

Abstract
The correct replication and repair of DNA is critical for a cell’s survival. Here we investigate the fidelity of mammalian DNA polymerase λ (pol λ) utilizing dynamics simulation of the enzyme bound to incorrect incoming nucleotides including A:C, A:G, A(syn):G, A:A, A(syn):A, and T:G, all of which exhibit differing incorporation rates for pol λ compared to A:T bound to pol λ. The wide range of DNA motion and protein residue side-chain motions observed in the mismatched systems demonstrates distinct differences when compared to the reference (correct base pair) system. Notably, Arg517’s interactions with the DNA template strand bases in the active site are more limited and Arg517 displays increased interactions with the incorrect dNTPs. This effect suggests that Arg517 helps provide a base-checking mechanism to discriminate correct from incorrect dNTPs. In addition, we find Tyr505 and Phe506 also play key roles in this base checking. A survey of the electrostatic potential landscape of the active sites and concomitant changes in electrostatic inter-action energy between Arg517 and the dNTPs reveals that pol λ binds incorrect dNTPs less tightly than the correct dNTP. These trends lead us to propose the following order for mismatch insertion by pol λ: A:C > A:G > A(syn):G > T:G > A(syn):A > A:A. This sequence agrees with available kinetic data for incorrect nucleotide insertion opposite template adenine with the exception of T:G, which may be more sensitive to the insertion context.
Keywords: pol λ, polymerase λ, mismatched base pair, Molecular Dynamics simulations, Protein/DNA complex, Conformational changes
1 Introduction
The maintenance of a cell’s genetic information is essential for its survival. DNA polymerases play a key role in this process by replicating and repairing DNA. Although all DNA polymerases catalyze the same nucleotidyl transfer reaction and have the same general conformation – a hand consisting of finger, palm, and thumb subdomains (1), they can exhibit very different error tendencies (2). One of the most basic types of errors that DNA polymerases make is the base substitution error. This occurs when the DNA polymerase inserts the wrong nucleotide opposite the DNA template base to form a non-standard base pair or mismatch (i.e., not an A:T or C:G Watson-Crick base pair). These errors can impair the integrity of a cell, especially if they occur within protein-coding regions of the DNA. Even when the error occurs in a non-coding region, mismatch incorporation can hinder or stall further DNA synthesis.
A number of interesting polymerases from the X- and Y-families have specialized functions such as lesion bypass and the ability to fill in only a few nucleotides at a time typically within the context of a DNA repair pathway (3, 4). These enzymes have lower fidelities than DNA polymerases involved in DNA replication and often display a very unusual base substitution error profile. For example, African Swine Fever Virus DNA polymerase X (pol X) inserts G:G with similar ability to Watson-Crick base pairs (5) and the Y-family DNA polymerase ι has a similar tendency to incorporate T:G (6, 7, 8, 9). To interpret these unusual error specificities, an understanding of atomic-level DNA polymerase/substrate interactions is essential.
Here we focus on understanding the base substitution error profile of mammalian DNA polymerase λ (pol λ). Understanding pol λ’s catalytic cycle is important because this enzyme resembles in both structure and function other members of the X-family. Pol λ has a moderate-fidelity in the range of 10−4–10−5 (10) like DNA polymerase β (pol β), another X-family enzyme. Pol λ’s polymerization ‘hand’ domain also resembles that of pol β (3) and both enzymes possess an additional 8-kDa domain with 5′-deoxyribose-5-phosphate lyase function (11, 12). Pol λ has a BRCT domain similar to some other X-family enzymes such as DNA polymerase µ (pol µ) and terminal deoxynu-cleotidyl transferase (TdT). This BRCT domain is connected to pol λ’s pol β-like core through a serine/threonine linker region (3).
Experimental studies suggest that pol λ, like pol β, is part of the main pathway for repairing small DNA lesions, the base excision repair (BER) pathway (13), and is employed to fill small gaps in DNA (12, 14, 15, 16, 17). In addition, pol λ, like pol µ, is hypothesized to participate in the nonhomologous end-joining (NHEJ) pathway, which serves to repair double-strand breaks in DNA and its BRCT domain is believed to facilitate interaction with other NHEJ pathway proteins (18, 19, 20, 21, 22).
Analyses of X-ray crystal structures and related computational studies (23, 24, 25, 26, 27, 28) have uncovered important differences and similarities regarding how pol β and pol λ incorporate a correct nucleotide into single-nucleotide gapped DNA. While pol βmoves between open (inactive) and closed (active) protein subdomain conformations upon binding the correct incoming nucleotide, pol λ remains in a closed subdomain conformation whether or not an incoming nucleotide is bound. Instead, the pol λ/DNA complex transitions from an inactive to active state, upon binding the correct incoming nucleotide, through a large shift of the DNA template strand that allows the templating base at the gap to align with the incoming nucleotide in the active site. Both pol λ and pol β utilize a sequence of active-site protein residue motions to help prepare the enzyme-substrate complex for the chemical reaction; these protein residues are hypothesized to act as “gate-keepers” regulating the assembly of the active site for the chemical reaction (29, 28). Computational studies of the chemical reaction with the correct substrate in both enzymes reveal an associative-like mechanism involving proton transfer to an active-site aspartate (30, 31, 32, 33, 34).
Recent studies suggest that the significant DNA motion in pol λ prior to chemistry may further reduce pol λ’s fidelity by providing an opportunity for deletion errors to occur through DNA template strand slippage (35, 36, 37). Significantly, pol λ generates even more single-base deletion errors than base substitution errors (38).
X-ray crystallographic studies have provided important clues in understanding why certain mismatches are more easily inserted than others by revealing structural differences between the mismatches and Watson-Crick base pairs in the polymerase active site. Pol β has been crystallized with mismatches in its active site both before and after their insertion (39, 40, 41). These structures demonstrate the dynamic nature of polymerase/mismatch interactions since the geometry of the mismatch changes depending on its position within the DNA helix. For example, in a crystallized binary pol β/DNA complex with an A:A mismatch at the primer terminus, the adenine bases stack, but a crystal structure obtained after the correct incoming nucleotide binds shows that the template adenine of the mismatch switches to the syn orientation (40). Furthermore, crystallized ternary pol β complexes with incorrect incoming nucleotides reveal a different mismatch geometry where the templating base shifts to create a transient ‘abasic’ site opposite the incorrect incoming nucleotide (41).
Computational studies of pol β and pol X have yielded insights into the process of incorrect nucleotide incorporation (42, 43, 44). Dynamics simulations of pol β bound to different incorrect nucleotides show varying amounts of active-site distortions that are mismatch dependent. Interestingly, the degree of active-site disorder mirrors trends in kinetic data for mismatch incorporation (42). Transition path sampling studies of pol β’s conformational closing when bound to correct G:C and incorrect G:A base pairs also reveal that the closed state is less stable with the mismatch than the correct nascent base pair (26, 43). Simulations of pol X suggest that G:G is more readily inserted when the incoming nucleotide is in the syn orientation, since full closing motion occurs with this mismatch geometry (44).
Although there are several key experimental studies of pol λ’s fidelity (45, 38, 46, 10, 47, 48), structural information regarding pol λ’s interactions with mismatches is more limited. Available are an X-ray crystal structure of a pol λ/DNA binary complex with a G:G mismatch at the primer terminus (48), as well as a complex with a T:T mismatch several base pairs upstream from the active site (35). In both structures, the mismatches fit within the DNA helix and cause minimal distortion. No structure of an incorrect incoming nucleotide bound to pol λ’s active site has yet been reported.
Here we investigate dynamics of pol λ bound to frequently incorporated mismatches (i.e., A:dCTP and T:dGTP) as well as others (i.e., A:dATP and A:dGTP) to determine the factors that contribute to insertion differences; reference studies of the correct A:dTTP base pair in pol λ’s active-site pocket are available for comparison (28). We also analyze simulations of the bulky purine-purine mismatches with the template base in both the anti and syn orientations to determine whether a particular base pair geometry might facilitate mismatch incorporation.
Clearly, all dynamics simulation data are subject to the approximations and limitations of an empirical force field which has been parameterized to reproduce experimental data. In particular, Mg2+ ions are modeled only through the parameterization of nonbonded interactions that are based on Lennard-Jones and Coulombic potentials (49, 50). This may result in shorter ion-ligand distances than observed in X-ray crystal structures (51, 28). Despite these approximations, related studies as done here for several mismatch systems with the same CHARMM (52) force field and conditions help identify important trends in pol λ mismatch interactions, dynamics, and energetics. Furthermore, we seek to identify the long-range effects of active-site Mg 2+ and not the specific lengths of the ion coordination distances.
Our combined studies assist in understanding pol λ’s unusual error profile. They show increased DNA motion as well as altered active-site geometries in the mismatches compared to the correct A:dTTP base pair system (28). The mismatches also display decreased stability. As observed previously (36, 37), the increased DNA motion in the mismatch simulations can be attributed to reduced Arg517/DNA interactions. Further analyses of the active-site electrostatic potential and energetic interactions between Arg517 and the incorrect incoming nucleotides provide evidence for Arg517’s base-checking role. This function is similar to that performed by Arg283 in pol β (53, 54). As in pol β and pol X simulations, the degree of active-site distortion in pol λ parallels kinetic data trends, except for T:G, which is more disordered than indicated by the data. This may be explained by different sequence contexts – the poorer stacking interactions between T:dGTP and the A:T primer terminus base pair than in mismatches where the templating base is adenine may cause the greater T:G instability in our case. Comparisons with error rate data also suggest that pol λ’s tendency for inserting this mismatch may depend on the context of its insertion.
2 Experimental Methods
2.1 Initial Models
Six initial models were prepared based on the X-ray crystal pol λ ternary complex (PDB entry 1XSN). In all models, the catalytic ion was positioned in the active site by superimposing the pol λ protein Cα atoms onto those of the pol β ternary complex (PDB entry 1BPY).
In the initial structures, missing protein residues 1–11 were added and mutant residue Ala543 was replaced with cysteine to reflect the natural amino acid sequence of pol λ. An oxygen atom was added to the 3′ carbon of the ddTTP sugar moiety in the ternary complex to form 2′-deoxythymidine 5′-triphosphate (dTTP). Similarly, an oxygen atom was added to the 3′ carbon of the primer terminus. Hydrogen atoms and other atoms from twenty protein residues located in the thumb, palm, and 8-kDa domain not resolved in the X-ray crystal structure were also added to the models. In each system, the active-site aspartate residues and triphosphate moiety of the dNTP were modeled in their unprotonated forms.
In each model, the A:dTTP nascent base pair was replaced with a different mismatch, namely: A:C, A:A. A:G, or T:G (in this notation, the template base’s symbol is written first followed by the incoming nucleotide’s symbol). Since purine bases can assume both anti and syn orientations, we modeled the template adenine of A:A and A:G mismatches in both orientations. In summary, the following mispairs were modeled (residues are in the normal anti form unless designated syn and an alternative notation appears in brackets): A:dCTP [A:C], A:dATP [A:A], A(syn):dATP [A(syn):A], A:dGTP [A:G], A(syn):dGTP [A(syn):G], and T:dGTP [T:G].
Optimized periodic boundary conditions in a cubic cell were introduced to all complexes using the PBCAID program (55). The smallest image distance between the solute, the protein complex, and the faces of the periodic cubic cell was 10 Å. To obtain a neutral system at an ionic strength of 150 mM, the electrostatic potential of all bulk water (TIP3 model) oxygen atoms was calculated using the Delphi package (56). Those water oxygen atoms with minimal electrostatic potential were replaced with Na+ and those with maximal electrostatic potential were replaced with Cl−. In placing the ions, a separation of at least 8 Å was maintained between the Na+ and Cl− ions and between the ions and protein or DNA atoms.
As shown by the model of the A:C system in, all initial models contain approximately Fig. 1 38,325 atoms, 278 crystallographically resolved water molecules, 10,481 bulk water molecules, two Mg2+ ions, an incoming nucleotide, and 39 Na+ and 29 Cl− counterions. The final dimensions of the box are: 73.93 Å × 77.78 Å × 72.43 Å.
Figure 1.

The pol λ/DNA/dCTP ternary system. A5: templating base at the gap (in T:G system, the templating base is T5); A6: adjacent template base that pairs with the primer terminus; T6: the primer terminus; T: DNA template strand; P: DNA primer strand. Blue protein side chains that rearrange in many mismatch systems. In (c), only the palm and thumb are shown for clarity.
2.2 Minimization, Equilibration and Dynamics Protocol
All six model systems were energy minimized and equilibrated using the CHARMM program (52) with the all-atom CHARMM27 force field (57). First, each system was minimized with fixed positions for all protein and nucleic heavy atoms except those from the added residues using SD for 5,000 steps followed by ABNR for 10,000 steps. Two cycles of further minimization were carried out for 10,000 steps using SD followed by 20,000 steps of ABNR. During these minimizations, the Cl−, Na+ and water relaxed around the protein/DNA complex. The equilibration process was started with a 30 ps simulation at 300 K using single-timestep Langevin dynamics and keeping the constraints used in the previous minimization step. The SHAKE algorithm was employed to constrain the bonds involving hydrogen atoms. This was followed by unconstrained minimization using 10,000 steps of SD followed by 20,000 steps of ABNR. A further 30 ps of equilibration at 300 K and minimization consisting of 2,000 steps of SD followed by 4,000 steps of ABNR were performed. The final equilibration step involved 130 ps dynamics at 300 K.
Production dynamics were performed using the NAMD program (58) with the CHARMM27 force field (57). First, the energy in each system was minimized using the Powell conjugate gradient algorithm. Systems were then equilibrated for 100 ps at constant pressure and temperature. Pressure was maintained at 1 atm using the Langevin piston method (59), with a piston period of 100 fs, a damping time constant of 50 fs, and piston temperature of 300 K. Temperature coupling was enforced by velocity reassignment every 2 ps. The water and ions were further energy minimized and equilibrated at constant temperature and volume for 30 ps at 300 K while holding all protein and DNA heavy atoms fixed. This was followed by minimization and 30 ps of equilibration at 300 K on the entire system. Then, production dynamics were performed at constant temperature and volume. The temperature was maintained at 300 K using weakly coupled Langevin dynamics of non-hydrogen atoms with damping coefficient γ = 10 ps−1 used for all simulations performed; bonds to all hydrogen atoms were kept rigid using SHAKE (60), permitting a time step of 2 fs. The system was simulated in periodic boundary conditions, with full electrostatics computed using the PME method (61) with grid spacing on the order of 1 Å or less. Short-range nonbonded terms were evaluated every step using a 12 Å cutoff for van der Waals interactions and a smooth switching function. The total simulation length for all systems was 20 ns.
Simulations using the NAMD package were run on local and NCSA SGI Altix 3700 Intel Itanium 2 processor shared-memory systems running the Linux operating system.
2.3 Active-site Electrostatic Potential Calculations
The equilibrated models of the pol λ A:C, A:G, A:A, and T:G mismatch systems and the correct A:T system, which was simulated in (28), were used for calculations of the active-site electrostatic potential with the QNIFFT program (62, 63). The A(syn):G and A(syn):A systems were not used since they are expected to be very similar to the other systems with either a dGTP or a dATP in the active-site. These calculations could help differentiate between the active-site environment when the incorrect incoming nucleotide is bound and the configuration when the correct incoming nucleotide is bound. This should help interpret the varied active-site motions in the mismatch simulations. The examination of the active-site electrostatic potential in tandem with electrostatic interactions between Arg517 and the incoming nucleotides could also elucidate Arg517’s role in pol λ’s fidelity.
3 Results
3.1 DNA and Protein Movement
The pol λ/DNA/dCTP complex is shown in Figure 1. In all mismatch systems, we monitor the motions of the DNA bound to pol λ since these may be indicators of how easily pol λ can proceed with misincorporation. Significant movement toward the inactive DNA position may signal the deactivation of the enzyme/substrate complex. As shown by the RMSD data with respect to active and inactive forms in Figure 2a (green showing active, red showing inactive), the DNA in the A:C system remains closest to its active position. However, the DNA and dCTP shift while staying in line with the DNA’s active position (Sup. Fig. S1a). The extreme opposite occurs in the A:A system where the DNA moves to the inactive position (Figure 2f).
Figure 2.

(a)–(f) Time evolution of the RMSD of DNA backbone atoms (P, O3′, C3′, C4′, C5′, O5′, O5T) in all trajectories relative to the active (green, PDB entry 1XSN) and inactive (red, PDB entry 1XSL) DNA positions. Superposition is performed with respect to protein Cα atoms. RMSD plots do not start at zero because each system was equilibrated prior to production dynamics. (g)– (m) Time evolution of Arg517/dNTP electrostatic interaction energy in correct and mismatched system trajectories.
In both the A:G and T:G systems, the DNA remains closer to its active position (Figure 2b,c), but small fluctuations toward the inactive DNA position occur, with those in the T:G system occurring more frequently. Substantially more extensive DNA motion occurs in the A(syn):A and A(syn):G systems (Figure 2d,e). In A(syn):A, a period of frequent DNA motion (i.e., during 2–13 ns of simulation) is bracketed by periods when the DNA stays mainly in the active position.
Despite the wide range of DNA motion, the overall protein conformation is very stable in all systems, but several subtle protein residue side-chain rearrangements occur, as described below.
3.2 Mismatch Geometry Changes and Associated Active-site Motions
In many of the mismatch systems, the active-site DNA rearranges significantly and new hydrogen bonding interactions form that indicate a lower active-site conformational stability than in the correct A:T system. The fewest rearrangements occur in the A:C and A:G systems. In the A:C system, although hydrogen bonds do not frequently occur between the templating adenine (A5) and dCTP, the dCTP is well-stabilized by stacking interactions with the primer terminus (T6), as shown in Figure 3a (refer to Sup. Fig. S1b for distance data). In the A:G system, the mismatched bases stack with one another for stability as shown in Figure 3b. In this arrangement, a water molecule frequently joins the A5:N1 and dGTP:O4′ atoms (Figure 3b, top), but when the water molecule is not present, Tyr505 provides additional support to A5 (Figure 3b, bottom, and Sup. Fig. S2a).
Figure 3.

(a) Key A:C geometry changes. Top and bottom parts show A:C system trajectory snapshots after 20 ns and 5.7 ns, respectively. Templating adenine (A5) and dCTP are colored by atom and the primer terminus (T6) is grey. The A5:dCTP mismatch is shown with respect to the position of the correct A:dTTP base pair (purple, PDB entry 1XSN). (b) Top and bottom show A:G system trajectory snapshots after 6.6 ns and 12.1 ns, respectively, that depict variations in A5:dGTP interactions and related Tyr505 and Arg514 changes. All A:G system atoms are colored by atom type. In the bottom image, dGTP position is shown with respect to correct dTTP (purple). (c) Time evolution of T5:dGTP geometry changes. All T:G system atoms are colored by atom type. Green and red structures represent the positions of residues in the pol λ ternary (active form, PDB entry 1XSN) and binary (inactive form, PDB entry 1XSL) complexes, respectively. A is the templating adenine of correct A:T system. In all parts, WAT is a water molecule, distances are indicated by dashed yellow lines, and green or blue arrows indicate movement.
More complex base pair changes occur within the T:G system as sketched in Figure 3c. The templating thymine, T5, and dGTP first transition from partially stacking to a form of wobble base pairing. Then, as the mismatch bases separate, motions in Arg517 and Tyr505 (discussed below) lead to a large rearrangement in dGTP (refer to Sup. Fig. S3a for relevant dGTP torsion data).
In the A(syn):A system, the dATP primarily interacts with A6, the template base adjacent to A(syn)5, without breaking the primer terminus base pair (refer to Sup. Fig. S4a–e for distance data). An examination of the equilibration phase of this system reveals that a movement of Arg517, similar to that in T:G, separates A(syn)5 from dATP (Sup. Fig. S5). Within the active site, the dATP geometry frequently changes beginning with a rotation of the sugar up to 90° as shown in Figure 4a,b. This is followed by another dATP change (Figure 4c,d) that corresponds to a period of frequent DNA motion toward the inactive position.
Figure 4.

Key dATP changes and their effects on DNA and interatomic distances within the active site of the A(syn):A system. (a) The dATP torsion change that repositions the sugar moiety as shown in (b). In (b), three different positions of the dATP sugar moiety (colored by atom) are shown. Blue arrow indicates range of motion. T6 is the primer terminus. (c) Another dATP torsion change (blue) shown with the RMSD in the DNA backbone atoms relative to the active (green, PDB entry 1XSN) and inactive (red, PDB entry 1XSL) DNA positions. (d) Position of dATP atoms involved in the torsion change shown in (c). (e) Active-site interatomic distance changes that correlate with torsion change in (c).
In the A(syn):G system, the mispaired bases also do not stably interact since dGTP flips between the two forms (straight and sideways) shown in Figure 5a that support different hydrogen bonds to the DNA template strand (refer to Sup. Fig. S6a for dGTP torsion data). These dGTP changes also cause the two upstream base pairs to break apart.
Figure 5.

(a) Key active-site DNA changes in the A(syn):G system. Trajectory snapshots are shown at 0, 8, and 20 ns. In the side-views (top), the DNA and dGTP are colored by atom, but, in the full-views (bottom), the mismatch is colored by atom and the primer terminus base pair (A6:T6) is colored grey. By 8 ns, the dGTP sugar has shifted upward (shown by blue arrow) when compared to the position of the correct dTTP (purple). (b) Main active-site changes in the A:A system shown by trajectory snapshots after 0, 0.08, 5, and 20 ns, arranged chronologically beginning in the upper left corner. Appearing in the snapshots are: the mismatch (A5:dATP; colored by atom), the primer terminus base pair (A6:T6; blue), Tyr505 (pink), and Phe506 (green). In both parts, arrows indicate motion and hydrogen bonding are indicated by dashed yellow lines.
In the A:A system, the mispaired adenines partially stack and slant (Figure 5b) and unusual hydrogen bonding occurs between the primer terminus (T6) and the templating base at the gap (A5), and this disrupts the primer terminus base pair (refer to Sup. Fig. S7a,b and other parts for hydrogen bonding data). Tyr505 intervenes by forming a hydrogen bond to A5 either directly or through a water molecule (Figure 5b and Sup. Fig. S7c). The very limited pairing of the DNA bases at the active site shifts the DNA toward its inactive position (Figure 2f), and movements in Phe506 lead to a rotation of the dATP, further distorting the active-site geometry (Sup. Fig. S8).
Additional information on the pol λ mismatch hydrogen bonding patterns and how they compare to structural data for mismatches in free DNA as well as other polymerase active sites is found in the Supplementary Material (Sup. Fig. S2, Sup. Fig. S4–Sup. Fig. S7, and Sup. Fig. S9).
3.3 Differences in Active-site Electrostatics and Arg517/dNTP Interactions between Mismatch and Correct Systems
From an examination of the electrostatic potential of pol λ’s active site with the correct A:dTTP base pair and with various mismatches, unfavorable protein/dNTP interactions emerge in the mismatch systems that destabilize the dNTP. As shown in Figure 6, the A:T system’s active site has mainly positive (blue) or neutral (white) electrostatic potential whereas the mismatch systems have more negative (red) electrostatic potentials near the sugar moiety of the dNTP. This stronger concentration of electron density in the active site destabilizes the sugar moiety. Indeed, an upward shift of the sugar occurs in the A:C, A:G, and A(syn):G systems and rotations of the sugar ring occur in the T:G, A(syn):A, and A:A systems (Figure 3–Figure 5).
Figure 6.

Electrostatic potential surfaces of the pol λ active site with either the correct or incorrect incoming nucleotides. Arg517’s position is marked in yellow.
The mismatch systems containing dGTP have an additional destabilizing factor due to the close proximity of the dGTP 2-amino group and N1 hydrogen atoms (i.e., dGTP:H21, H22, and H1 atoms) to a positive region of the electrostatic potential surface. To counterbalance this disruptive force, the dGTP, in A:G, forms two hydrogen bonds to the DNA backbone. Likewise, in T:G, the dGTP forms hydrogen bonds to T5 for stabilization, but when this interaction dissolves, the stabilizing effect vanishes and the guanine base flips away from the positive electrostatic potential region. A similar movement occurs in the the A(syn):G system where dGTP frequently bends “sideways” into the upstream DNA helix away from the positive electrostatic potential region.
The motions of the dNTPs also specifically correspond to electrostatic interaction energy changes with Arg517 (Figure 2g–m), one of the residues forming the positive region of the electrostatic potential surface near the dNTP base. Following the active-site electrostatic potential surface analysis, Arg517 interacts favorably with dTTP, dCTP, and dATP since the partially negatively charged dTTP:O2, dCTP:O2 and dATP:N3 atoms are closest to the Arg517 side chain. However, in the A:A and A:C systems, these interactions become less favorable after dATP and dCTP rearrange within the active site. In A(syn):A, the interactions frequently revert from being very favorable to being much less favorable.
As discussed above, the situation is different when dGTP is in the active-site pocket and the unfavorable electrostatic interactions appear to result from the proximity of the Arg517 side chain to the dGTP 2-amino group. In the T:G system, unfavorable interactions increase as Arg517 moves in closer proximity to dGTP (Figure 3c), but this is reversed by changes in both Arg517 and dGTP that produce more favorable electrostatic interactions (refer to Sup. Fig. S3 for data on the Arg517 and dGTP changes).
3.4 Further Details on Protein Side-chain Motions
Further rearrangements occur in the mismatch systems in active-site protein residues that increase active-site disorder. Summarized in Table 1 are the movements of key residues: Ile492, Tyr505, Phe506, Asn513, Arg514, and Arg517, with respect to those in the crystal ternary (active) and binary (inactive) pol λ complexes. Here, we elaborate on the most important motions and protein/DNA interactions.
Table 1.
Predominant Active-site Protein Residue Side-chain Orientations Assumed During Pol λ smatch Dynamicsa
| System | Protein Residue | |||||
|---|---|---|---|---|---|---|
| Tyr505 | Phe506 | IIe492 | Asn513 | Arg514 | Arg517 | |
| A:C | inactive | active | active | active | active | active |
| A:G | inactive | active | active | new | inactive | active |
| A(syn):G | inactive | active | active | new | active | active |
| T:G | mainly nactive |
mainly active, but briefly inactive |
inactive & new |
new | active | mainly active with small conformational changes |
| A(syn):A | inactive | inactive | inactive & new |
active | active | mainly active with small conformational changes |
| A:A | inactive, ctive, & new |
active and sideways variant |
active | active | intermediate | active |
Table Definitions: inactive, refers to side-chain orientation in binary pol λ /DNA complex (PDB entry 1XSL)
active, refers to side-chain orientation in ternary pol λ/DNA/dTTP complex (PDB entry 1XSN)
new, refers to side-chain orientation not captured crystallographically
intermediate, refers to side-chain orientation lying between the active and inactive positions
Arg517
As discussed above, electrostatic interactions between Arg517 and the dNTP are major determinants of dNTP stability within the active site. Incorrect dGTPs have the least favorable interactions with Arg517 and thus tend to reposition within the active site. In addition, specific movements in Arg517’s side chain, such as those in the T:G and A(syn):A systems, physically cause the separation of the mispaired bases. Since Arg517/DNA interactions also have a great impact on DNA stability, we summarize their hydrogen bonding in Table 2.
Table 2.
Arg517/DNA Hydrogen Bonding in the Mismatch and Correct Systems
| Simulation | T5:O2 or A5:53 |
A6:N3 | A6:O4′ | A6:O1P | Other |
|---|---|---|---|---|---|
| A:C | through a water molecule |
through a water molecule |
—— | through a water molecule |
with A5:O4′ through a water molecule |
| A:G | yes | through a water molecule |
yes | through water molecule |
—— |
| A(syn):G | —— | —— | yes, sometimes |
through a water molecule |
—— |
| T:G | yes, sometimes |
yes, sometimes directly & sometimes through a water molecule |
yes, sometimes |
through a water molecule |
with T7:O4′ through a water molecule; briefly with A6:O5′ & T5:O3′ |
| A(syn):A | —— | —— | —— | through a water molecule & briefly direct |
yes, sometimes to A6:O5′ |
| A:A | yes | —— | yes, briefly |
through a water molecule |
—— |
| A:Ta | yes | yes | yes | through a water molecule |
—— |
Data from [28]
Compared to the correct A:T system, fewer direct Arg517/DNA hydrogen bonds and more interactions mediated by water molecules occur in the mismatch systems. In the A:C system, all Arg517 interactions with the DNA occur through water molecules, yet the DNA stays close to its active position. The A:G system, which also favors the active DNA position, exhibits a combination of direct and indirect interactions between Arg517 and the DNA (Sup. Fig. S2b,c).
In the other mismatch systems we find that a greater number of transient Arg517/DNA interactions reduce the stability of the active DNA position. In T:G, the Arg517/DNA hydrogen bonding patterns rearrange and greater DNA motion occurs. While the T5 and dGTP form a wobble base pair, Arg517 forms one hydrogen bond to A6:O4′. After T5 and dGTP separate, Arg517’s interactions are principally with T5 (Figure 3c and Sup. Fig. S9a). Unfavorable interactions between dGTP and Arg517 lead to another rearrangement of dGTP and Arg517 (Figure 3c and Sup. Fig. S3b) resulting in multiple hydrogen bonds between Arg517 and T5, A6, and dGTP (Sup. Fig. S9a–d). Other indirect interactions with the DNA through water molecules also occur.
In the A(syn):G system, which shows frequent DNA motion, Arg517 only occasionally forms a hydrogen bond to A6:O4′ (Sup. Fig. S6b) and one indirect interaction through a water molecule. When the hydrogen bond does not occur with A6:O4′ (e.g., between 9–16 ns in Sup. Fig. S6b), significant DNA motion occurs (Figure 2e) that includes both A(syn)5 and A6 bases lifting to interact with Lys273 of the 8-kDa domain (Sup. Fig. S10).
In the A(syn):A system, Arg517 has even more limited DNA interactions because of dATP’s unusual hydrogen bonding with A6. Arg517 has brief interactions with the DNA backbone near A6 (Sup. Fig. S4f) and frequently forms a hydrogen bond to the dATP base (Sup. Fig. S4g). Since A(syn)5 is not constrained by hydrogen bonds to either Arg517 or dATP, it frequently interacts with Lys273 (Sup. Fig. S11) as in the A(syn):G system.
In A:A, Arg517 forms one steady hydrogen bond to A5:N3 and briefly forms a hydrogen bond to A6:O4′ (Sup. Fig. S7d) in addition to another interaction with the DNA backbone through a water molecule, but the lack of regular hydrogen bonding between the DNA in the active site leads to the full transition of the DNA to the inactive position. The A6 base, in particular, is poorly stabilized and it rearranges closer to Lys273 as in the A(syn):G and A(syn):A systems (Sup. Fig. S12).
Arg514
In the correct A:T system, Arg514 helps to stabilize the templating base at the gap through stacking interactions. This does not occur when the mispaired bases stack with one another. Thus, in the A:G and A:A systems, Arg514 moves closer to its inactive conformation (see Figure 3b for A:G system) and, in the T:G system, an Arg514 rearrangement occurs while T5 and dGTP move from stacking to pairing.
Asn513
Asn513 helps stabilize the dNTP in the active site by forming hydrogen bonds to the minor groove of the dNTP base in the correct and most incorrect systems. By contrast, in the A:C system Asn513 interacts with the dCTP sugar, which frees the dCTP base to move occasionally closer to A5 to form a hydrogen bond (Figure 3a, bottom). In systems with dGTP, Asn513’s side chain flips so that it can interact with guanine’s N3 atom, but the Asn513/dGTP link breaks when the dGTP rearranges in the A(syn):G and T:G systems. In the A(syn):A system, all interactions are mediated through water molecules.
Tyr505 and Phe506
Before the correct dNTP binds, Tyr505 interacts with the templating base at the gap. This sensitivity to dNTP binding is also seen in the mismatch systems, although Tyr505 continues to adopt its pre-dNTP binding (inactive) position in systems where the mismatched bases stack. In the T:G and A(syn):G systems, other changes in Tyr505 occur in response to dNTP movement. In the T:G system, Tyr505 is key in repositioning the dGTP along with Arg517. Following Arg517’s movement toward T5, Tyr505 forms a hydrogen bond to dGTP and helps reposition the dGTP as it transitions from its inactive to active position (Figure 3c and Sup. Fig. S9e). In the A(syn):G simulation, Tyr505 frequently rearranges to maintain a hydrogen bond to dGTP (Sup. Fig. S13).
In the A:A system, Phe506 moves between its active and a slanted or “sideways” orientation and this motion helps to separate dATP from the DNA and prevent its incorrect incorporation (Figure 5b). The back and forth motion of Phe506 stimulates a rotation in dATP that greatly elongates the distance between the T6:O3′ and dATP:Pα atoms (Sup. Fig S8).
3.5 Changes in Metal Ion Coordination andKey Distances for the Nucleotidyl Transfer Reaction
To understand the impact of the aforementioned DNA, dNTP, and protein changes on pol λ’s fidelity, we must consider how arrangements of the key atoms involved in the chemical reaction, namely T6:O3′, dNTP:Pα, and the Mg2+ ions and their coordinating atoms are affected. The mismatch active sites are shown in Figure 7 and key active-site geometry data are summarized in Table 3. Interestingly, the mismatch active sites can be classified into three types based on changes in the O3′–Pα distance that are: those that remain constant in length [A:C, A:G, and A(syn):G], those that switch between long and short values [A(syn):A], and those that gradually increase in length [T:G and A:A].
Figure 7.

Active site arrangements from all pol λ mismatch systems and the correct A:T system after 20 ns of simulation. Three active-site snapshots are shown from the A:A system trajectory to illustrate the evolving geometry of the active site. The coordination of the catalytic Mg2+ ion (A) and nucleotide-binding Mg2+ ion (B) with surrounding atoms is depicted by the dashed lines. Water molecules are included. Dashed green lines highlight the O3′–Pα distance.
Table 3.
Key Active-site Distance and Metal Ion Coordination Data
| System Feature | Systems | |||||
|---|---|---|---|---|---|---|
| A:C | A:G | A(syn):G | T:G | A(syn):A | A:A | |
 |
3.64 | 3.86 | 3.76 | 5.18 (decreases to 4.63 Å over 1– 18 ns) |
3.70 (increases to 5.96 Å over 3–13 ns) |
7.67 (decreases to 3.33 Å over 0– 5 ns and 4.18 Å over 5.5–16 ns) |
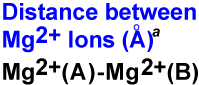 |
3.83 | 4.42 | 4.54 (decreases to 3.93 Å over 1.5– 10 ns) |
4.28 | 4.47 (decreases to 4.05 Å over 3 – 13ns)o |
3.50 |
 |
no | no | no | yes | no | bothb |
| Asp429:Oδ1 | yes | yes | yes | yes | yes | yes |
| Asp490:Oδ1 or Oδ2 | yes | yes | yes | both | no | no |
| T6:O3′ | yes | yes | yes | no | sometimes | noc |
| dNTP:O1α | yes | yes | yes | no | yes | yes |
|
Number of Water Molecules |
2 | 2 | 2 | 2 | 3 | 2 |
 |
yes | yes | yes | yes | yes | yes |
| Asp427:O | some-times | yes | some-times | no | yes | yes |
| Asp429:Oδ2 | yes | yes | yes | yes | yes | yes |
| dNTP:O1α | no | no | no | yes | no | no |
| dNTP:O1β | yes | yes | yes | yes | yes | yes |
| dNTP:O3γ | yes | yes | yes | yes | yes | yes |
|
Number of Water Molecules |
1 | 1 | 1 | 1 | 1 | 1 |
Average distances over the last nanosecond of the simulation
The second terminal Asp427 oxygen atom replaces the T6:O3′ ligand to the catalytic ion in A:A
The T6:O3′ atom briefly coordinates with the catalytic ion at the beginning of the A:A simulation
The short nucleotidyl transfer distances in the A:C, A:G, and A(syn):G systems result from the coordination of both the T6:O3′ and dNTP:O1α atoms to the catalytic ion. However, in A:G, the nucleotidyl transfer distance slips briefly to 5.8 Å, suggesting that its geometry is less stable than that in the A:C and A(syn):G systems. Longer O3′–Pα distances in the A(syn):A and A:A systems result from T6:O3′ only intermittently coordinating the catalytic ion, while in T:G, neither the T6:O3′ nor dGTP:O1α atoms coordinate the catalytic ion.
In addition, for the A(syn):A system, when the T6:O3′ atom does not coordinate the catalytic ion, the Mg2+−Mg2+ distance gets slightly smaller and the DNA tends to move more to the inactive position (Figure 4c,e). These changes are also associated with different dATP motions (Figure 4). The A(syn):A active site is also different in that it has one more active-site water molecule and an extra ion to coordinate dATP.
In most mismatch systems, the positions of the catalytic aspartate residues vary from the correct A:T system. In all systems except T:G and occasionally A:A, Asp427 is separated from the catalytic ion by a water molecule. In the A(syn):A and A:A simulations, Asp490 also does not coordinate the catalytic ion.
Although the A:A active site becomes very distorted, an unusual, but tight active-site geometry forms when the T6:O3′ atom loses its coordination to the catalytic ion and a hydrogen bond forms between T6:O3′ H and dATP:O2α (Figure 7g, middle, and Sup. Fig S8c). This active-site geometry suggests direct deprotonation of the 3′ -OH group to dATP during the chemical reaction (Figure 7g). Together, Phe506 and Arg488 help to foster this interaction. A change in the Phe506 side chain (Sup. Fig. S8a) pushes away the water molecules between T6:O3′ H and dATP:O2α and a bifurcated hydrogen bond formed between Arg488 and T6:O3′ (Sup. Fig. S7e) helps to align T6:O3′ H with dATP:O2α. After the hydrogen bond breaks between T6:O3′ H and dATP:O2α, the O3′ –Pα distance gradually increases (Sup. Fig. S8c,d).
4 Discussion
All our pol λ mismatch systems display weaker stabilization of the DNA by the enzyme, which we relate to a decrease in hydrogen bonding interactions between Arg517 and the active-site DNA. In the reference pol λ system bound to the correct A:T base pair, numerous hydrogen bonds form between Arg517 and the DNA that serve to stabilize the DNA in the active position (28, 36, 37). Not surprisingly, the reduced interactions result in some DNA motion in all mismatch systems. Previous studies have shown that changes in DNA position within the polymerase active site commonly occur when mismatches are present. X-ray crystal structures of pol β bound to incorrect dNTPs reveal a shift of the DNA template strand toward pol λ’s inactive DNA position as well as fewer interactions between the DNA and Arg283, the equivalent of Arg517 in pol λ (41). In X-ray crystal structures of A-family Bacillus DNA polymerase I fragment (BF) bound to mismatches, displacements of the DNA template strand and sometimes also the primer strand occur (64).
Depending on the pol λ mismatch system, the diminution in Arg517/DNA interactions impacts the stability of the active DNA conformation to differing degrees. For example, the loss of interactions translates into a major DNA shift from the active to inactive conformation for A:A, while it results in only a small DNA rearrangement within the active DNA conformation in the A:C system. We suggest that DNA motion is an indicator for how well pol λ will incorporate a mismatch. In this measure, increased DNA motion corresponds to decreased dNTP insertion. This is similar to the hampered thumb closing motion captured in simulations of pol β and pol X bound to mismatches (42, 44). Our analysis also suggests that the extent of DNA motion reflects the degree of variation in the active-site compared to the arrangement with the correct A:T base pair. Specifically, changes in the mismatch geometry, DNA pairing, and protein residue side-chain orientations are key factors in determining how much DNA motion occurs. Other unusual interactions such as those between DNA template-strand bases and Lys273 from the 8-kDa domain that occur in the A(syn):G, A(syn):A, and A:A systems may also serve to destabilize the DNA.
Using the number of dNTP and protein residue changes as a guide, we hypothesize why the DNA motion follows the trend: A:C < A:G < T:G < [A(syn):A ~ A(syn):G] < A:A. For the A:C system, minor dCTP and protein motions result in a small DNA shift from the active DNA position. For the A:G system, mismatch base stacking and larger protein rearrangements in Arg514 and Tyr505 result in slightly more DNA motion compared to the A:C case.
Unusual or unstable active-site DNA interactions produce much more DNA motion in the other mismatch systems. Within this group, the T:G system shows the least DNA motion because the active site remains intact while T5 and dGTP show wobble base pairing. When this base pairing ends, protein and dGTP changes occur that stimulate much more DNA motion toward the inactive position. The increased DNA motion in A(syn):A results from the unusual pairing in the active site between A6 and dATP. In addition, frequent changes in dATP occur and several active-site protein residues flip to their inactive positions. In the A(syn):G system, frequent dGTP rearrangement to a “sideways” orientation causes the two adjacent base pairs to break apart which brings about significant DNA motion. The full DNA movement to the inactive position in the A:A system corresponds to mismatch stacking and A5 pairing with the primer terminus which leads to a rotation in dATP within the active site. Protein side-chain motions also increase the active-site disorder.
Our results suggest that the motions of the dNTP are heavily influenced by the electrostatic potential surface contours of the active site. Generally, the dNTP sugar ring is less well stabilized in the context of a mismatch than a correct base pair. In addition, the guanine base of dGTP in the mismatch systems is also poorly supported because of the proximity of the base’s 2-amino group to a positive region in the active sites’s electrostatic potential surface, which includes Arg517. Similar unfavorable interactions in pol β, the Arg283Lys pol β mutant, and B-family polymerases, pol α and Herpes Simplex Virus 1 DNA polymerase, affect dGTP insertion by these enzymes (54, 65, 66).
In pol λ, unfavorable electrostatic interactions between Arg517 and the dNTP result in substantial dNTP rearrangements such as those in the T:G and A(syn):G systems. As is evident from the A:G system, hydrogen bond formation between the incorrect dNTP and the DNA can help to stabilize the dNTP within the active site. In this way, Arg517 acts in a base-checking role. In the T:G and A(syn):A systems, Arg517 approaches the dNTP the most closely as it moves toward the templating base position. Interestingly, Arg283 also appears to move close to the incorrect incoming nucleotides in X-ray crystal structures of pol β bound to G:A and C:A mismatches (41) and a new Arg283/dNTP hydrogen bonding is observed in binary pol β complexes bound to nicked DNA with mismatches located in the nascent base pair binding position (39). In relation to these pol β structures, it is hypothesized that the repositioning of Arg283 into the templating base position may serve to deactivate the system since it accompanies a shift in the DNA (41). Thus, Arg517 or Arg283 appear to play a strong role in dNTP insertion. A base-checking role for pol β’s Arg283 has been known for some time since the Arg283Ala pol β mutant exhibits a substantially reduced fidelity (53, 54, 67). Our prior studies of the Arg517Ala pol λ mutant also suggested that the mutant would have a reduced fidelity as compared to the wild-type polymerase because significant DNA shifting occurred between the active and inactive DNA positions (28).
Studies of pol β and pol X mismatch systems show that the more distortion in the active site, the less likely the mismatch will be incorporated (42, 44). Thus, the overall effect of the DNA motion and active-site rearrangements in pol λ lies in the resulting geometry of the atoms directly involved in the chemical reaction. A comparison with the correct A:T system indicates that the A:C active site is the tightest. However, it differs from the A:T active site in one important respect: it is missing Asp427 coordination to the catalytic ion. The A:G active site is very similar to A:C, but the O3′−Pα and Mg2+–Mg2+ distances are slightly longer. Surprisingly, the A(syn):G system’s active site is remarkably similar to these systems. The significantly more DNA motion and dGTP changes with A(syn):G make A:G preferred for insertion between these two forms. The T:G system has an ~ 1 Å longer O3′–Pα distance that increases to 5.18 Å after dGTP flips. This system also exhibits a break from the catalytic ion coordination pattern observed for A:C, A:G, and A(syn):G, and most closely resembles the active-site geometry in the pol λ simulation with the correct A:T base pair (28) since Asp427 and Asp490 coordinate the catalytic ion. The T:G mismatch is difficult to evaluate because less extensive DNA motion occurs although the active-site evolves into a more disordered state during the 20 ns trajectory. Since the DNA motion is somewhat more pronounced relative to A:G, but still close to the active position, we expect T:G to be incorporated less well than A:C, A:G, or A(syn):G.
Compared to the other mismatch systems, A(syn):A and A:A have the most unusual active-site organizations and display the least stability in their arrangements. Most noticeable are the large changes in the O3′–Pα distance. For A:A, the distance changes are directly related to Phe506 and dATP conformational changes. As the motions in these residues increase, so does the O3′–Pα distance. Prior to the motions, a tight active-site geometry forms where O3′H poses for transfer to the dATP:O2α atom. By contrast, the A(syn):A active site fluctuates between disorganized and more ordered active-site geometries depending on subtle changes in the dATP. These larger changes in the A:A and A(syn):A systems suggest that these mismatches will be much less efficiently incorporated by pol λ. However, between these adenine:adenine pairings, we expect the syn orientation of the template adenine to be preferred over the anti orientation since good active-site geometries are assumed more frequently and the DNA is not always in the inactive position.
Significantly our inferred trend sequence for nucleotide incorporation by pol λ (i.e., A:T > A:C > A:G > A(syn):G > T:G > A(syn):A > A:A) mirrors the observed trends in the reaction kinetics data (10) for nucleotide insertion opposite template adenine as summarized in Table 4. Indeed, the mismatches can be arranged similarly according to reaction rate constant, catalytic efficiency, and fidelity values. The notably different T:G kinetic data indicates that this mismatch is inserted faster and more efficiently than A:C. This difference may result from the mismatch insertion context. DNA polymerase selectivity has been known to depend not only on the mispair composition, but also on the surrounding sequence (68). For example, error rate data obtained from a short gap reversion assay (45) summarized in Table 4 suggest a different trend: A:C > T:G ≫ [A:G ~ A:A]. The nature of the primer terminus base pair is important because the stacking interactions between the T:G mismatch and adjacent base pairs have been shown to influence DNA duplex stability (69, 70). In the T:G system, the A:T primer terminus base pair does not stack as well with the T:dGTP mismatch as when the template base of the mismatch is an adenine since purine-purine interactions tend to be stronger (71).
Table 4.
Reaction Kinetics and Error Rate Data Pertaining to Base Substitution Errors
|
| |||
|---|---|---|---|
| Enzyme Complex |
Nucleotide Incorporation Rate Constant kp (s−1) |
Incorporation Efficiency kp/Kd (M−1s−1) |
Fidelityb |
| A:dTTP | 3.9 | 1.5×106 | 1 |
| A:dCTP | 0.0052 | 5.0×102 | 3.3×10−4 |
| A:dGTP | 0.00040 | 1.3×102 | 8.7×10−5 |
| A:dATP | 0.000034 | 1.8×101 | 1.2×10−5 |
| T:dGTP | 0.010 | 1.4×103 | 8.8×10−4 |
|
| |||
| Enzyme Complex |
Error Rate ×10−4 | ||
| A:dCTP | 5.3 | ||
| A:dGTP | 0.19 | ||
| A:dATP | 0.19 | ||
| T:dGTP | 4.5 | ||
Overall, our analyses show that fidelity in pol λ is a dynamic process involving not just rearrangements of the active site but changes in several important residues and variations in overall polymerase/DNA motion. The importance of active-site protein residue motions in the fidelity of polymerases has been previously highlighted. These motions are hypothesized to act as gate keepers that control the assembly of the active site for the chemical reaction (29). In pol λ, the altered interactions of Arg517 with the dNTP destabilize the active DNA conformation and lead to rearrangements in other protein side chains that disrupt the formation of a suitable active site for the chemical reaction. Tyr505 and Phe506 also emerge as being important “gates” or checkpoints in pol λ’s fidelity from their rearrangements in the A:A and T:G systems. A similar phenomenon occurs in pol β since structural data of pol β suggest that the side-chain conformations of Tyr271 and Phe272, analogous residues to pol λ’s Tyr505 and Phe506, respectively, determine the active or inactive state of the enzyme (39). Furthermore, Phe272Leu pol β mutant studies reveal increased base substitution errors compared to wild-type pol β as a result of a lower discrimination against incorrect dNTPs during ground-state binding (72). In sum, pol β discriminates between the correct and incorrect dNTP by more frequent DNA motion toward the inactive position and changeable active-site geometries. These changes occur when mismatches are present in the active site and deactivate the enzyme/substrate complex.
Supplementary Material
Acknowledgement
Research described in this article was supported in part by Philip Morris USA Inc. and Philip Morris International and by NSF grant MCB-0316771, NIH grant R01 ES012692, and the American Chemical Society’s Petroleum Research Fund award (PRF #39115-AC4) to T. Schlick. The computations are made possible by support for the SGI Altix 3700 by the National Center for Supercomputing Applications (NCSA) under grant MCA99S021, and by the NYU Chemistry Department resources under grant CHE-0420870. M. F. acknowledges funding from NYU’s Kramer fellowship for the 2008–2009 academic year. Molecular images were generated using the INSIGHTII package (Ac-celrys Inc., San Diego, CA), VMD (73), and the PyMOL Molecular Graphics System (DeLano Scientific LLC, San Carlos, CA).
List of Abbreviations
- pol λ
polymerase λ
- pol β
polymerase β
- dNTP
2′-deoxyribonucleoside 5′-triphosphate
- A6:T6
template adenine and primer thymine bases, respectively, of the primer terminus base pair
- A5
A(syn)5
- or T5
templating adenine or thymine base at the DNA gap
Footnotes
Supporting Information Available: Fourteen supplementary figures referenced in the main text of the paper and additional information regarding mismatch hydrogen bonding changes and their relationship to experimental data is included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Steitz TA. J. Biol. Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel TA. J. Biol. Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 3.Moon AF, Garcia-Diaz M, Batra VK, Beard WA, Bebenek K, Kunkel TA, Wilson SH, Pedersen LC. DNA Repair. 2007;6:1709–1725. doi: 10.1016/j.dnarep.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang W. Curr. Opin. Struct. Biol. 2003;13:23–30. doi: 10.1016/s0959-440x(02)00003-9. [DOI] [PubMed] [Google Scholar]
- 5.Lamarche BJ, Kumar S, Tsai M-D. Biochemistry. 2006;45:14826–14833. doi: 10.1021/bi0613325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson RE, Todd Washington M, Haracska L, Prakash S, Prakash L. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 7.Tissier A, McDonald JP, Frank EG, Woodgate R. Genes and Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Yuan F, Wu X, Wang Z. Mol. Cell. Biol. 2000;20:7099–7108. doi: 10.1128/mcb.20.19.7099-7108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Todd Washington M, Johnson RE, Prakash L, Prakash S. Mol. Cell. Biol. 2004;24:936–943. doi: 10.1128/MCB.24.2.936-943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiala KA, Duym WW, Zhang J, Suo Z. J. Biol. Chem. 2006;281:19038–19044. doi: 10.1074/jbc.M601178200. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto Y, Kim K. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Diaz M, Bebenek K, Kunkel TA, Blanco L. J. Biol. Chem. 2001;276:34659–34663. doi: 10.1074/jbc.M106336200. [DOI] [PubMed] [Google Scholar]
- 13.Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braithwaite EK, Kedar PS, Lan L, Polosina YY, Asagoshi K, Poltoratsky VP, Horton JK, Miller H, Teebor GW, Yasui A, Wilson SH. J. Biol. Chem. 2005;280:31641–31647. doi: 10.1074/jbc.C500256200. [DOI] [PubMed] [Google Scholar]
- 15.Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. J. Biol. Chem. 2005;280:18469–18475. doi: 10.1074/jbc.M411864200. [DOI] [PubMed] [Google Scholar]
- 16.Lebedeva NA, Rechkunova NI, Dezhurov SV, Khodyreva SN, Favre A, Blanco L, Lavrik OI. Biochim. Biophys. Acta. 2005;1751:150–158. doi: 10.1016/j.bbapap.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 17.Tano K, Nakamura J, Asagoshi K, Arakawa H, Sonoda E, Braithwaite EK, Prasad R, Buerstedde J-M, Takeda S, Watanabe M, Wilson SH. DNA Repair. 2007;6:869–875. doi: 10.1016/j.dnarep.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JW, Blanco L, Zhou T, Garcia-Diaz M, Bebenek K, Kunkel TA, Wang Z, Povirk LF. J. Biol. Chem. 2004;279:805–811. doi: 10.1074/jbc.M307913200. [DOI] [PubMed] [Google Scholar]
- 19.Fan W, Wu X. Biochem. Biophys. Res. Commun. 2004;323:1328–1333. doi: 10.1016/j.bbrc.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, Koiwai O, Hsieh C-L, Schwarz K, Lieber MR. Mol. Cell. 2004;16:701–713. doi: 10.1016/j.molcel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 21.Nick McElhinny SA, Havener JM, Garcia-Diaz M, Juarez R, Bebenek K, Kee BL, Blanco L, Kunkel TA, Ramsden DA. Mol. Cell. 2005;19:357–366. doi: 10.1016/j.molcel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Capp J-P, Boudsocq F, Bertrand P, Laroche-Clary A, Pourquier P, Lopez BS, Cazaux C, Hoffmann J-S, Canitrot Y. Nucleic Acids Res. 2006;34:2998–3007. doi: 10.1093/nar/gkl380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 24.Arora K, Schlick T. Biophys. J. 2004;87:3088–3099. doi: 10.1529/biophysj.104.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arora K, Schlick T. J. Phys. Chem. B. 2005;109:5358–5367. doi: 10.1021/jp0446377. [DOI] [PubMed] [Google Scholar]
- 26.Radhakrishnan R, Schlick T. Proc. Natl. Acad. Sci. USA. 2004;101:5970–5975. doi: 10.1073/pnas.0308585101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. Nat. Struct. Mol. Biol. 2005;12:97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 28.Foley MC, Arora K, Schlick T. Biophys. J. 2006;91:3182–3195. doi: 10.1529/biophysj.106.092080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radhakrishnan R, Arora K, Wang Y, Beard WA, Wilson SH, Schlick T. Biochemistry. 2006;45:15142–15156. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radhakrishnan R, Schlick T. Biochem. Biophys. Res. Commun. 2006;350:521–529. doi: 10.1016/j.bbrc.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin P, Pedersen LC, Batra VK, Beard WA, Wilson SH, Pedersen LG. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13294–13299. doi: 10.1073/pnas.0606006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bojin MD, Schlick T. J. Phys. Chem. B. 2007;111:11244–11252. doi: 10.1021/jp071838c. [DOI] [PubMed] [Google Scholar]
- 33.Alberts IL, Wang Y, Schlick T. J. Am. Chem. Soc. 2007;129:11100–11110. doi: 10.1021/ja071533b. [DOI] [PubMed] [Google Scholar]
- 34.Andres Cisneros G, Perera L, Garcia-Diaz M, Bebenek K, Kunkel TA, Pedersen LG. DNA Repair. 2008;7:1824–1834. doi: 10.1016/j.dnarep.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Cell. 2006;124:331–342. doi: 10.1016/j.cell.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 36.Bebenek K, Garcia-Diaz M, Foley MC, Pedersen LC, Schlick T, Kunkel TA. EMBO Reports. 2008;9:459–464. doi: 10.1038/embor.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foley MC, Schlick T. J. Am. Chem. Soc. 2008;130:3967–3977. doi: 10.1021/ja077982t. [DOI] [PubMed] [Google Scholar]
- 38.Bebenek K, Garcia-Diaz M, Blanco L, Kunkel TA. J. Biol. Chem. 2003;278:34685–34690. doi: 10.1074/jbc.M305705200. [DOI] [PubMed] [Google Scholar]
- 39.Krahn JM, Beard WA, Wilson SH. Structure. 2004;12:1823–1832. doi: 10.1016/j.str.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structure. 2005;13:1225–1233. doi: 10.1016/j.str.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Mol. Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arora K, Beard WA, Wilson SH, Schlick T. Biochemistry. 2005;44:13328–13341. doi: 10.1021/bi0507682. [DOI] [PubMed] [Google Scholar]
- 43.Radhakrishnan R, Schlick T. J. Am. Chem. Soc. 2005;127:13245–13252. doi: 10.1021/ja052623o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sampoli Benitez BA, Arora K, Balistreri L, Schlick T. J. Mol. Biol. 2008;384:1086–1097. doi: 10.1016/j.jmb.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Diaz M, Bebenek K, Sabariegos R, Dominguez O, Rodriguez J, Kirchhoff T, Garcia-Palomero E, Picher AJ, Juarez R, Ruiz JF, Kunkel TA, Blanco L. J. Biol. Chem. 2002;277:13184–13191. doi: 10.1074/jbc.M111601200. [DOI] [PubMed] [Google Scholar]
- 46.Fiala KA, Abdel-Gawad W, Suo Z. Biochemistry. 2004;43:6751–6762. doi: 10.1021/bi049975c. [DOI] [PubMed] [Google Scholar]
- 47.Maga G, Shevelev I, Villani G, Spadari S, Hubscher U. Nucl. Acids Res. 2006;34:1405–1415. doi: 10.1093/nar/gkl032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picher AJ, Garcia-Diaz M, Bebenek K, Pedersen LC, Kunkel TA. Nucleic Acids Res. 2006;34:3259–3266. doi: 10.1093/nar/gkl377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stote RH, Karplus M. Proteins. 1995;23:12–31. doi: 10.1002/prot.340230104. [DOI] [PubMed] [Google Scholar]
- 50.MacKerell AD., Jr J. Phys. Chem. B. 1997;101:646–650. [Google Scholar]
- 51.Yang L, Arora K, Beard WA, Wilson SH, Schlick T. J. Amer. Chem. Soc. 2004;126:8441–8453. doi: 10.1021/ja049412o. [DOI] [PubMed] [Google Scholar]
- 52.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 53.Beard WA, Osheroff WP, Prasad R, Sawaya MR, Jaju M, Wood TG, Kraut J, Kunkel TA, Wilson SH. J. Biol. Chem. 1996;271:12141–12144. doi: 10.1074/jbc.271.21.12141. [DOI] [PubMed] [Google Scholar]
- 54.Osheroff WP, Beard WA, Wilson SH, Kunkel TA. J. Biol. Chem. 1999;274:20749–20752. doi: 10.1074/jbc.274.30.20749. [DOI] [PubMed] [Google Scholar]
- 55.Qian X, Strahs D, Schlick T. J. Comput. Chem. 2001;22:1843–1850. doi: 10.1002/jcc.1135. [DOI] [PubMed] [Google Scholar]
- 56.Klapper I, Hagstrom R, Fine R, Sharp K, Honig B. Proteins. 1986;1:47–59. doi: 10.1002/prot.340010109. [DOI] [PubMed] [Google Scholar]
- 57.MacKerell A, Jr, Banavali NK. J. Comput. Chem. 2000;21:105–120. [Google Scholar]
- 58.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feller SE, Zhang Y, Pastor RW, Brooks BR. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 60.Ryckaert J-P, Ciccotti G, Berendsen HJC. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 61.Darden TA, York DM, Pedersen LG. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 62.Sharp KA, Honig B, Harvey SC. Biochemistry. 1990;29:340–346. doi: 10.1021/bi00454a006. [DOI] [PubMed] [Google Scholar]
- 63.Chin K, Sharp KA, Honig B, Pyle AM. Nature Struct. Biol. 1999;6:1055–1061. doi: 10.1038/14940. [DOI] [PubMed] [Google Scholar]
- 64.Johnson SJ, Beese LS. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 65.Patro JN, Urban M, Kuchta RD. Biochemistry. 2009;48:180–189. doi: 10.1021/bi801823z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cavanaugh NA, Urban M, Beckman J, Spratt TE, Kuchta RD. Biochemistry. 2009 doi: 10.1021/bi8022202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahn J, Werneburg BG, Tsai M-D. Biochemistry. 1997;36:1100–1107. doi: 10.1021/bi961653o. [DOI] [PubMed] [Google Scholar]
- 68.Kunkel TA, Bebenek K. Biochim. Biophys. Acta. 1988;951:1–15. doi: 10.1016/0167-4781(88)90020-6. [DOI] [PubMed] [Google Scholar]
- 69.Allawi HT, SantaLucia J., Jr Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 70.Allawi HT, SantaLucia J., Jr Nucleic Acids Research. 1998;26:4925–4934. doi: 10.1093/nar/26.21.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Browne DT, Eisinger J, Leonard NJ. J. Am. Chem. Soc. 1968;90:7302–7323. doi: 10.1021/ja01028a023. [DOI] [PubMed] [Google Scholar]
- 72.Li SX, Vaccaro JA, Sweasy JB. Biochemistry. 1999;38:4800–4808. doi: 10.1021/bi9827058. [DOI] [PubMed] [Google Scholar]
- 73.Humphrey W, Dalke A, Schulten K. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.