

Abstract
A Brønsted base activation mode for oxidative, Pd(II)/sulfoxide catalyzed, intermolecular C—H allylic amination is reported. N,N-diisopropylethylamine was found to promote amination of unactivated terminal olefins, forming the corresponding linear allylic amine products with high levels of stereo-, regio-, and chemoselectivity. The predictable and high selectivity of this C—H oxidation method enables late-stage incorporation of nitrogen into advanced synthetic intermediates and natural products.
Introduction
The ubiquitous C—H bond has emerged as a viable precursor to many functional groups through several transition metal-catalyzed reactions.1 The practical utility of such reactions for complex molecule synthesis is governed by their ability to operate with predictable and high levels of chemo-, stereo-, regio- and site selectivity. The strategic application of selective C—H oxidation reactions at late stages of syntheses has been demonstrated to increase product diversity1d as well as eliminate functional group interconversions and/or oxidation state changes, which are often required when working with oxidized intermediates.2
 |
(1) |
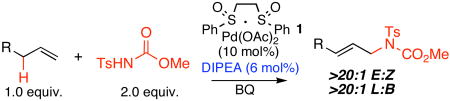
The prevalence of nitrogen functionality in biologically important small molecules, accompanied by the extensive functional group manipulations (FGM) commonly employed to install nitrogen underscores the utility of direct C—H to C—N bond transformations.3 For example, routine synthesis of linear allylic amines involves oxidative, nucleophilic, and often reductive conditions to access the requisite allylic alcohol, which is generally converted into a leaving group before displacement with nitrogen.4,5 We recently reported Pd(II)/bis-sulfoxide catalyzed allylic C—H amination reactions that furnish either branched6 or linear7 allylic amines directly from terminal olefins with predictable and high regioselectivities.8 Moreover, we have demonstrated that direct installation of nitrogen functionality from C—H bonds can have a powerful streamlining effect on synthetic routes. For example, the intermolecular C—H to C—N bond forming route to a rigidified (+)-deoxynegamycin analogue proceeded with five fewer steps, five fewer FGM, and a higher overall yield than the alternative C—O to C—N bond forming route.7 Despite this, the intermolecular allylic C—H amination reaction suffered from several limitations associated with the requirement for a Lewis acid activator [Cr(salen)Cl 2] to effect functionalization. Incompatibilities with Lewis basic functionality, and isomerization of terminal olefins on electron-deficient substrates could limit the applicability of this otherwise useful and general method. With the goal of further developing this significant transformation, herein we report a novel nucleophile activation strategy for promoting intermolecular allylic C—H amination that operates via electrophilic Pd(II)-catalysis with acidic N-tosylcarbamate nucleophiles (pKa ∼ 3.5).9 This strategy employs exogenous catalytic amine base to elevate the concentration of anionic carbamate nucleophile that is not associated with the Pd counterion, resulting in one of the most mild and selective C—H oxidation reactions reported to date (Equation 1).
Results and Discussion
Design Principles
We previously introduced Pd(II)/sulfoxide catalysts (e.g. 1) as electrophilic promoters of a heterolytic, allylic C—H cleavage event to generate π-allylPd intermediates; this eliminates the need for pre-oxidized allylic precursors, as required in Pd(0) substitution reactions (Figure 1, A).
Figure 1.

Activation Strategies for Catalytic, Allylic C—H Oxidation Reactions
Although both Pd(0) and Pd(II) manifolds proceed through a common π-allylPd intermediate, they require very different strategies for promoting functionalization (Figure 1, B). The basic, reducing conditions of Pd(0)-mediated allylic substitution reactions are compatible with strong nucleophiles (i.e. stoichiometric anionic nucleophiles) and σ-donating ligands.10 However, both of these strategies for promoting functionalization are incompatible with electrophilic Pd(II)-mediated allylic C—H oxidation reactions that proceed under acidic conditions. We have alternatively delineated two general activation strategies for functionalization with acidic N-tosylcarbamate pro-nucleophiles. The first involves the use of π-acidic ligands [benzoquinone (BQ)11] acting with a Lewis acid co-catalyst [Cr(III)(salen)Cl, 2] to promote intermolecular functionalization, possibly by activating electrophilic π-allylPd intermediates towards nucleophilic attack.7 The second strategy harnesses palladium carboxylate counterions as a source of endogenous base to deprotonate and thereby activate tethered pro-nucleophiles (Figure 1, B).6 Unfortunately, this mild strategy proved ineffective for promoting intermolecular functionalization. We hypothesized that the addition of an exogenous base in catalytic amounts would increase the concentration of deprotonated nitrogen nucleophile in solution, thereby promoting functionalization without requiring a tether.12 In addition to providing a novel Brønsted base activation mode for intermolecular oxidative Pd(II) systems, this strategy has the potential to overcome several challenges faced by the Lewis acid/BQ system, such as incompatibilities with Lewis basic functionality and isomerization of terminal olefins on electron-deficient substrates.
Reaction Optimization
Two features of the catalytic base additive were predicted to be critical to the success of this activation mode: (1) sufficiently high basicity to generate an elevated concentration of deprotonated nucleophile in solution, (2) a weak binding affinity for the highly electrophilic Pd(II) catalyst.
Table 1 provides a summary of the experiments exploring these criteria. As previously demonstrated, only a trace amount (ca. 1%) of intermolecular allylic C—H amination product was detected under the endogenous catalytic base conditions that promote intramolecular C—H amination with this type of nucleophile (Table 1, entry 1). In the presence of Lewis acid activator and quinone [i.e. Cr(salen)Cl 2 (6 mol%) and 1,4-benzoquinone (BQ, 2 equiv.)], Pd/sulfoxide catalyst 1 (10 mol%) furnished intermolecular allylic C—H amination product 4 with good yields and selectivities (Table 1, entry 2). To test the hypothesis that an external base could replace Lewis acid activation and alternatively promote functionalization by direct deprotonation of the nitrogen pro-nucleophile, we surveyed a series of common organic Brønsted bases as catalytic additives. Pyridine (6 mol%) exhibited low reactivity, potentially due to its ability to serve as a ligand for Pd (Table 1, entry 3). In an effort to prevent a possible binding event, sterically congested 2,6-di-tert-butylpyridine was screened; even this additive failed to promote the reaction (Table 1, entry 4), supporting the hypothesis that a minimum level of basicity may also be needed for this activation strategy. Coordinating, primary amines with an increased level of basicity such as N-isopropylamine gave similarly poor results (Table 1, entry 5). Significantly, however, strongly basic yet sterically encumbered amines such as N,N-diisopropylamine, triethylamine, and N,N-diisopropylethylamine (DIPEA) generated 4 in good yields (Table 1, entries 6-8) and selectivities for the linear (E)-amine product. Although a certain concentration of deprotonated nucleophile is clearly needed for reactivity, addition of stoichiometric amounts of pre-formed anionic nucleophile, MeOC(O)NTs-HDIPEA, generated only small amounts of product (Table 1, entry 9). This suggests that one or more steps in the catalytic cycle are incompatible with high concentrations of even a weakly coordinating anionic nucleophile. Accordingly, the concentration of Brønsted base additive was found to have a dramatic effect on reaction yields (Figure 2). Adding between 3 and 10 mol% DIPEA dramatically increases the yield of 4, whereas increasing the amount of DIPEA to 25 mol% or higher was deleterious to product formation. The observed trend is most likely due to attenuation of the electrophilicity of the Pd/bis-sulfoxide catalyst 1 at high concentrations of base, although inhibition of acid-mediated catalyst reoxidation cannot be ruled out.13
Table 1.
Exogenous, Catalytic Brønsted Base Additives for Direct Allylic C—H Amination
 | ||||
|---|---|---|---|---|
| entry | additive | yield(%)a | L:Bb | E:Zc |
| 1 | none | 1d | - | - |
| 2 | (S,S)-Cr(salen)Cl 2 | 50 | 11:1 | 19:1 |
| 3 | pyridine | 9 | 12:1 | 14:1 |
| 4 | 2,6-di-tert-butylpyridine | - | - | - |
| 5 | N-isopropylamine | - | - | - |
| 6 | N,N-diisopropylamine | 66 | 12:1 | 15:1 |
| 7 | triethylamine | 60 | 14:1 | 15:1 |
| 8 | N,N-diisopropylethylamine | 66 | 11:1 | 17:1 |
| 9 | MeOC(O)NTsH-DIPEAe | 9 | 6:1 | 10:1 |
isolated yield of linear product.
linear/branched ratio determined by HPLC of crude reaction mixture.
determined by 1H NMR after chromatography.
1H NMR yield by comparison with internal standard.
2.0 equiv. of the pre-formed salt was used instead of MeOC(O)NHTs.
Figure 2.
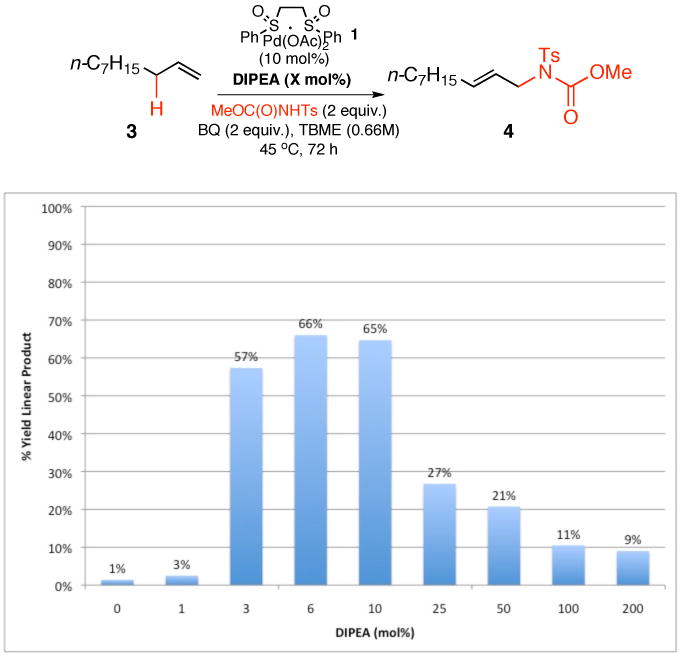
Effect of DIPEA Concentration on Reactivity for Linear Allylic C—H Amination
Reaction Scope
These newly discovered DIPEA-base amination conditions provide an improvement in product yield and chemoselectivity with N-tosylcarbamate nucleophiles, which enabled a substantial broadening in substrate scope (Table 2). Generally, we found that the Cr conditions led to low yields of product for substrates possessing isomerization-prone olefins. In contrast, aliphatic and aromatic substrates containing these types of olefins worked well under the DIPEA conditions (Table 2, entries 1 and 5). Gratifyingly, aromatic substrates containing widely used synthetic handles such as esters, aldehydes, nitriles, and phenols all underwent efficient reaction to their corresponding allylic amine products under the DIPEA conditions (Table 2, entries 2-6). Products 6-10 represent powerful bifunctional aryl building blocks, with the potential for subsequent manipulation. Aryl triflates are also well tolerated, displaying the orthogonality of this Pd(II) reaction to traditional Pd(0) methodologies (Table 2, entry 5).
Table 2.
Scope and Comparison of the Brønsted Base vs. Lewis Acid Promoted Allylic C—H Amination
 | |||||
|---|---|---|---|---|---|
| entry | allylic amination product | isolated yield | |||
| DIPEAa | Cr(salen)Cl | ||||
| 1 | 5 | 61%b | 37%c | ||
| 2 | 6 | R = CHO | 77% | 76% | |
| 3 | 7 | R = CN | 79% | 59% | |
| 4 | 8 | R = H | 81% | 53% | |
| 5 | 9 | R = Tf | 89% | 36% | |
| 6 | (-)-10 | 54% | <1% | ||
| 7 | (+)-11 | 48%e | <1% | ||
| 8 | (-)-12 | 64% | <1% | ||
| 9 | (+)-13 | 59% | 45% | ||
| 10 | 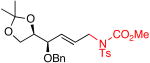 |
(+)-14 | 76% | 63%d | |
| 11 | 15 | R = Me | 84% | 53%d | |
| 12 | 16 | R = Bn | 87% | 65%d | |
| 13 | 17 | R = t-Bu | 69%f | 40%d | |
| 14 | 18 | R = Fm | 55%f | 40%d | |
Average of at least two runs at 0.3 mmol. Products were isolated as one regio- and olefin isomer (1H NMR).
18:1 E/Z.
13:1 E/Z.
Reported in ref. 7.
48 h reaction time.
10 mol% DIPEA, CCl4 solvent, 24 h reaction time; Fm = 9-fluorenylmethyl.
Epoxides are valuable intermediates in complex molecule synthesis, because they can be readily elaborated. For example, chiral (S,S)-Cr(salen) complex 2 is known to catalyze asymmetric epoxide ring opening reactions.14 Expectedly, terminal epoxide substrates exposed to 2 led to complex mixtures, providing none of the desired products (Table 2, entries 6-8). In contrast, DIPEA conditions yielded allylic amination products with high selectivities and good yields (Table 2, entries 6-9). Even highly reactive glycidol moieties are preserved under these reaction conditions (Table 2, entry 6). Products such as (-)-10 can be ring-opened to generate several important structural motifs, such as those found in beta-blocker15 and cystic fibrosis drugs.16 Di- and trisubstituted epoxides have the advantage of being capable of regioselective ring opening as a means of generating structural diversity (Table 2, entries 8 and 9).17 Site-selective olefin epoxidations of di- and trisubstituted olefins followed by allylic C—H amination of terminal olefins enables the streamlined construction of bifunctional building blocks as exemplified by (-)-12 and (+)-13. As previously shown with the Cr/BQ system, amino sugar precursor (+)-14 under the DIPEA conditions was isolated as a single diastereomer, indicating no change in configuration at the adjacent stereocenter (Table 2, entry 10).7
Additionally, a variety of different N-tosylcarbamate esters were examined to evaluate nucleophile scope. Using allyl cyclohexane as a substrate, substantial increases in yield were found for both the methyl- and benzyl N-tosylcarbamate under the DIPEA conditions relative to the Cr/BQ system (Table 2, entries 11 and 12). Yields with tert-butyl- and 9-fluorenylmethyl N-tosylcarbamates could also be increased to above 50%, however, at this time optimal conditions utilize 10 mol% DIPEA and carbon tetrachloride solvent to achieve good yields with these nucleophiles (Table 2, entries 13 and 14).18 Overall, product yields under the DIPEA conditions were consistently higher than conditions employing Cr/BQ.
As a final point, the intermolecular linear allylic amination presents a convenient method for rapidly accessing a variety of valuable, nitrogen-containing compounds. N-Tosylcarbamate nucleophiles can be easily synthesized on a multi-gram scale by reacting commercially available N-tosylisocyanate with the desired alcohol to afford crystalline, bench stable reagents (Figure 3, A). Moreover, the allylic N-tosyl carbamate moiety can be transformed into a variety of diverse compounds in high yields (Figure 3, B). For example, both the carbamate and the tosyl groups can be selectively removed by employing either mild base or a one-electron reductant, respectively. Upon removal of the tosyl moiety, the carbamate can either be hydrolyzed to yield the free allylic amine, or reduced to the corresponding methylamine. It is significant to note that all of these transformations can be performed on densely functionalized molecules while preserving the valuable internal olefin.
Figure 3.

Versatility of the N-Tosyl carbamate Functional Group
a) K2CO3, MeOH, 25°C, 3.5 h; (b) Na-C10H8, -78°C, DME, 10 min; (c) NaOH, H2O/EtOH, 110°C, 4 h; (d) LiAlH4, THF, reflux, 5 h.
Late-stage C—H Amination of Natural Product Derivatives
Late stage C—H activation approaches can be particularly powerful when applied to the diversification of complex natural products or the functionalization of late-stage intermediates in a total synthesis. We targeted estrone derivatives19 (+)-24/(+)-26 for the amination reaction, as linear amines have been used to generate steroid-antibiotic hybrid compounds capable of selectively destroying estrogen receptor-rich cancer cells (Figure 4, A).20
Figure 4.

Late-Stage Oxidation of Natural Product Derivatives via Direct Allylic C—H Amination
(a) 1 (10 mol%), DIPEA (6 mol%), TsNHCO2Me (2.0 equiv.), BQ (2.0 equiv.), 0.66 M TBME, 45°C, 72 h.
Protected (+)-23 underwent smooth conversion to the corresponding linear allylic amine product with outstanding selectivity (Figure 4, A, >20:1 E/Z, L/B). Remarkably, the unprotected diol (+)-25 was also an excellent substrate for the Brønsted base-promoted amination reaction.21 α-Cedrene derived (-)-27, also containing an unprotected secondary alcohol moiety as well as a sterically hindered all-carbon quaternary center in the homoallylic position, underwent efficient amination to give product (-)-28 in high yield with exceptional selectivity (Figure 4, B). Tolerance of unprotected alcohol moieties on substrates (+)-25 and (-)-27 further distinguishes this versatile amination method, since most C—H amination systems face chemoselectivity issues with free alcohols.8b These examples highlight the potential of highly chemoselective C—H oxidation reactions to directly diversify natural products, without the need for functional group manipulations characteristic of traditional carbon-heteroatom bond forming processes.
Mechanistic Considerations
On the basis of the data described above and our previous work, we favor a catalytic cycle for Brønsted base promoted intermolecular allylic C—H amination as illustrated in Figure 5, A.
Figure 5.

Proposed Catalytic Cycle for Brønsted Base-Catalyzed, Linear Allylic C—H Amination
The DIPEA base initially deprotonates the N-tosylcarbamate to generate nucleophilic species A that participates in π-allylPd functionalization to afford product and trialkylammonium acetate species B (Figure 5, A). Under these acidic conditions, it is unlikely that DIPEA base is regenerated during the course of the catalytic cycle. Accordingly, we suggest that after the first turnover, species B serves as the base for subsequent deprotonations.22 Consistent with this proposal, increasing the concentration of acetate anion in solution by addition of (n-Bu)4NOAc or [(i-Pr)2EtH]NOAc afforded the product in comparable yield to that obtained with addition of DIPEA (Figure 5, B).23
These results clearly establish that intermolecular allylic C—H amination can be promoted via nucleophile activation. Regarding the Cr system, however, two modes of Lewis acid-promoted functionalization could still be operating: activation of the electrophilic π-allyl intermediate towards nucleophilic attack, or activation of the N-tosylcarbamate nucleophile towards deprotonation. Seminal studies performed in our labs on an asymmetric allylic oxidation reaction indicated that Cr(III)(salen) complexes increase the rate of BQ-mediated π-allylPd functionalization by binding to a proposed π-allylPd(BQ) complex and increasing its electrophilicity (Equation 2).24
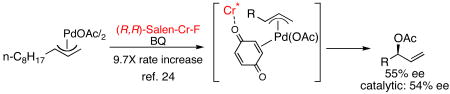 |
(2) |
Based on this model, we hypothesized that if a promoter is acting via an electrophile activation mode that is mediated by BQ, the use of more sterically encumbered quinones (which do not bind well to the π-allylPd intermediate) should impede product formation.24,25 Alternatively, a promoter that proceeds by activating the nucleophile through deprotonation should show little or no dependence on the quinone's steric environment. To investigate the role of a Pd-quinone interaction for promoting amination, we subjected both sets of reaction conditions (Cr & DIPEA) to a variety of methyl substituted benzoquinones and evaluated the trends in reactivity. As expected, the Brønsted base-promoted system shows no dependence on the nature of the quinone for reactivity; even when methyl groups shield both olefins of the quinone, high yields for the linear allylic amine product are observed (Table 3, entries 1-6). This data is consistent with our proposed mechanism in which Brønsted base-promoted intermolecular allylic C—H amination proceeds by nucleophile activation. In stark contrast, the Lewis acid system [Cr(salen)Cl 2] requires unhindered quinones for promoting product formation (Table 3, entries 1-4). Even quinones with only a single olefin blocked (such as 2-methylbenzoquinone and 2,3-dimethylbenzoquinone) lead to diminished product yields under Lewis acid conditions. When both olefins of the quinone are sterically blocked from binding to the π-allylPd complex, very low reactivity corresponding to low conversion is observed (Table 3, entries 5 and 6). Additionally, while both systems use quinone as a terminal oxidant, suprastoichiometric loadings of BQ (2 equiv.) are needed for optimal reactivity with the Cr system, whereas the DIPEA system operates equally well with no excess quinone (Table 3, entry 2). All of this data is consistent with the Lewis acid working with a π-allylPd(BQ) complex to promote functionalization and suggests that Lewis acid/BQ and Brønsted base activation proceed via distinct pathways for promoting allylic amination.24 Mechanistic studies are ongoing to elucidate the details of the Lewis acid/BQ activation mode.
Table 3.
Effect of Quinone Sterics on Lewis Acid vs. Brønsted Base C—H Amination Systems
 | |||
|---|---|---|---|
| DIPEA | Cr(salen)Cl 2 | ||
| Entry | Quinone | Yield(%)a | Yield(%)a |
| 1 |  |
66 | 50 |
| 2 | 67b | 28b | |
| 3 | 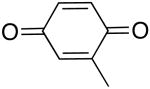 |
71 | 35 |
| 4 | 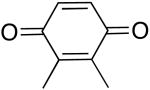 |
60 | 39 |
| 5 | 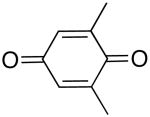 |
60 | 11 |
| 6 |  |
75 | 7 |
Average isolated yield of linear product after at least two runs at 0.3 mmol.
BQ (1.0 equiv.) was used.
Conclusion
In summary, we report the first catalytic Brønsted base strategy for nucleophile activation under the acidic, electrophilic conditions of oxidative Pd(II) catalysis. This discovery provides a novel way to activate acidic pro-nucleophiles for intermolecular π-allylPd functionalization without attenuating the electrophilicity of the catalyst (1) required for C—H cleavage. Significantly, this mild activation mode affords one of the most chemoselective C—H oxidations reported to date, enabling sensitive moieties such as terminal epoxides and unprotected alcohols to be present in the allylated starting materials. This advance should further the use of the direct C—H allylic amination reaction towards the streamlined synthesis and diversification of complex molecules. Moreover, we anticipate that this novel catalytic base strategy will find general use for promoting functionalization via pro-nucleophile activation in electrophilic transition metal-catalyzed processes that are not tolerant of strongly basic conditions.
Experimental Procedures
Synthesis of Methyl N-tosylcarbamate
To a 1 L flame-dried round bottom flask (under nitrogen) was added dry methanol (500 mL) and a stir bar. The flask was cooled in an ice bath cooled at 0°C, and p-toluenesulfonylisocyanate (100 mL, 0.657 mol, 1.0 equiv.) was added dropwise via syringe. The reaction was then allowed to warm to room temperature. After stirring 2 h, removal of the solvent in vacuo produced a colorless syrup that crystallized spontaneously. The solids were then triturated with pentane (2 × 50 mL) and ether (1 × 50 mL), followed by drying under high vacuum (0.5 torr) to yield white plates (141 g, 615 mmol, 94% yield).
General Procedure for the Allylic C—H Amination
A 1 dram, screw-top vial under air atmosphere was charged with terminal olefin (0.3 mmol, 1.0 equiv.), followed by tert-butylmethyl ether (0.450 mL). Catalyst 1 (15.1 mg, 0.03 mmol, 0.10 equiv.), 1,4-benzoquinone (64.9 mg, 0.6 mmol, 2.0 equiv.), N-methyl-tosylcarbamate (137.6 mg, 0.6 mmol, 2.0 equiv.), and a magnetic stir bar were then sequentially added. N,N-Diisopropylethylamine (3.14 μL, 0.018 mmol, 0.06 equiv.) was added last via syringe. The vial was fitted with a Teflon cap, magnetically stirred, and placed in a 45 °C oil bath. After 72 h, the vial was removed, allowed to cool to room temperature, and thoroughly rinsed into a 125 mL separatory funnel with ether (ca. 30 mL). The organic phase was washed with 5% aq. K2CO3 (6 × 10 mL), and the combined aqueous rinses were back-extracted with ether (2 × 30 mL). The combined organic extracts were dried over MgSO4 or Na2SO4 (epoxides & aldehydes), filtered through a 1:1 mixture of Celite/silica gel, and evaporated to dryness (30-40 °C, 30 torr). The crude mixture was purified by flash chromatography on silica gel (ethyl acetate/hexanes), and evaporated to dryness to afford the aminated product.
Supplementary Material
Acknowledgments
Financial support was provided by NIH/NIGMS (grant no. GM076153) and kind gifts were received from Eli Lilly, Bristol-Myers Squibb, Pfizer, Amgen, a R.C. Fuson graduate fellowship, and a James R. Beck graduate fellowship (to S.A.R.). We thank the Aldrich Chemical Company and Strem Chemicals, Inc. for generous gifts of commercial bis(sulfoxide)/Pd(OAc)2 catalyst 1. We thank Mr. N. Vermeulen for checking the experimental procedure outlined in Table 2, entry 2.
Footnotes
Supporting Information Available. Detailed experimental procedures, full compound characterization, 1H NMR and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Muller P, Fruit C. Chem Rev. 2003;103:2905. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]; (b) Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. and references therein. [Google Scholar]; (c) Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]; (d) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (e) Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lewis JC, Bergman RG, Ellman JA. Acc Chem Res. 2008;41:1013. doi: 10.1021/ar800042p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Labinger JA, Bercaw JE. Nature. 2002;417:507. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (h) Crabtree RH. J Organomet Chem. 2004;689:4083. [Google Scholar]
- 2.(a) Fraunhoffer KJ, Bachovchin DA, White MC. Org Lett. 2005;7:223. doi: 10.1021/ol047800p. [DOI] [PubMed] [Google Scholar]; (b) Hoffmann RW. Synthesis. 2006;21:3531. [Google Scholar]; (c) Covell DJ, Vermeulen NA, Labenz NA, White MC. Angew Chem Int Ed. 2006;45:8217. doi: 10.1002/anie.200603321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aliphatic/Benzylic: Liang C, Collet F, Peillard FR, Müller P, Dodd RH, Dauban P. J Am Chem Soc. 2007;130:343. doi: 10.1021/ja076519d.Lebel H, Huard K, Lectard S. J Am Chem Soc. 2005;127:14198. doi: 10.1021/ja0552850.Olson DE, Du Bois J. J Am Chem Soc. 2008;130:11248. doi: 10.1021/ja803344v.Milczek E, Boudet N, Blakey S. Angew Chem Int Ed. 2008;47:6825. doi: 10.1002/anie.200801445. Aryl: Li JJ, Mei TS, Yu JQ. Angew Chem Int Ed. 2008;47:6452. doi: 10.1002/anie.200802187.Jordan-Hore JA, Johansson CCC, Gulias M, Beck EM, Gaunt MJ. J Am Chem Soc. 2008;130:16184. doi: 10.1021/ja806543s.Thu HY, Yu WY, Che CM. J Am Chem Soc. 2006;128:9048. doi: 10.1021/ja062856v.Tsang WC, Zheng N, Buchwald SL. J Am Chem Soc. 2005;127:14560. doi: 10.1021/ja055353i.Brasche G, Buchwald SL. Angew Chem Int Ed. 2008;47:1932. doi: 10.1002/anie.200705420.
- 4.Linear allylic amination via oxygenated intermediates: Pd(0) catalysis, Trost BM, Keinan E. J Org Chem. 1979;44:3451. Overman rearrangement: Overman LE. Acc Chem Res. 1980;13:218. Mitsunobu amination: Mulzer J, Funk G. Synthesis. 1995;1:101.
- 5.Amination reviews: Cheikh RB, Chaabouni R. Synthesis. 1983;9:685.Johannsen M, Jørgensen KA. Chem Rev. 1998;98:1689. doi: 10.1021/cr970343o.
- 6.Fraunhoffer KJ, White MC. J Am Chem Soc. 2007;129:7274. doi: 10.1021/ja071905g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reed SA, White MC. J Am Chem Soc. 2008;130:3316. doi: 10.1021/ja710206u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For seminal work on allylic C—H amination see: Pd(II), Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584. doi: 10.1021/jo952088i. Nitrenes, (b) refs. 3a-d. Ene reactions, Kalita B, Nicholas KM. Tetrahedron Lett. 2005;46:1451.
- 9.Taylor LD, Pluhar M, Rubin LE. J Polym Sci, Part B: Polym Phys. 1967;5:77. [Google Scholar]
- 10.Pd(0)-catalyzed allylic alkylations with neutral enamines have also been reported: Ibrahem I, Córdova A. Angew Chem Int Ed. 2006;45:1952. doi: 10.1002/anie.200504021.Bihelovic F, Matovic R, Vulovic B, Saicic RN. Org Lett. 2007;9:5063. doi: 10.1021/ol7023554.
- 11.Chen MS, White MC. J Am Chem Soc. 2004;126:1346. doi: 10.1021/ja039107n.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198. For a single report of BQ/Pd(II) binding: Albéniz AC, Espinet P, Martín-Ruiz B. Chem Eur J. 2001;7:2481. doi: 10.1002/1521-3765(20010601)7:11<2481::aid-chem24810>3.0.co;2-2.
- 12.For catalytic base strategies for direct arylation reactions see: Lafrance M, Lapointe D, Fagnou K. Tetrahedron. 2008;64:6015. and references therein.
- 13.A similar parabolic behavior between nitrogen base concentration and reactivity has been observed for several Pd(II)/Pd(0) alcohol oxidation systems, although these reactivity trends were largely attributed to ligand effects. See: Steinhoff BA, Guzei I, Stahl SS. J Am Chem Soc. 2004;126:11268. doi: 10.1021/ja049962m.Schultz MJ, Adler RS, Zierkiewicz W, Privalov T, Sigman MS. J Am Chem Soc. 2005;127:8499. doi: 10.1021/ja050949r.
- 14.Chiral (S,S)-Cr(salen) complex 2 is known to catalyze asymmetric epoxide ring-opening reactions, see: Hansen KB, Leighton JL, Jacobsen EN. J Am Chem Soc. 1996;118:10924.Jacobsen EN, Kakiuchi F, Konsler RG, Larrow JF, Tokunaga M. Tetrahedron Lett. 1997;38:773.
- 15.Kitaori K, Furukawa Y, Yoshimoto H, Otera J. Tetrahedron. 1999;55:14381. [Google Scholar]
- 16.Hirsh AJ, Molino BF, Zhang J, Astakhova N, Geiss WB, Sargent BJ, Swenson BD, Usyatinsky A, Wyle MJ, Boucher RC, Smith RT, Zamurs A, Johnson MR. J Med Chem. 2006;49:4098. doi: 10.1021/jm051134w. [DOI] [PubMed] [Google Scholar]
- 17.Davis CE, Bailey JL, Lockner JW, Coates RM. J Org Chem. 2003;68:75. doi: 10.1021/jo026506c. [DOI] [PubMed] [Google Scholar]
- 18.The NsNHCO2Me nucleophile was also tested under the following sets of conditions: Cr(salen)Cl: 38% yield; DIPEA (6 mol%)/TBME: 16% yield; DIPEA (10 mol%)/CCl4: 24% yield (average of 2 runs).
- 19.Fevig TL, Katzenellenbogen JA. J Org Chem. 1987;52:247. [Google Scholar]
- 20.Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ. Bio Med Chem Lett. 1999;9:1233. doi: 10.1016/s0960-894x(99)00185-7. [DOI] [PubMed] [Google Scholar]
- 21.The reaction using our previous Cr(salen)Cl conditions gave product (+)-24 in 38% yield.
- 22.In contrast to the Cr(salen)Cl conditions, no acetate (or any other) byproducts were observed under the DIPEA conditions by crude 1H NMR. Given this and the high conversion of the starting material, the remainder of the mass balance may be polymeric material.
- 23.The hygroscopic nature of (n-Bu)4NOAc led to variability under our air atmosphere/wet solvent conditions, leading us to pursue DIPEA for its operational simplicity.
- 24.Covell DJ, White MC. Angew Chem Int Ed. 2008;47:6448. doi: 10.1002/anie.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Previous mechanistic studies indicated that BQ acts as a π-acidic ligand to promote carboxylate functionalization of π-allyl intermediates; 2,6-DMBQ does not promote carboxylate functionalization of π-allylPdOAc dimer to branched allylic acetate, presumably due to an inability to bind as a π-acidic ligand, see ref. 11b.
- 24.At this time, we cannot rule out the possibility that BQ acts to promote formation of the active Cr species that subsequently activates the N-tosylcarbamate nucleophile.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.