

Abstract
Retro iminonitroso Diels-Alder reactions were investigated in both solution and solid phase. In thermal or Cu(I)-mediated reactions, interconversion of various nitroso cycloadducts occurred in the presence of separate dienes. Up to 99% of conversion was observed. Use of chiral ligands in the Cu(I)-medicated reactions gave new cycloadducts enantioselectively.
Since its discovery in 1947, the nitroso Diels-Alder reaction (NDA) has become a powerful synthetic tool for the formation of 1-amino-4-hydroxy-2-ene derivatives in one step.1 Many groups have continued to develop and apply nitroso Diels-Alder reactions in organic syntheses.2 We successfully used nitroso Diels-Alder reactions to obtain cycloadducts as versatile building blocks for a variety of synthetic applications, including preparation of carbocyclic nucleoside analogs and novel natural product scaffolds.3
The retro Diels-Alder reaction (retro DA) has been known since the discovery of the forward DA itself and has been used to generate a wide variety of reactive species in a synthetically useful manner.4 The retro DA strategy has been of considerable utility in promoting acylnitroso cycloaddition reactions. For example, it has been used to store in situ generated transient and unisolable acylnitroso agents 1 by cycloaddition with dimethylanthracene (DMA).5 To avoid the direct use of oxidant, the resultant cycloadducts can be thermolized at relatively low temperature to release the free acylnitroso species 1 (Scheme 1). However, very few examples have been reported regarding retro nitroso Diels-Alder reactions (retro NDA) related to aryl or heteroaryl nitroso agents.6 Studies of retro NDA reactions would benefit understanding of reaction mechanisms and extend their synthetic use. We envisioned that the dissociation of nitroso cycloadducts might occur under thermal or Lewis acid-mediated conditions to generate new nitroso cycloadduct scaffolds in the presence of added dienes. Herein we describe the preliminary results of this study in both solution and solid phase.
Scheme 1.

Nitroso Diels-Alder and retro acylnitroso Diels-Alder reaction.
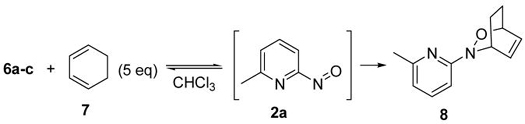
Three structurally differentiated nitroso cycloadducts 6a–c were obtained from NDA reactions between 6-methyl-2-nitrosopyridine 2a and colchicine 3, 1,3-cyclopentadiene 4 and ergosterol 5, respectively (Scheme 2).3e We chose those cycloadducts for initial investigations of the retro NDA reactions since, in our early report, we noticed that cycloaddition with colchicine was a reversible process.3e The experiments were conducted by treating cycloadducts 6a–c with 5.0 equivalents of 1,3-cyclohexadiene 7 at both 25 °C and 50 °C.7 As shown in Table 1, dissociations of colchicine adduct 6a and ergosterol adduct 6c at 25 °C were observed and in situ trapping of the released nitroso species 2a with 7 gave adduct 8 in 43% and 20% yield, respectively (entries 1 and 5). Not surprisingly, at elevated temperature (50 °C), complete retro NDA reactions of 6a and 6c occurred within several hours, affording adduct 8 in good yields (entries 2 and 6). In the presence of a large excess of 7, the net reaction of 6c was accelerated and compound 8 was obtained in 54% yield at 25 °C (entry 7). In contrast, no retro NDA reaction was detected for adduct 6b at 25 °C or even at high temperature (entries 3 and 4). These observations clearly indicated that structure and configuration played important roles in the related retro NDA reactions. Compared to their parent natural products or adduct 6b, NDA adducts 6a and 6c are more sterically congested.
Scheme 2.

Nitroso Diels-Alder reaction with various dienes.
Table 1.
Retro NDA reaction of iminonitroso cycloadducts 6a–c
 | ||||
|---|---|---|---|---|
| Entry | Adduct | Temp. (°C) | Time (h) | 8 (%)a |
| 1 | 6a | 25 | 24 | 43 |
| 2 | 6a | 50 | 5 | 89 |
| 3 | 6b | 25 | 24 | 0 |
| 4b | 6b | >50 | - | 0 |
| 5 | 6c | 25 | 24 | 20 |
| 6 | 6c | 50 | 2 | 86 |
| 7c | 6c | 25 | 24 | 54 |
Isolated yields reported.
Temperature was increased from 50 °C to 70 °C (reflux).
CHCl3/1,3-cyclohexadiene 7 (10% v:v) as solvent.
Heating 6a and 6b in the presence of 7 at 50 °C produced only 89% and 86% of adduct 8 even though no starting compounds (6a and 6c) were recovered (entries 2 and 6, Table 1). This encouraged further study and revealed that a small portion of released nitroso 2a underwent reaction to form azo-oxy compound 9.8 To the best of our knowledge, the formation of 9 directly from a pyridinylnitroso precursor has not been reported. Further analyses of solutions of 6-methyl-2-nitrosopyridine, 2a, showed that while 2a existed in equilibrium with the corresponding azodioxy dimer,9 azo-oxy compound 9 slowly formed upon standing in pure organic solvent10 (Figure 1).
Figure 1.

Solution state equilibrium of 6-methyl-2-nitrosopyridine 2a and generation of 9.
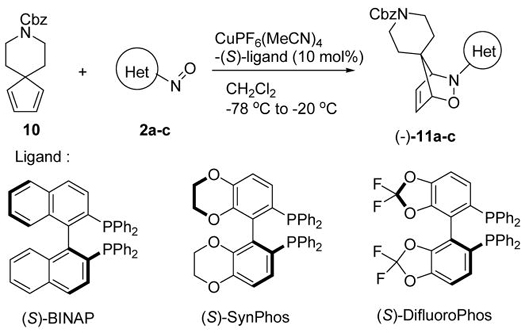
The retro iminonitroso Diels-Alder reaction was also observed when we subjected racemic spirocyclic adduct 11b3d to the catalytic asymmetric iminonitroso Diels-Alder reaction conditions. Trapping of nitroso agent 2b generated from the degradation of 11b with 1,3-cyclohexadiene 7 in the presence of Cu(I)PF6(MeCN)4 and (S)-BINAP produced cycloadduct 12 with moderate enantioselectivity (19% yield, 52% ee, Scheme 3).11 The absolute stereochemistry was proposed based on the literature-based mechanistic model.12
Scheme 3.

Retro NDA reaction of spirocyclic adduct 11b.
To minimize the competing non-catalytic asymmetric iminonitroso Diels-Alder reaction process, work-up temperature was modified from room temperature to −20 °C. By doing this, cycloadduct 11b was obtained in 74% yield and with an improved ee value (entries 1 and 2, Table 2). Under these optimized conditions, an extended study was performed using different iminonitroso reagents 2a–c and chiral phosphine ligands.13 Use of chiral ligand (S)-DifluoroPhos and 6-methyl-2-nitrosopyridine, 2a, provided spirocyclic adduct 11a with the highest ee value (96% ee, entry 5). A dramatically decreased ee value was observed when 5-methyl-3-nitrosoisoxazole, 2c, was used (28% ee, entry 6).
Table 2.
NDA Reaction with various iminonitroso agents and chiral phosphine ligands
 | ||||
|---|---|---|---|---|
| Entry | Nitroso | Ligand | 11c (%) | ee (%)d |
| 1a |
2b 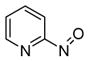
|
(S)-BINAP | 11b, 71 | 51 |
| 2b |
2b 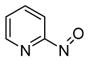
|
(S)-BINAP | 11b, 74 | 65 |
| 3b |
2b 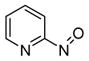
|
(S)-DifluoroPhos | 11b, 75 | 86 |
| 4b |
2a 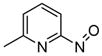
|
(S)-SynPhos | 11a, 85 | 92 |
| 5b |
2a 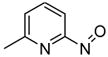
|
(S)-DifluoroPhos | 11a, 74 | 96 |
| 6b |
2c 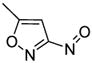
|
(S)-DifluoroPhos | 11c, 65 | 28 |
Reaction was worked up at room temperature.
Reaction was worked up at −20 °C.
Isolated yields reported.
Determined by Chiral HPLC.
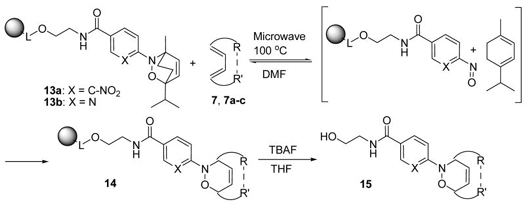
Retro Diels-Alder reactions were also attempted in solid phase using NDA adducts attached to resin. The adduct resins 13a and 13b,14 derived from α-terpinene, were exposed to various dienes in a microwave cavity for 5 min, then products 15 were cleaved from the resin with TBAF and analyzed by LC/MS.15 The results are summarized in Table 3. While the nitrophenyl NDA adduct 13a provided quantitative retro-NDA adducts with all tested dienes, the pyridyl derivative 13b was less reactive under the same conditions (entries 1–6, Table 3). Reactions with unsymmetric dienes such as 2,4-hexadien-1-ol, 7b, and 1-methoxy-1,3-cyclohexadiene, 7c, gave cycloadducts as mixtures of two regioisomers in good to excellent yields (entries 4–6).
Table 3.
Retro HDA reaction of resin-bounded α-terpinene adducts with various dienes
 | |||
|---|---|---|---|
| Entry | Adduct | Diene | 15 (%)a |
| 1 | 13a |
7 |
99 |
| 2 | 13b |
7 |
81 |
| 3 | 13a |
7a |
99 |
| 4 | 13a |
7b |
99 (6:4) |
| 5 | 13a |
7c |
99 (> 9:1) |
| 6 | 13b |
7c |
75 (6:4) |
Determined by LC/MS.
In summary, we found that various iminonitroso cycloadducts can undergo retro nitroso Diels-Alder reactions in both solution and solid phase under thermal or Cu(I)-mediated conditions. By trapping the released nitroso dienophiles with separate dienes, new adduct scaffolds were generated in an efficient fashion. Use of chiral ligands in the Cu(I)-medicated reactions gave new cycloadducts enantioselectively. We hope that the chemistry we describe here can help expand the use of retro iminonitroso Diels-Alder reactions in organic syntheses.
Acknowledgments
We gratefully acknowledge support from the NIH (GM 075885). We thank the Lizzadro Magnetic Resonance Research Center at Notre Dame for NMR facilities and Nonka Sevova for mass spectrometry analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Wichterle O. Collect Czech Chem Commun. 1947;12:292. [Google Scholar]; (b) Arbuzov YA. Dokl Akad Nauk SSSR. 1948;60:993. [Google Scholar]
- 2.For reviews, see: Streith J, Defoin A. Synthesis. 1994;11:1107.Vogt PF, Miller MJ. Tetrahedron. 1998;54:1317.Yamaoto H, Momiyama N. Chem Commun. 2005:3514. doi: 10.1039/b503212c.Yamaoto Y, Yamamoto H. Eur J Org Chem. 2006:2031.Samarakoon T, Hanson RR. Chemtracts. 2007;20:220.
- 3.For relevant publications, see: Ghosh A, Ritter A, Miller MJ. J Org Chem. 1995;60:5808.Zhang DY, Sueling C, Miller MJ. J Org Chem. 1998;63:885. doi: 10.1021/jo971696q.Li FZ, Brogan JB, Gage JL, Zhang DY, Miller MJ. J Org Chem. 2004;69:4538. doi: 10.1021/jo0496796.Lin WM, Gupta A, Kim KH, Mendel D, Miller MJ. Org Lett. 2009;11:449. doi: 10.1021/ol802553g.Li FZ, Yang BY, Miller MJ, Zajicek J, Noll BC, Möllmann U, Dahse HM, Miller PA. Org Lett. 2007;7:2923. doi: 10.1021/ol071322b.Krchnak V, Waring KR, Noll Br C, Möllmann U, Dahse H-M, Miller MJ. J Org Chem. 2008;73:4559. doi: 10.1021/jo8004827.
- 4.For reviews, see: Kwart H, King K. Chem Rev. 1968;68:415.Ripoll JL, Rouessac A, Rouessac F. Tetrahedron. 1978;34:19.Rickborn B. Org React. 1998;53:223.Stajer G, Csede F, Fulop F. Current Org Chem. 2003;7:1423.For recent examples, see: Boul PJ, Reutenauer P, Lehn JM. Org Lett. 2005;7:15. doi: 10.1021/ol048065k.Snyder SA, Kontes F. J Am Chem Soc. 2009;131:1745. doi: 10.1021/ja806865u.
- 5.For an early example, see: Horsewood P, Kirby GW, Sharma RP, Sweeny JG. J Chem Soc Perkin Trans I. 1983:1802.
- 6.(a) Gamenara D, Dias E, Tancredi N, Herinzen H, Moyna P, Forbes E. J Braz Chem Soc. 2001;4:489. [Google Scholar]; (b) Wanner MJ, Koomen GJ. J Chem Soc Perkin Trans I. 2001;16:1908. [Google Scholar]
- 7.General procedures: To a solution of cycloadduct 6 (0.05 mmol) in CHCl3 (2 mL) was added 1,3-cyclohexadiene 7 (24 uL, 0.25 mmol). The reaction mixture was stirred at room temperature for 24 h, or heated at 50 °C and monitored by TLC until 6 was consumed. The solvent was removed under reduced pressure and the crude product was purified by silica gel chromatography (hexanes:EtOAc = 5:1) to yield compound 8 as a yellowish solid.
- 8.Spectral data of 9 (yellowish solid): mp: 85–87 °C; 1H NMR (500 MHz, CDCl3) δ 8.22 (d, J = 8.2 Hz, 1 H), 8.17 (d, J = 8.2 Hz, 1 H), 7.80 (m, 1 H), 7.76 (m, 1 H), 7.42 (d, J = 7.6 Hz, 1 H), 7.17 (d, J = 7.6 Hz, 1 H), 2.70 (s, 3 H), 2.63 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 158.8, 158.7, 157.1, 155.8, 139.2, 138.2, 127.0, 123.8, 115.6, 114.9, 24.6, 24.5; (FAB) MS: 229 [M+1]+.
- 9.Gowenlock BG, Maidment MJ, Orrell KG, Sik V, Giuseppe M, Vasapollo G, Hursthouse MN, Malik KMA. J Chem Soc Perkin Trans II. 2000:2280. [Google Scholar]
- 10.For detailed discussions of formation of azo-oxy compounds from nitrosobenzene, see: Zuman P, Shah B. Chem Rev. 1994;94:1621.
- 11.Cu(I)-mediated retro NDA reaction with spirocyclic adduct 11b: To an oven dried round-bottomed flask was added anhydrous CH2Cl2 (5 mL), Cu(I)PF6(CH3CN)4 (9 mg, 0.025 mmol,) and (S)-BINAP (16 mg, 0.025 mmol) in order. After stirring for 0.5 h under Ar, the reaction mixture was cooled to −78 °C, racemic compound 11b (94 mg, 0.25 mmol) and 1,3-cyclohexadiene (0.1 mL) were added. The solution was gradually warmed to room temperature over 2 h. The crude product was chromatographed on silica gel (hexanes:EtOAc = 5:1) to afford 11b (70 mg, yield 75%, ee = 24%) as a white solid and 12 (9 mg, yield 19%, ee = 52%) as a colorless oil, respectively. Enantiomeric excess was determined by HPLC with Chiralcel AD-H column (95:5 hexanes:2-propanol), 1.0 mL/min, for 11b, major enantiomer tr = 19.6 min, minor enantiomer tr = 18.0 min; for 12, major enantiomer tr = 14.8 min, minor enantiomer tr = 10.9 min.
- 12.(a) Yamamoto Y, Yamamoto H. J Am Chem Soc. 2004;41:4128. doi: 10.1021/ja049849w. [DOI] [PubMed] [Google Scholar]; (b) Yamamoto Y, Yamamoto H. Angew Chem, Int Ed Engl. 2005;44:7082. doi: 10.1002/anie.200501345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.General procedures: To a oven-dried round-bottomed flask was added anhydrous CH2Cl2 (4 mL), Cu(I)PF6(CH3CN)4 (9 mg, 0.025 mmol,), and chiral phosphine ligand (0.025 mmol) in order under Ar. After stirring for 1 h, the reaction mixture was cooled to −78 °C, nitroso compound 2 (0.30 mmol) was added. The resulting dark blue mixture was stirred for 10 min, then spriocyclic diene 10 (0.10 g, 0.36 mmol) in CH2Cl2 (0.5 mL) was added dropwise. The solution was gradually warmed to −20 °C over 2 h and chromatographed immediately on silica gel to afford nitroso-Diels-Alder adduct 11. Compound 11a (colorless oil): 1H NMR (300 MHz, CDCl3) δ 7.31 (m, 6 H), 6.64 (d, J = 6.9 Hz, 1 H), 6.58 (d, J = 7.2 Hz, 1 H), 5.14 (s, 3 H), 4.74 (s, 1 H), 3.59 (m, 4 H), 2.43 (s, 3 H), 2.03 (m, 4 H); 13C NMR (75 MHz, CDCl3) δ 137.7, 134.3, 130.2, 128.5, 128.0, 127.9, 116.4, 108.7, 86.2, 70.4, 67.0, 60.1, 42.2, 41.7, 29.2; HPLC: Chiralcel AD-H column (95:5 hexanes:2-propanol), 1.0 mL/min, major enantiomer tr = 14.6 min, minor enantiomer tr = 15.6 min; Compound 11b (white solid, mp: 123–124 °C): 1H NMR (300 MHz, CDCl3) δ 8.19 (dd, J = 3.9, 4.8, 1 H), 7.49 (m, 1 H), 7.36 (m, 6 H), 6.78 (d, J = 12 Hz, 1 H), 6.21 (m, 1 H), 6.04 (m, 1 H), 5.14 (s, 3 H), 4.74 (s, 1 H), 3.52 (m, 4 H), 2.04 (m, 4 H); 13C NMR (75 MHz, CDCl3) δ 147.1, 146.8, 139.2, 137.3, 134.1, 130.3, 128.3, 127.8, 127.6, 125.5, 118.5, 116.8, 111.7, 86.2, 69.9, 66.8, 60.0, 42.0, 41.4; Compound 11c (colorless oil): 1H NMR (500 MHz, CDCl3) δ 7.35 (m, 5 H), 6.33 (m, 1 H), 6.22 (m, 1 H), 5.65 (s, 1 H), 5.13 (s, 2 H), 4.69 (s, 1 H), 4.64 (s, 1 H), 3.44 (m, 4 H), 2.30 (s, 3 H), 1.97 (m, 1 H), 1.90 (m, 1 H), 1.63 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 169.4, 169.0, 136.7, 134.2, 131.7, 128.5, 128.0, 95.6, 85.7, 67.1, 61.4, 42.2, 41.5; HPLC: Chiralcel AD-H column (88:12 hexanes:2-propanol), 1.0 mL/min, major enantiomer tr = 26.1 min, minor enantiomer tr = 31.0 min.
- 14.(a) Krchnak V, Möllmann U, Dahse HM, Miller MJ. J Comb Chem. 2008;10:94. doi: 10.1021/cc700140h. [DOI] [PubMed] [Google Scholar]; (b) Krchnak V, Möllmann U, Dahse HM, Miller MJ. J Comb Chem. 2008;10:104. doi: 10.1021/cc7001414. [DOI] [PubMed] [Google Scholar]
- 15.Gerneral procedures: Resin-bounded α-terpinene HDA adduct 13 (~ 50 mg) was washed three times with THF, then 2 mL of a 0.25 M solution of diene in DMF was added. The resin slurry was transfered to a vial and exposed to 100 °C in a microwave cavity for 5 min. The resultant resin was washed five times with THF and the product was released by treatment with 0.1 M TBAF in 1 mL of THF for 30 min. The obtained product was analyzed by LC/MS. For analytical data of compound 15, sec ref. 14.