

Abstract
Alzheimer's Disease (AD) is the most common age-related dementia, with a current prevalence in excess of five million individuals in the United States. The aggregation of amyloid-beta (Aβ) into fibrillar amyloid plaques is a key pathological event in the development of the disease. Microglial proinflammatory activation is widely known to cause neuronal and synaptic damage that correlates with cognitive impairment in AD. However, current pharmacological attempts at reducing neuroinflammation mediated via microglial activation have been largely negative in terms of slowing AD progression. Previously, we have shown that microglia express proinflammatory cytokines and a reduced capacity to phagocytose Aβ in the context of CD40, Aβ peptides and/or lipopolysaccharide (LPS) stimulation, a phenomenon that can be opposed by attenuation of p44/42 mitogen-activated protein kinase (MAPK) signaling. Other groups have found that blueberry (BB) extract both inhibits phosphorylation of this MAPK module and also improves cognitive deficits in AD model mice. Given these considerations and the lack of reduced Aβ quantities in behaviorally improved BB-fed mice, we wished to determine whether BB supplementation would alter the microglial proinflammatory activation state in response to Aβ. We found that BB significantly enhances microglial clearance of Aβ, inhibits aggregation of Aβ1–42, and suppresses microglial activation, all via suppression of the p44/42 MAPK module. Thus, these data may explain the previously observed behavioral recovery in PSAPP mice and suggest a means by which dietary supplementation could mitigate an undesirable microglial response toward fibrillar Aβ.
Introduction
Approximately 5.1 million people have Alzheimer's disease (AD) in the United States.1 It is the most common age-related progressive dementia, and widespread synaptic injury and loss has been consistently correlated with its characteristic neurocognitive deficits. indeed as the disease progresses, so does neuron cell loss.2 Amyloid-beta (Aβ) is a 37-to 43-amino-acid peptide generated by cleavage from a large transmembrane precursor, the amyloid precursor protein (APP). The aggregation of Aβ into fibrillar amyloid plaques is a key pathological event in the development of AD.3 Importantly, these amyloid structures have been demonstrated both in vitro and in vivo to be neurotoxic and synaptotoxic.4–6 Thus, neuroinflammation may be one of the most important mediators of subsequent histopathologic changes resulting in affective dysregulation and cognitive decline in AD. Although several mechanisms may be crucial in amyloid-induced neurtoxicity and neuroinflammation, in vitro studies indicate that a self-potentiating cycle of Aβ peptide-associated brain inflammation is critical.
Indeed aggregated forms of Aβ peptide activate microglial intracellular signaling cascades, leading to production of cytokines and chemokines or “pro-inflammatory microglial activation,” a central phenomenon in AD neuroinflammation.7 These activated cells have caused synaptic pruning and neuronal damage.8,9 In addition many proinflammatory cytokines also confer increased amyloidogenic processing of App by neurons,10 further fueling the inflammatory cycle in the AD brain. Indeed, the major cellular mediators associated with inflammation in and around amyloid plaques seem largely to be activated microglia, and to a lesser extent reactive astrocytes.11,12 Compared to the healthy average brain, abundant activated microglia are typically localized in or around amyloid plaques of the AD neocortex, and microglial processes both border and penetrate plaque cores.13,14
These activated microglia express proinflammatory cell-surface markers, including major histocompatibility complex II (MHC II).15 MHC II is overexpressed in areas of microglial proliferation around plaques in AD brain, while downregulated in control brain.16–18 The mechanism underlying this fibrillar, Aβ-induced activation into an MHC II-expressing, proinflammatory phenotype is not clear presently. However evidence exists that Aβ interacts with several receptors present in microglia plasma membranes, including integrins, receptor of advanced glycation end products (RAGE), the serpin enzyme complex (SEC) receptor,19 and scavenger receptors (SR), which mediate activation of intracellular tyrosine kinase pathways.20,21 This, in turn, results in mitogen-activated protein kinase (MAPK) activation, which causes phosphorylation of cAMP response element-binding protein (CREB). This terminal event then increases transcription of genes related to cytokine and complement components synthesis as well as release of intracellular Ca++ stores and reactive oxygen species (ROS).22 Taken together, these data suggest Aβ-induced activation of microglia involves clustering of cell-surface receptors, which then cause MAPK activation, ultimately leading to synaptic and neuronal damage.
Previously, it was demonstrated that blueberry (BB) supplementation reversed the deleterious effects of aging on motor behavior and neuronal signaling in senescent rodents.23 It has also been demonstrated that BB-fed (from 4 months of age) PSAPP mice (a transgenic line carrying mutant APP and presenilin 1 transgenes) showed no deficits in Y-maze performance (at 12 months of age) with no alterations in Aβ’” burden.24,25 Because the BB supplementation yielded these beneficial effects on several deficits in behavior as well as neuronal signaling in the senescent and AD mouse models, an addition of BB may be of benefit in reducing the proinflammatory Aβ-mediated microglial activation. Findings from our laboratory suggest that this activation occurs through a p44/42 MAPK-dependent pathway.26,27 Given these considerations and the lack of reduced Aβ quantities in behaviorally improved BB-fed mice,25 we wished to determine whether BB supplementation would alter the microglial proinflammatory activation state in response to Aβ. We found that BB enhances microglial clearance of Aβ, inhibits aggregation of Aβ1–42, and suppresses microglial activation, all via suppression of the p44/42 MAPK module. Thus, these data may explain the previously observed behavioral recovery in PSAPP mice25 and suggest a means by which dietary supplementation could mitigate an undesirable microglial response toward fibrillar Aβ.
Materials and Methods
Materials
Mouse anti-human Aβ monoclonal antibody (BAM-10) was purchased from Sigma (St. Louis, MO). Aβ1–42 and fluorescein isothiocyanate (FITC)-conjugated Aβ1–42 were obtained from Biosource International (Camarillo, CA). Orange fluorescing cyanine dye, Cy3, was purchased from Amersham (Piscataway, NJ) for conjugation with the Aβ1–42 peptide. Blueberry (freeze-dried power) was obtained from Van Drunen Farm (Momence, IL). Purified FITC-anti-mouse MHC class II antibodies were obtained from PharMingen (San Diego, CA). DuoSet™ mouse tumor necrosis factor-α (TNF-α) enzyme-linked immunosorbent assay (ELISA) kit was obtained from R&D systems (Minneapolis, MN). Mouse interleukin-6 (IL-6) ELISA kit was obtained from eBioscience (San Diego, CA). Antibodies for phospho-p44/42 (pp44/42, Thr202/Tyr204) MAPK and total p44/42 MAPK were obtained from Cell Signaling Technology (Beverly, MA) as well as cell lysis buffer and sodium dodecyl sulfate (SDS) blue loading buffers. PD98059 (a specific MEK1/2 inhibitor) was obtained from Calbiochem (La Jolla, CA). PD98059 was dissolved in dimethylsulfoxide (DMSO) before adding to complete cell medium. DMSO alone was used as a solvent control, which did not differ from the untreated controls presented. Thioflavin T and bacterial lipopolysaccharide (LPS) were obtained from Sigma (St. Louis, MO), and LPS was dissolved in complete cell culture medium. Anti-rabbit horseradish peroxidase (HRP)-conjugated immunoglobulin G (IgG) secondary antibodies and western blotting luminol reagent were obtained from Pierce (Rockford, IL). Immun-Blot polyvinylidene difluoride (PVDF) membranes were purchased from Bio-Rad systems (Minneapolis, MN).
Murine primary cell culture
Breeding pairs of BALB/c mice (Jackson Laboratory, Bar Harbor, ME) were housed in the animal facility at the University of South Florida Health Science Center. Murine primary culture microglia were isolated from mouse cerebral cortices and grown in complete RPMI 1640 medium according to previously described methods.26 Briefly, cerebral cortices from newborn mice (1–2 days old) were isolated under sterile conditions and kept at 4°C prior to mechanical dissociation. Cells were grown in RPMI 1640 medium supplemented with 5% heat-inactivated fetal calf serum (FCS), 2 mM glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μM 2-mercaptoethanol. Primary cultures were kept for 14 days so that only glial cells remained. Microglial cells were isolated by shaking flasks at 200 rpm in a Lab-Line™ Incubator-Shaker. More than 98% of these glial cells stained positive for Mac-1 (Boehringer Mannheim, Indianapolis, IN).
MTT assay
For cell proliferation analysis, primary mouse microglia were seeded at 2 × 104 cells/well (n = 6 for each condition) in 96-well tissue culture plates containing 100 μL of complete RPMI 1640 medium. These cells were treated for 24, 48, and 72 hr with BB extract at different concentrations (25 μg/mL, 50 μg/mL, and 100 μ/mL). Cell proliferation as determined by MTT assay (MTT kit, Sigma). BB extract did not show high activity on proliferation of primary mouse microglial cells in culture (data not shown).
Thioflavin T measurement
Thioflavin T measurement was performed by according to previously described methods28 with slight modifications. For Aβ aggregate-formation assay, Aβ (20 μM) dissolved in 50 mM Tris-HCl buffer (pH 7.4) with a test BB extract (25 μg/mL, 50 μg/mL, and 100 μ/mL) was incubated at 37°C for 0, 12, 24, 48, and 72 hr. At the end of the incubation, 10 μM thioflavin T in 50 mM glycine/NaOH (pH 9.0) was added to the mixture. Fluorescence of thioflavin T bound to Aβ aggregates was measured with a microplate reader (SpectraMax®, Molecular Devices) with an excitation wavelength of 442 nm and emission wavelength of 485 nm after incubation for 30 min at room temperature. Percentage inhibition was calculated by comparing these fluorescence values with those found in control solutions with no BB.
Microglial phagocytosis assays
Primary mouse microglia were seeded at 1 × 105 cells/well (n = 6 for each condition) in 24-well tissue culture plates containing 0.5 mL of complete RPMI 1640 medium. “Aged” FITC-Aβ1–42 is created by preaggregating Aβ1–42 prior to adding to microglial cells. Specifically, FITC-Aβ1–42 was first diluted in complete medium to a final concentration of 20 μM and vortexed. The mixture was then pH-adjusted to 5.0 with 1 M HCl and incubated at 37°C for 24 hr, as previously described.29 These cells were then treated for 2 hr with 500 nM “aged” FITC-Aβ1–42. In the presence of FITC-Aβ1–42, microglial cells were then co-treated with BB (50 μg/mL). Some of these cells were treated with PD98059 (5 μM), BB (50 μg/mL), or PD98059 and BB for 1 hr prior to LPS (50 ng/mL), or BAM-10 (2.5 μg/mL), in the presence of FITC-Aβ1–42 for 2 hr. Microglial cells were then rinsed three times in Aβ-free complete medium, and the media were exchanged with fresh Aβ-free complete medium for 10 min to allow for removal of nonincorporated Aβ and promote concentration of Aβ into phagosomes. Extracellular and cell-associated FITC-Aβ were quantified using an MSF (Spectra-Max®, Molecular Devices) with an emission wavelength of 538 nm and an excitation wavelength of 485 nm. A standard curve from 0 to 600 nM of FITC-Aβ was run for each plate. Total cellular proteins were quantified using the Micro BCA Protein Assay (Pierce, Rockford, IL). The mean fluorescence values for each sample at 37°C and 4°C at the 2-hr time point were determined by fluorometric analysis. Relative fold change values were calculated as: mean fluorescence value for each sample at 37°C/mean fluorescence value for each sample at 4°C. In this manner, both extracellular and cell-associated FITC-Aβ were quantified. Considering nonspecific adherence of Aβ to the plastic surface of culture plates, an additional control without cells was carried out through all of experiments above, and the mean values have been normalized to these controls. An incubation time of less than 4 hr did not change the amount of Aβ peptide detected in the supernatant, which is consistent with a previous report.30 To determine the extent to which cell death might have influenced the phagocytic activity in the various treatment groups, we performed the lactate dehydrogenase (LDH) assay on the relevant supernatant. Data showed that there was no significant cell death occurring over the 3-hr time frame in any of the treatment groups (data not shown, p < 0.05).
Fluorescence microscope examination
“Aged” FITC-Aβ1–42 was prepared according to methods described above. Microglial cells were cultured at 1 × 105 cells/well in 24-well tissue culture plates with glass inserts. In the presence of FITC-Aβ1–42 (1 μM), microglial cells were co-treated with BB (50 μg/mL) at 37°C for 2 hr. Additionally, in parallel 24-well tissue culture plates, microglial cells were incubated at 4°C with the same treatment as above. Following treatment, these cells were washed five times with ice-cold phosphate buffered saline (PBS) to remove extracellular Aβ and fixed for 10 min at 4°C in 4% (wt/vol) paraformaldehyde (PFA) diluted in PBS, followed by staining with 4′,6-diamidino-2-phenylindole (DAPI) at 4°C for 15 min. Finally, sections were mounted with fluorescence mounting medium containing Slow Fade antifading reagent (Molecular Probes, Eugene, OR), and then viewed under an olympus IX71/IX51 microscope equipped with a digital camera system (40×).
Fluorescence confocal microscopy
In parallel with the fluorescence microscope examination, slides were viewed with a Leica DMI6000 inverted microscope, TCS SP5 confocal scanner, and 63X/1.4NA Plan Apochromat oil immersion objectives (Leica Microsystems, Germany). Excitation wavelengths of 488 nm (for FITC) and 405 nm (for DAPI) were used to generate fluorescence emission in green (for Aβ1–42) and blue (for nuclei), respectively. Images were captured with photomultiplier detectors at 3× zoom and were prepared with the LAS AF software, version 1.6.0, build 1016 (Leica Microsystems, Germany). Differential interference contrast (DIC) images were also captured using the 488 laser line.
Primary microglial cells (seeded in 24-well tissue culture plates at 5 × 104 cells/well) were treated with Cy3-Aβ1–42 peptide (300 nM) in the presence or absence of BB (50 μg/mL) at 37°C for 48 hr, Cy3-Aβ1–42 was prepared according to previous methods.29 Following treatment, these cells were washed, fixed, and permeabilized in 0.2% Triton X-100, 5% horse serum for 1 hr. This was followed by staining with FITC-anti-mouse MHC class II antibody (2 μg/mL) incubated overnight at 4°C. Slides were analyzed using the same fluorescence confocal microscope equipped as above.
TNF-α and IL-6 ELISA
Primary cultured microglial cells were plated in 24-well tissue culture plates (Costar, Cambridge, MA) at 5 × 104 cells/well for 24 hr. They were then stimulated for 16 hr with LPS (50 ng/mL), Aβ peptides (1 μM), or LPS + Aβ peptides in the presence or absence of pretreatment (1 hr) with BB (50 μg/mL), PD98059 (5 μM), or appropriate controls. Cell-free supernatants were collected and assayed by TNF-α (R&D Systems, Minneapolis, MN), and IL-6 (San Diego, CA) ELISA kits in strict accordance with the manufacturer's instruction. The BCA Protein Assay (Pierce, Rockford, IL) was performed to measure total cellular protein from each of the cell groups under consideration just before quantification of cytokine release.
Western immunoblotting
Murine primary culture microglia were plated in six-well tissue culture plates at a density of 8 × 105 cells/well. These cells were incubated for 30 min with or without LPS (50 ng/mL), Aβ1–42 (1 μM), in the presence or absence of pre-treatment (1 hr) with BB (50 μg/mL), PD98059 (5 μM), or appropriate controls. After treatment, microglial cells were washed in ice-cold phosphate-buffered saline (PBS) three times. Next the cells were lysed with 1× SDS sample buffer (62.5 mM Tris-HCI [pH 6.8], 2% SDS, 10% glycerol, 50 mM dithiothreitol), sonicated for 15 sec, and then heated at 100°C for 5 min. The cell lysates were centrifuged at 12,000 rpm (4°C) for 5 min, and the protein concentration of the supernatant was measured by BCA Protein Assay System (Pierce). Western blotting of phosphorylated p44/42 MAPK was performed according to the manufacturer's instructions using phospho-specific antibodies. Briefly, proteins were electrophoresed on a 10% sodium dodecyl sulfate–olyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to immunoblotting PVDF membranes (Bio-Rad). The membranes were blocked for 1 hr at room temperature in Tris-buffered saline (TBS, Bio-Red) and 0.1% Tween-20 with 5% nonfat dry milk, and then were incubated with primary antibodies overnight at 4°C. After incubation with horseradish peroxidase (HRP)-conjugated secondary antibody, the protein bands were detected with a Super Signal west Femto Maximum Sensitivity Substrate (Pierce) and BIOMAX-MR film (Eastman Kodak Co.). For detection of total p44/42, the membranes were stripped with Restore Western blot Stripping Buffer (Pierce) followed by incubation with specific antibodies.
Statistical Analysis
All experiments were performed at least three times, and the representative results were shown. Data were analyzed using analysis of variance (ANOVA) followed by post hoc comparisons of means by Bonferroni, or were analyzed using the Student t-test. A value of p < 0.05 was considered to be significant.
Results
BB enhances microglial phagocytosis of Aβ1–42 peptide
Microglial phagocytosis of Aβ is most likely a terminal event leading to removal of β-amyloid from the brain parenchyma.31 We have found that microglia demonstrating this phagocytic phenotype are polarized toward an anti-inflammatory state characterized by attenuated p44/42 MAPK activity.7,26,27 Because BB was previously shown to enhance cognitive performance in rodent models of AD and without coincident reduction of parenchymal amyloid, we sought to determine whether microglial phagocytosis was modulated by BB.24 Thus “aged” FITC-tagged Aβ1–42 (500 nM) was added to primary cultured microglial cells for 2 hr in the absence (control), presence of BB, positive control BAM-10 (2.5 μg/mL) or IgG isotype-control (2.5 μg/mL). As a control for nonphagocytic incorporation of Aβ by microglia, microglial cells were incubated at 4°C in parallel cell culture plates under the same treatment conditions described above. Cell supernatants and lysates were analyzed for extracellular and cell-associated FITC-Aβ using a fluorometer. As shown in Fig. 1A (top and bottom panel), BB enhances microglia phagocytosis of Aβ1–42 peptide, a result that was further verified by quantitative immunofluorescence assay (Fig. 1B). In a parallel experiment, results further showed microglial phagocytosis of Aβ1–42 peptide was localized within the cytoplasm of microglial cells (Fig. 1C).
FIG. 1.
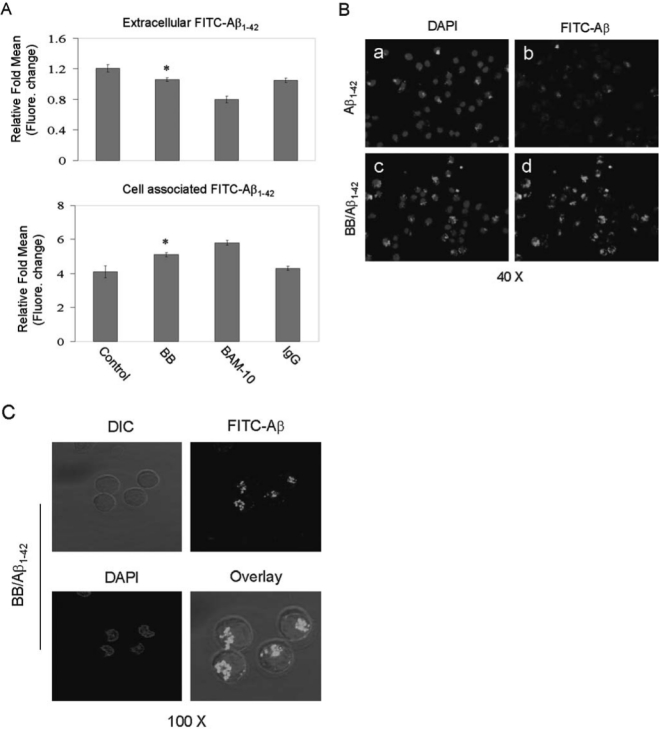
BB enhances microglia phagocytosis of Aβ1–42 peptide. (A) Cell supernatants and lysates were analyzed for extracellular (top panel) and cell-associated (bottom panel) FITC-Aβ1–42 using a fluorometer. Data are represented as the relative fold of mean fluorescence change (mean ± SD), calculated as the mean fluorescence for each sample at 37°C divided by mean fluorescence at 4°C (n = 6 for each condition presented). One-way ANOVA followed by post hoc comparison showed a significant between-group difference (*p < 0.05, compared with control). (B) Subsequently, fluorescence microscope examination was performed using a 40× objective with appropriate filter selection. The darkfield images (a and c) show the DAPI-labeled nuclear stain, whereas, b and d show only the FITC Aβ1–42 stain of the same fields. (C) In parallel experiments, microglial cells were treated with 1 μM “aged” FITC-Aβ1–42 and BB for 2 hr. Following treatment, these cells were fixed and stained with DAPI. The images were analyzed by confocal microscope and show FITC-Aβ1–42 (green staining) localized within the cytoplasm of microglial cells. (Color version is available online at www.liebertonline.com/rej).
BB inhibits the formation of Aβ1–42 aggregation
With regard to the inhibitory effect of BB on Aβ fibril formation, time dependencies and concentration dependencies were examined by using the thioflavin T method (Fig. 2A), which detects mainly mature β-pleated sheet amyloid fibrils.32 Figure 2 shows data for the thioflavin T assay, with incubation of 20 μM Aβ1–42 peptides in 50 mM Tris-HCl buffer (pH 7.4) with or without BB extract (25 μg/mL, 50 μg/mL, and 100 μg/mL). After incubation at 37°C for different time points, BB (50 μg/mL and 100 μg/mL) significantly inhibited Aβ aggregation (p < 0.05, or p < 0.001, Student t-test) at the 48 hr point by approximately 75% and 60% respectively, whereas BB (25 μg/mL, 50 μg/mL, and 100 μg/mL) further inhibited Aβ aggregation (p < 0.05, or p < 0.001, Student t-test) at the 72-hr point by approximately 82% (25 μg/mL), 65% (50 μg/mL), and 57% (100 μg/mL), respectively. In this experiment, BB was shown to inhibit peptide aggregation in a clear, time- and concentration-dependent manner.
FIG. 2.

BB inhibits formation of Aβ1–42 aggregation. Effect of different BB concentration on Aβ1–42 aggregation was assessed by the thioflavin T method. Reaction mixtures containing 20 μM of Aβ1–42, 50 mM Tris-HCl buffer (pH 7.4), and different concentrations of BB extract were incubated at 37°C for indicated time points. Aβ aggregation was expressed as a percentage of control, which was observed in the absence of BB extract. Values represent the means ± SD from three independent experiments. (*) p < 0.05, (**) p < 0.001 compared with the BB-untreated control.
BB suppresses microglial activation and enhances microglial phagocytosis of Aβ1–42 peptide through a p44/42 MAPK-dependent pathway
Previous studies have shown that activation of MEK1/2 and downstream p44/42 MAPK is involved in TNF-α and IL-6 production in macrophages, monocytes, and microglia after activation of these cells with a variety of stimuli, including LPS and CD40 ligand.27,33–36 Moreover, BB extract inhibited the production of inflammatory mediators from LPS-activated BV2 microglia.37 These data led us to ask whether the observed effect of BB on opposing microglial activation might be mediated via activation of the MAPK module. Thus, we analyzed p44/42 MAPK phosphorylation status in microglial cell lysates after treatment with LPS (50 ng/mL) or Aβ1–42 (1 μM) or appropriate controls for 30 min. Results showed that phosphorylation of p44/42 was induced within 30 min after treatment with LPS or Aβ1–42. Furthermore, we observed that treatment microglial cells with PD98059 (PD, 5 μM; a selective inhibitor of MEK1/2) or BB (50 μg/mL) for 1 hr prior to treatment with LPS (50 ng/mL) or Aβ1–42 (1 μM) for 30 min, result in significant reduction of activation of LPS or Aβ -induced p44/42 MAPK (Fig. 3A, B). To determine whether BB and activation of p44/42 MAPK was responsible for TNF-α and IL-6 production after co-treatment of microglia with LPS and Aβ1–42 peptide, we treated microglia with BB or PD98059 before stimulation with LPS and Aβ1–42 peptide. Production of TNF-α and IL-6 were markedly decreased compared with appropriate controls within 16 hr after treatment with BB, PD98059, and LPS plus Aβ1–42 peptides. Namely, either BB or PD abrogated LPS- or LPS/Aβ1–42-induced production of TNF-α or IL-6 (Fig. 3C, top and bottom panels). These observations prompted us to investigate the role of p44/42 MAPK signaling in the BB- mediated microglial phagocytosis of Aβ1–42 peptide. Microglial cells were pretreated with BB or PD98059 (5 μM) alone, or a combination of PD98059 and BB for 1 hr, then co-treated with “aged” FITC-tagged Aβ1–42 (500 nM) for 2 hr in the absence (control) or presence of LPS. Cell culture supernatants were collected and cell lysates were prepared to measure Aβ by fluorometer (Fig. 3D, top and bottom panels). Results show that cross-linking BB boosts microglial phagocytosis of Aβ1–42 peptide, which was enhanced by inhibition of p44/42 activation. Whereas, the combination of BB and LPS/PD did not confer additional effects on microglial phagocytosis of Aβ1–42 peptide compared with LPS/PD98059. These data collectively suggests that BB suppresses microglial activation and enhances microglial phagocytosis of Aβ1–42 peptide through a p44/42 MAPK-dependent pathway.
FIG. 3.
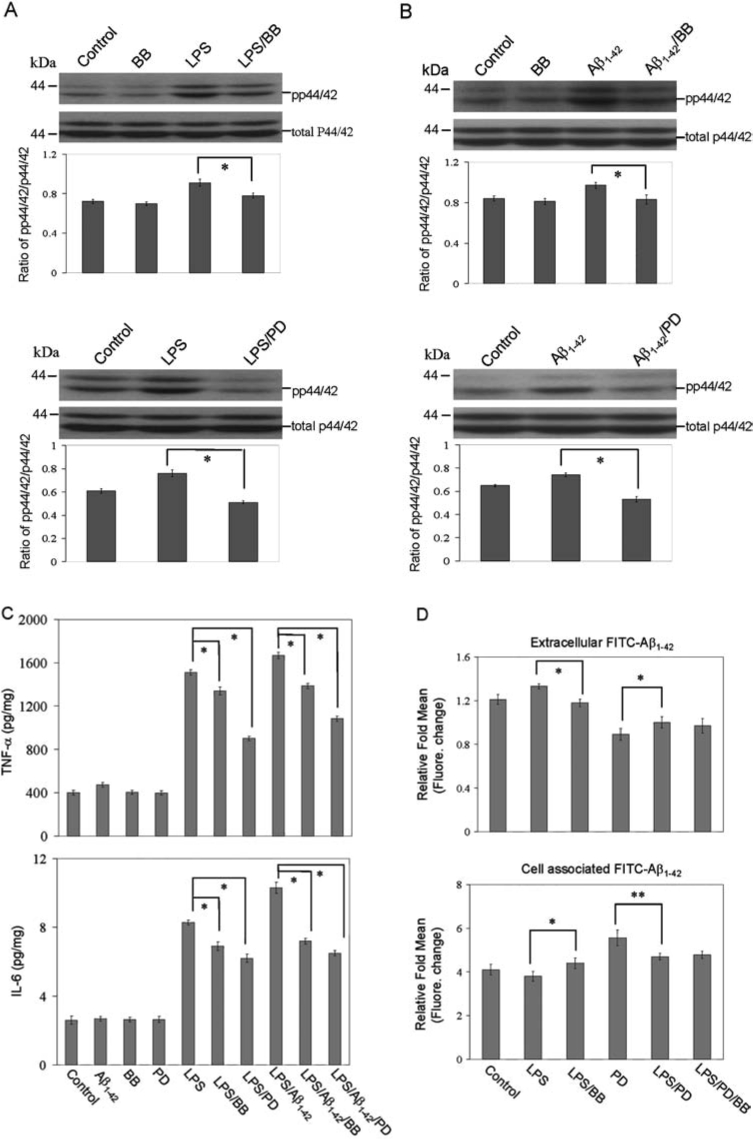
BB suppresses microglial activation and enhances microglial phagocytosis of Aβ1–42 peptide through a p44/42 MAPK-dependent pathway. Microglial treatment conditions are indicated and are further described in the Materials and Methods section. Cell lysates were analyzed by western immunoblotting using specific antibodies that recognize phosphorylated or total p44/42 MAPK at the indicated time point (A or B, top panels). Phosphorylation of p44/42 MAPK after treatment with LPS or Aβ1–42 peptides was inhibited by PD98059 (A or B, bottom panels). Histograms below the immunoblots represent the mean band density ratio ± 1 SD (pp44/42/total p44/42 MAPK; n = 3 for each condition presented, *p < 0.05). (C) Microglial activation is evidenced by mean TNF-α and IL-6 release ± 1 SD (n = 3 for each condition presented; *p < 0.001). (D) Microglial phagocytosis of Aβ1–42 peptide after pretreatment with PD98059, BB or PD98059, and BB for 1 hr then co-treated with “aged” FITC-tagged Aβ1–42 or LPS. Supernatants and cell lysates were analyzed for extracellular (top panel) and cell-associated (bottom panel) FITC- Aβ1–42 using a fluorometer (*p < 0.05, **p < 0.001). For A–D, one-way ANOVA followed by post hoc Bonferroni testing was used. Note: PD, PD98059; pp, phosphorylated.
BB inhibits co-localization of microglial MHC class II and Aβ1–42 peptide
MHC class II is expressed by microglial cells in the frontal cortex and hippocampus of normally aging individuals, and levels of expression are markedly increased in these brain regions in AD cases. The impairment of MHC II function results in a significant reduction of microglia-associated central nervous system (CNS) inflammation,38 suggesting the elevated level of MHC class II expression is strongly associated with a microglial proinflammatory response. We have shown that CD40 ligation increases MHC II- Aβ peptide complexes35 or cross-linking of CD45RB inhibits MHC class II- Aβ co-localization, as detected by fluorescence microscopy.27 To examine further whether BB could inhibit formation of immunogenic MHC II-Aβ peptide complexes on the cell surface, we treated microglia with BB (50 μg/mL) in the presence or absence of “aged” Cy3- Aβ1–42 peptide (300 nM) for 48 hr, followed by immunofluorescence staining with FITC-conjugated anti-mouse MHC class II antibody. Results show that BB inhibits MHC class II- Aβ co-localization as detected by fluorescence confocal microscope equipped (Fig. 4).
FIG. 4.
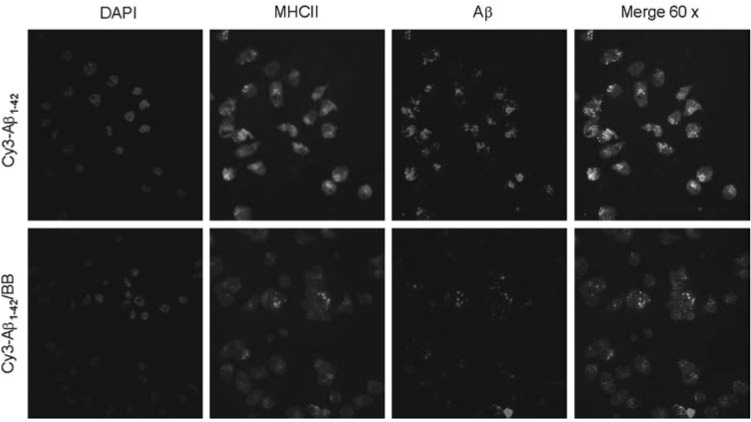
BB inhibits microglia MHC class II-Aβco-localization. To examine microglia MHC II-Aβpeptide complex formation on the cell surface, microglia were treated with “aged” Cy3- Aβ1–42 peptide (300 nM) in the presence or absence of BB for 48 hr followed by staining with FITC-anti-mouse MHC class II antibody and confocal microscopy. Note: Red indicates Aβ-positive; green indicates MHC class II-positive; yellow indicates the co-localization of MHC class II and Aβ; blue indicates DAPI nuclear stain of the same fields. Original magnification, 60× for top and bottom panels. (Color version is available online at www.liebertonline.com/rej).
Discussion
The proinflammatory activation state of microglia seems to be a pervasive link between the majority of neurodegenerative diseases, including human immunodeficiency virus (HIV)-associated dementia (HAD),39,40 multiple sclerosis (MS),41,42 amytropic lateral sclerosis (ALS),43,44 stroke,45,46 and AD.9,47 Because it is a common link, the study of compounds, especially those that can be easily taken as a part of diet, is attractive as most of these diseases come with advancing age, suggesting a significant epidemiologic problem.
However, current pharmacological attempts at reducing neuroinflammation mediated via microglial activation have been largely negative in terms of slowing AD progression.48 These anti-inflammatory drugs have included nonsteroidal anti-inflammatory drugs (NSAIDS), prednisone, COX-2 inhibitors (celecoxib, rofecoxib, and nimesulide), and hydroxychloroquine.49–52 These trials have failed for various reasons. The COX-2 inhibitors have not been in clinical use long enough for epidemiological data to accumulate, but their failure could be fairly well forecasted because COX-2 is highly expressed in normal pyramidal neurons, including those of AD cases.53 Thus, these drugs will largely affect vulnerable pyramidal neurons and not microglia. Also animal neurotoxicity was observed in models following COX-2 in-hibition.54 Hydroxychloroquine confers side effects of toxicity, neuronal, and retinal damage.55 Indeed no epidemiological or immunohistochemical evidence exists to support a role for this 4-aminoquinoline in neuroinflammation. NSAIDs have been shown in vitro to reduce the neurotoxicity of activated microglia toward neuronal cells,56 and the original NSAID study by Rogers and colleagues seemed to reflect this notion. This trial demonstrated an arrest of mental deterioration over a 6-month period in AD patients compared to nontreated AD patients.48 However, follow-up studies using NSAIDS have not been as promising. For example, a trial of the mixed COX inhibitor diclofenac combined with misoprostyl, demonstrated nonstatistically significant arrest of mental deterioration in drug compared with placebo patients.57 However, a Japanese trial did show a significantly slower cognitive decline compared to placebo where cases were treated with a combination of estrogen and vitamin E, in addition to the NSAID.58 It is also important to note that gastrointestinal and adverse side effects can be a barrier to patient compliance with such agents, particularly with chronic use in the elderly.59
Thus, natural compounds specifically aimed at blocking microglial activation may be more efficacious at ameliorating microglial-associated neuropathology in AD. In this study, we focused on identifying a specific intracellular signaling target, p44/42 MAPK, which when activated, could enhance microglial activation downstream of the extracellular pro-inflammatory mediators Aβ peptide and/or LPS. Our rationale for such investigation was three fold. First, it has been reported that in macrophages, LPS induces protein tyrosine phosphorylation of several proteins, including the p42 and p44 MAPKs in mice, similar to the manner in which fibrillar Aβ activates human moncytes.22 As mice require a proinflammatory stimulus to mimic human microglia responses to Aβ, LPS was co-administered. Second, if we could inhibit microglial activation by Aβ peptide in the presence of LPS, the self-potentiating inflammatory cycle resulting from intracellular signal transduction cascades could be suppressed. Third, our previous experiments indicate that inhibition of p44/42 MAPK attenuated the microglial proinflammatory MHC II-expressing phenotype.36
Our data show that microglia can be activated after treatment with Aβ peptides and LPS into a proinflammatory state as evidenced by significantly increased secretion of TNF-α IL-6 (Fig 3C), decreased Aβ phagocytosis (Fig 3D), and increased phosphorylation of p44/42 MAPK (Fig 3A,B). Because BB conferred behavioral improvement in PSAPP without mitigation of amyloid plaques in previous studies, we next determined if BB was a negative regulator of the proinflammatory response of microglia to Aβ and LPS. Data showed that BB markedly reduced microglial of p44/42 MAPK activation resulting from Aβ and LPS co-treatment (Fig 3A,B).
These data are in line with previous experiments in our7 and other60 laboratories that strongly suggest an additional role in microglial Aβ responses, which may act to balance the proinflammatory microglial activation described above.61 Consistent with this hypothesis, we did observe increased microglial Aβ cell association activity under these conditions, suggesting that BB stimulates non-MHC-II-expressing (Fig. 4), pro-phagocytic microglial phenotype (Figs. 1B,C and 3D).7 In accordance with this, co-treatment of Aβ-and LPS-activated microglia with BB resulted in statistically significant reduction of microglial IL-6 and TNF-α secretion (Fig. 3C), other important mediators of neuronal damage in AD.62,63 As an additional possible anti-amyloid mechanism, we sought to establish whether BB could also inhibit Aβ aggregation in vitro and found that all doses tested (25, 50, and 100 μg/mL) aggregation was significantly inhibited after 48 and 72 hr (Fig. 2). Together, these results suggest that BB is a viable approach for both downregulating Aβ-induced microglial activation and possibly attenuating Aβ conglomeration into plaques.
It is important to note, however, that use of microglial expression of MCH II as a marker of activation is limited. Microglia usually remain in a ramified resting form, but age may alter this quiescent state, as is seen by upregulated MHC II expression.64,65 It has already been suggested that MCH II expression is a marker of cell aging, in addition to, or even instead of, being an activation marker.66,67 Future studies will be required to correlated CD45 (another marker for microglial activation)-immunostaining, in addition to the effect of aging which may be expressed by the MHC class II immunoreactivity.
Previously, it has been shown that dietary supplements with BB extracts reduce some neurological deficits in aged animal models.68,69 Catechin is the major flavonoid found in blueberries (Vaccinium ashei Reade), with 387 mg/100 grams fresh weight. Blueberry extract also contains epicatechin, ranging from 34 to 129 mg/100 grams fresh weight, whereas total anthocyanins range from 84 to 113 mg/100 grams fresh weight.70 Joseph and colleagues showed that dietary supplementation for 8 weeks with BB extracts reversed cognitive deficits in Morris water maze performance tests in 19-month-old rats.23 This effect seemed to occur via downregulaton of the proinflammatory nuclear factor-κB (NF-κB) module, as the aged rats with BB extracts diet had significantly lower levels than aged control diet.64 Additional evidence was seen in PSAPP mice, which develop age-dependent AD-like senile plaques.24 These mice, when supplemented with BB extracts (2% of diet) from 4 to 12 months of age, displayed Y-maze performance similar to that of nontransgenic mice and significantly better than that of nonsupplemented transgenic mice.25 However in contrast to our in vitro microglial phagocytosis experiments, the examination of the brain of these mice revealed blueberry extracts had no affect on Aβ peptide production, deposition, or quantity.25 This would suggest that the blueberry induced prophagocytic property of microglia may not be enough to compensate for the strong amyloidogenic APP processing in the PSAPP mouse strain.
Thus, taking previous these previous studies and current findings into account, we suggest that BB extract supplementation might prevent cognitive deficits most likely through an antimicroglial activation mechanism, which results in the protection of neurons and synapses from toxic inflammatory mediators, without a necessary reduction in parenchymal Aβ.
Acknowledgments
This work is supported by grants to J.T. from the National Institutes of Health/National Institute of Aging (NIH/NIA; AG04418). B.G. is supported by an National Institute of Mental Health (NIMH) K08 Clinical Scientist Career Development Award. We thank Mr. Jun Tian for his technical assistance in murine primary microglial cell cultures.
References
- 1.Sparks MB. Inpatient care for persons with Alzheimer's disease. Crit Care Nurs Q. 2008;31:65–72. doi: 10.1097/01.CNQ.0000306399.09777.24. [DOI] [PubMed] [Google Scholar]
- 2.Tiraboschi P. Hansen LA. Alford M. Masliah E. Thal LJ. Corey-Bloom J. The decline in synapses and cholinergic activity is asynchronous in Alzheimer's disease. Neurology. 2000;55:1278–1283. doi: 10.1212/wnl.55.9.1278. [DOI] [PubMed] [Google Scholar]
- 3.Sambamurti K. Greig NH. Lahiri DK. Advances in the cellular and molecular biology of the beta-amyloid protein in Alzheimer's disease. Neuromolecular Med. 2002;1:1–31. doi: 10.1385/NMM:1:1:1. [DOI] [PubMed] [Google Scholar]
- 4.Kim HJ. Chae SC. Lee DK. Chromy B. Lee SC. Park YC. Klein WL. Krafft GA. Hong ST. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J. 2003;17:118–120. doi: 10.1096/fj.01-0987fje. [DOI] [PubMed] [Google Scholar]
- 5.Klein WL. Abeta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 6.Town T. Vendrame M. Patel A. Poetter D. DelleDonne A. Mori T. Smeed R. Crawford F. Klein T. Tan J. Mullan M. Reduced Th1 and enhanced Th2 immunity after immunization with Alzheimer's beta-amyloid(1–42) J Neuroimmunol. 2002;132:49–59. doi: 10.1016/s0165-5728(02)00307-7. [DOI] [PubMed] [Google Scholar]
- 7.Townsend KP. Town T. Mori T. Lue LF. Shytle D. Sanberg PR. Morgan D. Fernandez F. Flavell RA. Tan J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol. 2005;35:901–910. doi: 10.1002/eji.200425585. [DOI] [PubMed] [Google Scholar]
- 8.McGeer PL. Rogers J. McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- 9.Shie FS. Woltjer RL. Manipulation of microglial activation as a therapeutic strategy in Alzheimer's disease. Curr Med Chem. 2007;14:2865–2871. doi: 10.2174/092986707782359981. [DOI] [PubMed] [Google Scholar]
- 10.Gitter BD. Boggs LN. May PC. Czilli DL. Carlson CD. Regulation of cytokine secretion and amyloid precursor protein processing by proinflammatory amyloid beta (A beta) Ann N Y Acad Sci. 2000;917:154–164. doi: 10.1111/j.1749-6632.2000.tb05379.x. [DOI] [PubMed] [Google Scholar]
- 11.Itagaki S. McGeer PL. Akiyama H. Zhu S. Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 12.McGeer PL. Akiyama H. Itagaki S. McGeer EG. Immune system response in Alzheimer's disease. Can J Neurol Sci. 1989;16:516–527. doi: 10.1017/s0317167100029863. [DOI] [PubMed] [Google Scholar]
- 13.Bamberger ME. Landreth GE. Microglial interaction with beta-amyloid: implications for the pathogenesis of Alzheimer's disease. Microsc Res Tech. 2001;54:59–70. doi: 10.1002/jemt.1121. [DOI] [PubMed] [Google Scholar]
- 14.Perlmutter LS. Barron E. Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett. 1990;119:32–36. doi: 10.1016/0304-3940(90)90748-x. [DOI] [PubMed] [Google Scholar]
- 15.Togo T. Akiyama H. Kondo H. Ikeda K. Kato M. Iseki E. Kosaka K. Expression of CD40 in the brain of Alzheimer's disease and other neurological diseases. Brain Res. 2000;885:117–121. doi: 10.1016/s0006-8993(00)02984-x. [DOI] [PubMed] [Google Scholar]
- 16.Haga S. Akai K. Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol. 1989;77:569–575. doi: 10.1007/BF00687883. [DOI] [PubMed] [Google Scholar]
- 17.Rogers J. Luber-Narod J. Styren SD. Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging. 1988;9:339–349. doi: 10.1016/s0197-4580(88)80079-4. [DOI] [PubMed] [Google Scholar]
- 18.Styren SD. Civin WH. Rogers J. Molecular, cellular, and pathologic characterization of HLA-DR immunoreactivity in normal elderly and Alzheimer's disease brain. Exp Neurol. 1990;110:93–104. doi: 10.1016/0014-4886(90)90054-v. [DOI] [PubMed] [Google Scholar]
- 19.Boland K. Behrens M. Choi D. Manias K. Perlmutter DH. The serpin-enzyme complex receptor recognizes soluble, nontoxic amyloid-beta peptide but not aggregated, cytotoxic amyloid-beta peptide. J Biol Chem. 1996;271:18032–18044. doi: 10.1074/jbc.271.30.18032. [DOI] [PubMed] [Google Scholar]
- 20.El Khoury J. Hickman SE. Thomas CA. Cao L. Silverstein SC. Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 21.Yan SD. Chen X. Fu J. Chen M. Zhu H. Roher A. Slattery T. Zhao L. Nagashima M. Morser J. Migheli A. Nawroth P. Stern D. Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 22.McDonald DR. Bamberger ME. Combs CK. Landreth GE. Beta-amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joseph JA. Shukitt-Hale B. Denisova NA. Bielinski D. Martin A. McEwen JJ. Bickford PC. Reversals of age-related declines in neuronal signal transduction, cognitive, and motor behavioral deficits with blueberry, spinach, or strawberry dietary supplementation. J Neurosci. 1999;19:8114–8121. doi: 10.1523/JNEUROSCI.19-18-08114.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arendash GW. King DL. Gordon MN. Morgan D. Hatcher JM. Hope CE. Diamond DM. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001;891:42–53. doi: 10.1016/s0006-8993(00)03186-3. [DOI] [PubMed] [Google Scholar]
- 25.Joseph JA. Denisova NA. Arendash G. Gordon M. Diamond D. Shukitt-Hale B. Morgan D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–162. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- 26.Tan J. Town T. Mori T. Wu Y. Saxe M. Crawford F. Mullan M. CD45 opposes beta-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase. J Neurosci. 2000;20:7587–7594. doi: 10.1523/JNEUROSCI.20-20-07587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu Y. Hou H. Nikolic WV. Ehrhart J. Rrapo E. Bickford P. Giunta B. Tan J. CD45RB is a novel molecular therapeutic target to inhibit Abeta peptide-induced microglial MAPK activation. PLoS ONE. 2008;3:e2135. doi: 10.1371/journal.pone.0002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujiwara H. Iwasaki K. Furukawa K. Seki T. He M. Maruyama M. Tomita N. Kudo Y. Higuchi M. Saido TC. Maeda S. Takashima A. Hara M. Ohizumi Y. Arai H. Uncaria rhynchophylla, a Chinese medicinal herb, has potent antiaggregation effects on Alzheimer's beta-amyloid proteins. J Neurosci Res. 2006;84:427–433. doi: 10.1002/jnr.20891. [DOI] [PubMed] [Google Scholar]
- 29.Chung H. Brazil MI. Soe TT. Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- 30.Mitrasinovic OM. Murphy GM., Jr. Accelerated phagocytosis of amyloid-beta by mouse and human microglia overexpressing the macrophage colony-stimulating factor receptor. J Biol Chem. 2002;277:29889–29896. doi: 10.1074/jbc.M200868200. [DOI] [PubMed] [Google Scholar]
- 31.Familian A. Eikelenboom P. Veerhuis R. Minocycline does not affect amyloid beta phagocytosis by human microglial cells. Neurosci Lett. 2007;416:87–91. doi: 10.1016/j.neulet.2007.01.052. [DOI] [PubMed] [Google Scholar]
- 32.LeVine H., 3rd Thioflavine T interaction with synthetic Alzheimer's disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hambleton J. McMahon M. DeFranco AL. Activation of Raf-1 and mitogen-activated protein kinase in murine macrophages partially mimics lipopolysaccharide-induced signaling events. J Exp Med. 1995;182:147–154. doi: 10.1084/jem.182.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suttles J. Milhorn DM. Miller RW. Poe JC. Wahl LM. Stout RD. CD40 signaling of monocyte inflammatory cytokine synthesis through an ERK1/2-dependent pathway. A target of interleukin (il)-4 and il-10 anti-inflammatory action. J Biol Chem. 1999;274:5835–5842. doi: 10.1074/jbc.274.9.5835. [DOI] [PubMed] [Google Scholar]
- 35.Tan J. Town T. Paris D. Mori T. Suo Z. Crawford F. Matt-son MP. Flavell RA. Mullan M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- 36.Tan J. Town T. Saxe M. Paris D. Wu Y. Mullan M. Ligation of microglial CD40 results in p44/42 mitogen-activated protein kinase-dependent TNF-alpha production that is opposed by TGF-beta 1 and IL-10. J Immunol. 1999;163:6614–6621. [PubMed] [Google Scholar]
- 37.Lau FC. Bielinski DF. Joseph JA. Inhibitory effects of blueberry extract on the production of inflammatory mediators in lipopolysaccharide-activated BV2 microglia. J Neurosci Res. 2007;85:1010–1017. doi: 10.1002/jnr.21205. [DOI] [PubMed] [Google Scholar]
- 38.Perlmutter LS. Scott SA. Barron E. Chui HC. MHC class II-positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res. 1992;33:549–558. doi: 10.1002/jnr.490330407. [DOI] [PubMed] [Google Scholar]
- 39.Giunta B. Obregon D. Hou H. Zeng J. Sun N. Nikolic V. Ehrhart J. Shytle D. Fernandez F. Tan J. EGCG mitigates neurotoxicity mediated by HIV-1 proteins gp120 and Tat in the presence of IFN-gamma: role of JAK/STAT1 signaling and implications for HIV-associated dementia. Brain Res. 2006;1123:216–225. doi: 10.1016/j.brainres.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giunta B. Zhu Y. Hou H. Rrapo E. Fernandez F. Tan J. HIV-1 TAT inhibits microglial phagocytosis of Aa peptide. Int J Clin Exp Pathol. 2008:260–275. [PMC free article] [PubMed] [Google Scholar]
- 41.Gray E. Thomas TL. Betmouni S. Scolding N. Love S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008;18:86–95. doi: 10.1111/j.1750-3639.2007.00110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koning N. Bo L. Hoek RM. Huitinga I. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann Neurol. 2007;62:504–514. doi: 10.1002/ana.21220. [DOI] [PubMed] [Google Scholar]
- 43.McGeer PL. McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- 44.Robertson J. Beaulieu JM. Doroudchi MM. Durham HD. Julien JP. Mushynski WE. Apoptotic death of neurons exhibiting peripherin aggregates is mediated by the proinflammatory cytokine tumor necrosis factor-alpha. J Cell Biol. 2001;155:217–226. doi: 10.1083/jcb.200107058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaushal V. Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petcu EB. Sfredel V. Platt D. Herndon JG. Kessler C. Popa-Wagner A. Cellular and molecular events underlying the dysregulated response of the aged brain to stroke: a mini-review. Gerontology. 2008;54:6–17. doi: 10.1159/000112845. [DOI] [PubMed] [Google Scholar]
- 47.Mrak RE. Griffin WS. Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J Neuropathol Exp Neurol. 2007;66:683–686. doi: 10.1097/nen.0b013e31812503e1. [DOI] [PubMed] [Google Scholar]
- 48.Rogers J. Kirby LC. Hempelman SR. Berry DL. McGeer PL. Kaszniak AW. Zalinski J. Cofield M. Mansukhani L. Willson P, et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 49.Aisen PS. Davis KL. Berg JD. Schafer K. Campbell K. Thomas RG. Weiner MF. Farlow MR. Sano M. Grundman M. Thal LJ. A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study. Neurology. 2000;54:588–593. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- 50.Aisen PS. Schmeidler J. Pasinetti GM. Randomized pilot study of nimesulide treatment in Alzheimer's disease. Neurology. 2002;58:1050–1054. doi: 10.1212/wnl.58.7.1050. [DOI] [PubMed] [Google Scholar]
- 51.Soininen H. West C. Robbins J. Niculescu L. Long-term efficacy and safety of celecoxib in Alzheimer's disease. Dement Geriatr Cogn Disord. 2007;23:8–21. doi: 10.1159/000096588. [DOI] [PubMed] [Google Scholar]
- 52.Van Gool WA. Weinstein HC. Scheltens P. Walstra GJ. Effect of hydroxychloroquine on progression of dementia in early Alzheimer's disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet. 2001;358:455–460. doi: 10.1016/s0140-6736(01)05623-9. [DOI] [PubMed] [Google Scholar]
- 53.Yasojima K. Schwab C. McGeer EG. McGeer PL. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999;830:226–236. doi: 10.1016/s0006-8993(99)01389-x. [DOI] [PubMed] [Google Scholar]
- 54.McGeer PL. Cyclo-oxygenase-2 inhibitors: rationale and therapeutic potential for Alzheimer's disease. Drugs Aging. 2000;17:1–11. doi: 10.2165/00002512-200017010-00001. [DOI] [PubMed] [Google Scholar]
- 55.Stein M. Bell MJ. Ang LC. Hydroxychloroquine neuromyotoxicity. J Rheumatol. 2000;27:2927–2931. [PubMed] [Google Scholar]
- 56.Klegeris A. Walker DG. McGeer PL. Toxicity of human THP-1 monocytic cells towards neuron-like cells is reduced by non-steroidal anti-inflammatory drugs (NSAIDs) Neuropharmacology. 1999;38:1017–1025. doi: 10.1016/s0028-3908(99)00014-3. [DOI] [PubMed] [Google Scholar]
- 57.Scharf S. Mander A. Ugoni A. Vajda F. Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. 1999;53:197–201. doi: 10.1212/wnl.53.1.197. [DOI] [PubMed] [Google Scholar]
- 58.Arai H. Suzuki T. Sasaki H. Hanawa T. Toriizuka K. Yamada H. [A new interventional strategy for Alzheimer's disease by Japanese herbal medicine] Nippon Ronen Igakkai Zasshi. 2000;37:212–215. doi: 10.3143/geriatrics.37.212. [DOI] [PubMed] [Google Scholar]
- 59.Capell HA. Disease modifying antirheumatic drugs: longterm safety issues. J Rheumatol Suppl. 2001;62:10–15. [PubMed] [Google Scholar]
- 60.Matter ML. Zhang Z. Nordstedt C. Ruoslahti E. The alpha5beta1 integrin mediates elimination of amyloid-beta peptide and protects against apoptosis. J Cell Biol. 1998;141:1019–1030. doi: 10.1083/jcb.141.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bamberger ME. Harris ME. McDonald DR. Husemann J. Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laws SM. Perneczky R. Wagenpfeil S. Muller U. Forstl H. Martins RN. Kurz A. Riemenschneider M. TNF polymorphisms in Alzheimer disease and functional implications on CSF beta-amyloid levels. Hum Mutat. 2005;26:29–35. doi: 10.1002/humu.20180. [DOI] [PubMed] [Google Scholar]
- 63.Tarkowski E. Liljeroth AM. Minthon L. Tarkowski A. Wallin A. Blennow K. Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Res Bull. 2003;61:255–260. doi: 10.1016/s0361-9230(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 64.Goyarzu P. Malin DH. Lau FC. Taglialatela G. Moon WD. Jennings R. Moy E. Moy D. Lippold S. Shukitt-Hale B. Joseph JA. Blueberry supplemented diet: effects on object recognition memory and nuclear factor-kappa B levels in aged rats. Nutr Neurosci. 2004;7:75–83. doi: 10.1080/10284150410001710410. [DOI] [PubMed] [Google Scholar]
- 65.Sloane JA. Hollander W. Moss MB. Rosene DL. Abraham CR. Increased microglial activation and protein nitration in white matter of the aging monkey. Neurobiol Aging. 1999;20:395–405. doi: 10.1016/s0197-4580(99)00066-4. [DOI] [PubMed] [Google Scholar]
- 66.Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 67.Croisier E. Moran LB. Dexter DT. Pearce RK. Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation. 2005;2:14. doi: 10.1186/1742-2094-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gemma C. Mesches MH. Sepesi B. Choo K. Holmes DB. Bickford PC. Diets enriched in foods with high antioxidant activity reverse age-induced decreases in cerebellar beta-adrenergic function and increases in proinflammatory cytokines. J Neurosci. 2002;22:6114–6120. doi: 10.1523/JNEUROSCI.22-14-06114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y. Chang CF. Chou J. Chen HL. Deng X. Harvey BK. Cadet JL. Bickford PC. Dietary supplementation with blueberries, spinach, or spirulina reduces ischemic brain damage. Exp Neurol. 2005;193:75–84. doi: 10.1016/j.expneurol.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 70.Sellappan S. Akoh CC. Krewer G. Phenolic compounds and antioxidant capacity of Georgia-grown blueberries and blackberries. J Agric Food Chem. 2002;50:2432–2438. doi: 10.1021/jf011097r. [DOI] [PubMed] [Google Scholar]