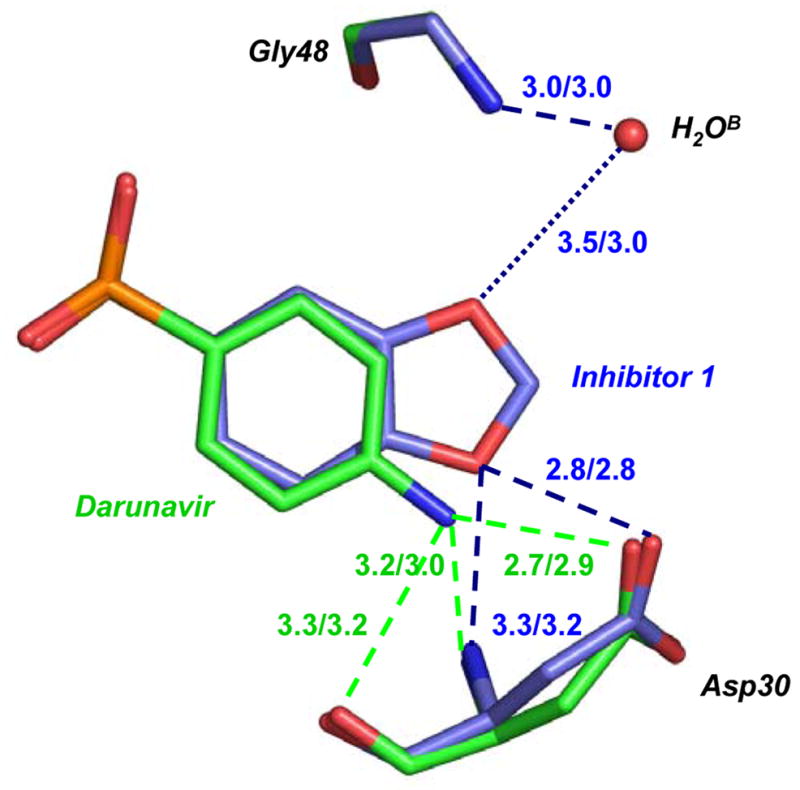
Figure 3. Protease interactions with inhibitor 1 and darunavir.

3A. Polar interactions of inhibitor 1 with PR
The major conformation of inhibitor 1 is shown with interacting PR residues. Water is shown as red spheres. Hydrogen bond interactions are indicated by dotted lines, and CH…O interactions with main chain PR atoms by dashed lines. The H2OC O-H…π interaction with the P2′ aromatic ring is shown as a gray dotted line. The interactions of the alternate conformation of inhibitor are essentially the same.
3B. Comparison of PR interactions with inhibitor 1 and darunavir.
Carbon atoms in PR/inhibitor 1 are colored in blue and those in PR/darunavir are green. The hydrogen bond interactions are indicated by the dashed lines, with distances in Å for the major/minor conformation of inhibitor, and colored blue for PR/inhibitor 1 and green for PR/darunavir. The dotted line indicates the water-mediated interaction with a weaker hydrogen bond for the major conformation.
Comparison of the PR interactions with inhibitor 1 and darunavir
It is important to compare the details of the PR interactions with inhibitor 1 to those of the chemically related darunavir. The chemical difference is that the P2′ aniline group of darunavir has been replaced by the 1, 3-benzodioxole group in inhibitor 1 (Figure 1). The atoms of the two inhibitors (excluding the different P2′ groups) superimpose with an RMSD of 0.16 Å for the major conformation and 0.23 Å for the minor conformation. Therefore, the inhibitor structures are very similar for the equivalent atoms. Inhibitor 1 and darunavir formed very similar hydrogen bond, C-H…O, and C-H…π interactions with the PR; the interatomic distances are almost identical. However, the interactions of the chemically distinct P2′ groups differ as shown in Figure 3B. The three direct hydrogen bonds formed by the aniline group of darunavir to Asp 30 were substituted by the two direct hydrogen bonds and one water-mediated interaction of the 1, 3-benzodioxole group of inhibitor 1 with Gly 48 (Figure 3B). Importantly, darunavir had no equivalent interaction with the flap residue Gly 48. The interaction of inhibitor 1 with Gly 48 may stabilize the flexible flap region, as observed for the peptide substrates. Both inhibitors share very similar hydrophobic groups, and form similar hydrophobic interactions with PR. PR/inhibitor 1 showed slightly more van der Waals interactions between the protein and the inhibitor (100 with distance less than 4.0 Å) than observed for the PR/darunavir complex (96 with distance less than 4.0 Å).
Comparison of inhibitor interactions in PR and mutants
Overall, inhibitor 1 formed similar hydrogen bond interactions in the four mutant complexes and in the wild type PR complex, with the exception of PRD30N (Figure 4). Generally, the mutants showed altered hydrophobic contacts of inhibitor with the mutated residues. The significance of the small structural differences in the mutant complexes relative to wild type PR are best demonstrated in comparison to the changes described for the same mutant complexes with darunavir 10, 11. Both inhibitor 1 and darunavir showed similar hydrogen bond interactions with PR, PRV82A, and PRI84V, while small differences were observed for the other two mutants (Figure 4). The interactions of each mutant will be considered separately.
Figure 4. Structural changes in mutants compared to wild type PR.






The major and minor conformations of side chains in PR are shown in dark and light grey ball-and-stick, respectively. The alternate conformations in mutant complexes are shown in red and pink, and the alternate conformations of inhibitor are in blue and cyan. The polar interactions are indicated by the dashed lines, van der Waals interactions by the dotted lines, and C-H…π interactions in dot-and-dash line. The interatomic distances are shown in Å for both subunits.
4A. Interactions of P1′ of inhibitor 1 with residue 82′in PRV82A and PR. The minor conformation of inhibitor 1 and alternate conformation of Val82 in PR are omitted for clarity. The shift in position of Cα 82 between PR and mutant is shown in blue dotted lines. 4B. Van der Waals interactions of P1′ in inhibitor 1 with residue 82′ in PRV82A and PR. View and representation are similar to 4A.
4C. PRI84V and PR interactions with inhibitor 1.
4D. PRI50V and PR interactions with inhibitor 1. Only a single conformation is shown for clarity.
4E. PRD30N and PR interactions with inhibitor 1.
4F. PRD30N and PR interactions with darunavir.
The complex with PRV82A had a single conformation of inhibitor and Ala 82, whereas Val 82 and inhibitor 1 had two alternate conformations in the other structures. The side chain of Val 82 in the PR/inhibitor 1 structure formed 3 C-H…π interactions with the phenyl group at P1 and a van der Waals contact with the P1′ group. The interactions of Ala 82 in the mutant are very similar, because the main chain of residue 82 has moved by 0.5 and 0.8 Å for the Cα atoms of the two subunits, respectively, compared to the wild type (Figure 4A and B). This structural change has repositioned the side chain of Ala 82 to form 3 C-H…π interactions with the P1 phenyl, and one van der Waals interaction with the P1′ leucine-like group of inhibitor. A similar shift of Ala 82 compared to Val 82 was observed for the corresponding complexes with darunavir 11 and indinavir 26.
Reduced van der Waals contacts and loss of a C-H…O interaction were apparent in the PRI84V/inhibitor 1 structure. The side chain of the mutated Val 84 had one interaction with the P1 phenyl group, while in the wild type PR/inhibitor 1 complex, Ile 84/84′ had a weak CH…O interaction with a sulfonamide oxygen of inhibitor 1, and 3 hydrophobic contacts with the phenyl group (Figure 4C). In comparison, Val 84 in the PRI84V/darunavir structure had lost two contacts for both conformations of darunavir 11. For both inhibitors the mutant showed fewer interactions than in the wild type enzyme.
The mutant PRI50V showed fewer changes in the interactions with inhibitor 1 than were observed for darunavir. A good hydrophobic C-H…π contact of Ile 50/50′ with the P2′ group of inhibitor 1 was lost in the PRI50V mutant (Figure 4D), which is similar to the changes observed in the equivalent complexes with darunavir. The side chain of Val 50 had moved to form a different C-H…O interaction with the sulfonamide oxygen compared to that in the wild type complex. However, inhibitor 1 retained the strong hydrogen bond interactions of the P2′ group with Asp 30 in the PRI50V complex, unlike darunavir where the interaction with the carbonyl oxygen of Asp 30 was absent in the mutant complex 10. Therefore, darunavir loses a strong hydrogen bond interaction in the PRI50V complex, while inhibitor 1 loses an energetically weaker hydrophobic contact.
The most notable differences were observed in the inhibitor complexes with mutant PRD30N (Figure 4E and F). The structure of PRD30N/darunavir showed a water-mediated interaction of the P2′ aniline group with the side chain of Asn 30, instead of the direct hydrogen bond observed for one conformation of Asp 30 in the wild type PR/darunavir structure 10. In contrast, the polar interactions of the P2′ 1, 3-benzodioxole group of inhibitor 1 with Asn/Asp 30 were similar in PRD30N and in PR for the major conformations of inhibitor and Asn 30. However, the minor conformation of the Asn 30 side chain formed a new hydrogen bond with an oxygen atom of the bis-THF in the minor conformation of inhibitor 1 (Figure 4E). Therefore, the interactions with inhibitor 1 were maintained in the mutant and wild type PR, while darunavir had replaced a direct hydrogen bond with a water-mediated interaction for one conformation of Asp 30.
The interactions of inhibitor 1 with the mutants may be more similar to those with the wild type PR due to the water-mediated interaction with Gly 48, which was not seen in the darunavir complexes. This interaction with Gly 48 in the flap will stabilize the closed flap conformation of the protease dimer, and more closely mimics the protease interaction with substrates 27. The absence of a hydrogen bond interaction of darunavir with Gly 48 may facilitate the observed structural changes in its interactions with mutants PRD30N and PRI50V.
DISCUSSION
In preliminary studies inhibitor 1 has shown extremely potent antiviral activity on HIV-1 9. Inhibitor 1 showed about 10-fold higher potency for wild type strains of HIV than the chemically related darunavir. Moreover, inhibitor 1 is also chemically related to the protease inhibitor brecanavir, which is in Phase III clinical trials 28. Brecanavir differs from inhibitor 1 in the presence of a large substituted tyrosine at P1 instead of the P1 phenyl group. The structural and kinetic analyses are reported here for inhibitor 1 with wild type PR and the drug resistant mutants PRD30N, PRI50V, PRV82A and PRI84V. The inhibition constants and the protease-inhibitor crystal structures showed relatively small changes compared to the wild type PR, except for PRD30N. Both PRD30N and PRV82A showed compensating interactions with inhibitor 1 relative to those of PR, while reduced hydrophobic contacts were observed with PRI50V and PRI84V. These structural data suggest that inhibitor 1 will maintain inhibitory activity on the resistant variants of HIV containing these mutations. Importantly, inhibitor 1 retained similar polar interactions with PRD30N and PRI50V, unlike the significantly reduced interactions and inhibition of darunavir. Previously, we predicted that darunavir would be less effective on HIV variants containing the D30N or I50V mutations. Our crystallographic, kinetic, and antiviral data suggest that inhibitor 1 will be a valuable addition to the antiviral inhibitors with high potency against resistant strains of HIV. The detailed structural analysis and very similar Ki values for darunavir and inhibitor 1 suggest that the higher antiviral potency of inhibitor 1 arises from better bioavailability and/or better pharmacokinetic behavior.
EXPERIMENTAL METHODS
Protein expression, purification, and inhibition assays
The HIV-1 PR and mutants, which include 5 mutations Q7K, L33I, L63I, C67A and C95A to optimize protein stability 29 were constructed, expressed and purified as described in 29–31. Mutations were confirmed by nucleic acid sequencing and protein mass spectrometry. The inhibition constants were determined by a fluorescence assay using the substrate RE(Edans)SQNY*PIVRK(Dabcyl)R (synthesized by Dr. Ivo Blaha, Ferring Leciva, Prague) as described in 18. 8 μl of protease (final concentration of 7–57 nM) was added to 100 μl solution of 500 mM phosphate buffer, pH = 5.6, 1 M NaCl, 2 mM EDTA, 10 mM dithiothreitol, then 2 μl of inhibitor was added. The inhibitors were dissolved in dimethylsulfoxide (DMSO). Their concentrations were determined from peak areas on HPLC (LaChrom, Hitachi, Merck) based on their high structural similarities for amprenavir. The mixture was pre-incubated at 37 °C for 5 min. 90 μl substrate dissolved in water (final concentration of 6 μM) was then added to the mixture to initiate the reaction. The reaction solution was assayed over 5 min for the increase in fluorescence using 355 nm excitation and 460 nm emission wavelengths in a Wallac 1420 Victor2 fluorimeter-luminometer (Wallac Oy, Turku, Finland). Inhibition constants were obtained from the IC50 values estimated from a dose-response curve using the equation Ki = (IC50 − 0.5[E])/(1+[S]/Km), where [E] and [S] are the PR and substrate concentrations, respectively.
Crystallographic analysis
The inhibitor 1 was dissolved in dimethylsulfoxide (DMSO) and centrifuged briefly to remove any insoluble material. The inhibitor 1 was mixed with protease in a ratio of 2:1 to 5:1. Crystals were grown by the hanging-drop vapor-diffusion method at room temperature. Crystals of the PRD30N complex grew from a 2.5 mg/ml protein solution at pH 4.8 with 25mM sodium acetate, 10% (w/v) sodium chloride, 0.5% dioxane and 1.5% (v/v) DMSO. The PRI50V complex crystallized from a 2.3 mg/ml protein solution at pH 4.6 with 25mM sodium acetate, 9.4% (w/v) sodium chloride and 6.7% (v/v) DMSO. The PRV82A complex crystallized from a 3.3 mg/ml protein solution at pH 5.0 with 25mM citrate phosphate, 10% (w/v) sodium chloride and 6–7% (v/v) DMSO. Crystals of the PRI84V complex grew from a 2.5 mg/ml protein solution buffered at pH 5.0 with 25mM sodium acetate, 8% (w/v) sodium chloride and 10% (v/v) DMSO.
A single crystal was mounted in a fiber loop with 20–30% (v/v) glycerol as cryoprotectant in the reservoir solution. X-ray diffraction data were collected at Advanced Photon Source, SER-CAT beamline. The data were integrated, scaled and merged using the HKL2000 package 32. The structures were solved by molecular replacement with AMoRe 33 using the starting model 1FS65 from the Protein Data Bank 34 for PRV82A and PR for the other structures. Refinement was carried out using SHELX-97 35 and manual adjustment used the molecular graphics program O 36. Structural figures were made using Bobscript 37, Ras3D and PyMol 38.
The crystal structures have been deposited in the Protein Data Bank with the codes: 2Z4O for PR/inhibitor 1, 2QD7 for PRV82A/inhibitor 1, 2QD8 for PRI84V/inhibitor 1, 2QCI for PRD30N/inhibitor 1, and 2QD6 for PRI50V/inhibitor 1
Acknowledgments
Yunfeng Tie was supported in part by a Molecular Basis of Disease Fellowship, and Irene Weber and Robert Harrison are Distinguished Cancer Scholars. The research was supported in part by the Molecular Basis of Disease Program, the Georgia Research Alliance, the Georgia Cancer Coalition, the National Institute of Health grants GM62920 and GM53386. We thank the staff at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02–06CH11357.
Abbreviations
- PR
protease
- HIV-1
human immunodeficiency virus type 1
- HAART
highly active antiretroviral therapy
- AIDS
acquired immunodeficiency syndrome
- PI
protease inhibitor
- THF
tetrahydrofurane
Footnotes
PDB IDs: 2Z4O for PR/inhibitor 1, 2QD7 for PRV82A/inhibitor 1, 2QD8 for PRI84V/inhibitor 1, 2QCI for PRD30N/inhibitor 1, and 2QD6 for PRI50V/inhibitor 1
References
- 1.Barbaro G, Lucchini A, Barbarini G. Highly active antiretroviral therapy in HIV-associated pulmonary hypertension. Minerva Cardioangiol. 2005;53:153–4. [PubMed] [Google Scholar]
- 2.Barlett JA, DeMasi R, Quinn J, Moxham C, Rousseau F. Overview of the Effectiveness of Triple Combination Therapy in Antiretroviral-Naïve HIV-1 Infected Adults. AIDS. 2001;15:1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 3.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–60. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 4.Grabar S, Pradier C, Le Corfec E, Lancar R, Allavena C, Bentata M, Berlureau P, Dupont C, Fabbro-Peray P, Poizot-Martin I, Costagliola D. Factors associated with clinical and virological failure in patients receiving a triple therapy including a protease inhibitor. Aids. 2000;14:141–9. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]
- 5.Hertogs K, Bloor S, Kemp SD, Van den Eynde C, Alcorn TM, Pauwels R, Van Houtte M, Staszewski S, Miller V, Larder BA. Phenotypic and genotypic analysis of clinical HIV-1 isolates reveals extensive protease inhibitor cross-resistance: a survey of over 6000 samples. AIDS. 2000;14:1203–1210. doi: 10.1097/00002030-200006160-00018. [DOI] [PubMed] [Google Scholar]
- 6.Wu TD, Schiffer CA, Gonzales MJ, Taylor J, Kantor R, Chou S, Israelski D, Zolopa AR, Fessel WJ, Shafer RW. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–4847. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhee SY, Liu T, Ravela J, Gonzales MJ, Shafer RW. Distribution of human immunodeficiency virus type 1 protease and reverse transcriptase mutation patterns in 4,183 persons undergoing genotypic resistance testing. Antimicrob Agents Chemother. 2004;48:3122–6. doi: 10.1128/AAC.48.8.3122-3126.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Clercq E. New approaches toward anti-HIV chemotherapy. J Med Chem. 2005;48:1297–313. doi: 10.1021/jm040158k. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Bis-tetrahydrofuran: A privileged ligand for a new generation of HIV-protease inhibitors that combat drug-resistance. Chem Med Chem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 10.Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang YF, Leshchenko S, Ghosh AK, Harrison RW, Weber IT. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J Med Chem. 2006;49:1379–87. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2004;338:341–52. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 12.Jarvis B, Faulds D. Nelfinavir. A review of its therapeutic efficacy in HIV infection. Drugs. 1998;56:147–67. doi: 10.2165/00003495-199856010-00013. [DOI] [PubMed] [Google Scholar]
- 13.Mahalingam B, Louis JM, Reed CC, Adomat JM, Krouse J, Wang YF, Harrison RW, Weber IT. Structural and kinetic analysis of drug resistant mutants of HIV-1 protease. Eur J Biochem. 1999;263:238–245. doi: 10.1046/j.1432-1327.1999.00514.x. [DOI] [PubMed] [Google Scholar]
- 14.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: High resolution crystal structures of the mutant protease/substrate analogue complexes. Proteins: Structure, Function, and Genetics. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 15.Tisdale M, Myers R, Randall S, Maguire M, Ait-Khaled M, Elston R, Snowden W. Resistance of the HIV Protease Inhibitor Amprenavir In Vitro and in Clinical Studies. Clin Drug Invest. 2000;20:267–285. [Google Scholar]
- 16.Johnson VA, Brun-Vezinet F, Clotet B, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD. Update of the Drug Resistance Mutations in HIV-1: Fall 2006. Top HIV Med. 2006;14:125–130. [PubMed] [Google Scholar]
- 17.Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) GRL-98065 potent against multi-PI-resistant HIV in vitro. Antimicrob Agents Chemother. 2007;51:2143–2155. doi: 10.1128/AAC.01413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagossi P, Kadas J, Miklossy G, Boross P, Weber IT, Tozser J. Development of a Microtiter Plate Fluorescent Assay for Inhibition Studies on the HTLV-1 and HIV-1 Proteinases. J Virol Met. 2004;119:87–93. doi: 10.1016/j.jviromet.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Reiling KK, Endres NF, Dauber DS, Craik CS, Stroud RM. Anisotropic dynamics of the JE-2147-HIV protease complex: drug resistance and thermodynamic binding mode examined in a 1.09 A structure. Biochemistry. 2002;41:4582–94. doi: 10.1021/bi011781z. [DOI] [PubMed] [Google Scholar]
- 20.Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J Mol Biol. 1996;256:228–34. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- 21.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114. J Mol Biol. 2006;363:161–73. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gustchina A, Sansom C, Prevost M, Richelle J, Wodak SY, Wlodawer A, Weber IT. Energy calculations and analysis of HIV-1 protease-inhibitor crystal structures. Protein Eng. 1994;7:309–317. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- 23.Hodge CN, Aldrich PE, Bacheler LT, Chang CH, Eyermann CJ, Garber S, Grubb M, Jackson DA, Jadhav PK, Korant B, Lam PY, Maurin MB, Meek JL, Otto MJ, Rayner MM, Reid C, Sharpe TR, Shum L, Winslow DL, Erickson-Viitanen S. Improved cyclic urea inhibitors of the HIV-1 protease: synthesis, potency, resistance profile, human pharmacokinetics and X-ray crystal structure of DMP 450. Chem Biol. 1996;3:301–14. doi: 10.1016/s1074-5521(96)90110-6. [DOI] [PubMed] [Google Scholar]
- 24.Lam PY, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, et al. Rational design of potent, bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. Science. 1994;263:380–4. doi: 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- 25.Lam PY, Ru Y, Jadhav PK, Aldrich PE, DeLucca GV, Eyermann CJ, Chang CH, Emmett G, Holler ER, Daneker WF, Li L, Confalone PN, McHugh RJ, Han Q, Li R, Markwalder JA, Seitz SP, Sharpe TR, Bacheler LT, Rayner MM, Klabe RM, Shum L, Winslow DL, Kornhauser DM, Hodge CN, et al. Cyclic HIV protease inhibitors: synthesis, conformational analysis, P2/P2′ structure-activity relationship, and molecular recognition of cyclic ureas. J Med Chem. 1996;39:3514–25. doi: 10.1021/jm9602571. [DOI] [PubMed] [Google Scholar]
- 26.Mahalingam B, Wang YF, Boross PI, Tozser J, Louis JM, Harrison RW, Weber IT. Crystal structures of HIV protease V82A and L90M mutants reveal changes in the indinavir-binding site. Eur J Biochem. 2004;271:1516–1524. doi: 10.1111/j.1432-1033.2004.04060.x. [DOI] [PubMed] [Google Scholar]
- 27.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 angstroms resolution crystal structures of HIV-1 protease mutants with substrate analogs. Febs J. 2005;272:5265–77. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ford SL, Reddy YS, Anderson MT, Murray SC, Fernandez P, Stein DS, Johnson MA. Single-dose safety and pharmacokinetics of brecanavir, a novel human immunodeficiency virus protease inhibitor. Antimicrob Agents Chemother. 2006;50:2201–6. doi: 10.1128/AAC.01490-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wondrak EM, Louis JM. Influence of flanking sequences on the dimer stability of human immunodeficiency virus type 1 protease. Biochemistry. 1996;35:12957–12962. doi: 10.1021/bi960984y. [DOI] [PubMed] [Google Scholar]
- 30.Wlodawer A, Erickson JW. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 31.Louis JM, Wondrak EM, Copeland TD, Smith CA, Mora PT, Oroszlan S. Chemical synthesis and expression of the HIV-1 protease gene in E. coli. Biochem Biophys Res Commun. 1989;159:87–94. doi: 10.1016/0006-291x(89)92408-x. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Navaza J. AMoRe: An automated package for molecular replacement. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- 34.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheldrick GM, Schneider TR. SHELXL: High resolution refinement. Methods in Enzymology. 1997;277:319–343. [PubMed] [Google Scholar]
- 36.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 37.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D Biol Crystallogr. 1999;55:938–40. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]