

Summary
Intermittent administration of parathyroid hormone (iPTH) is used to treat osteoporosis as it improves bone architecture and strength, but the underlying cellular and molecular mechanisms are unclear. Here we show that iPTH increases the production of Wnt10b by bone marrow CD8+ T cells, and induces these lymphocytes to activate canonical Wnt-signaling in pre-osteoblasts. Accordingly, in responses to iPTH, T cell null mice display diminished Wnt signaling in pre-osteoblasts and blunted osteoblastic commitment, proliferation, differentiation and lifespan which result in decreased trabecular bone anabolism and no increase in strength. Demonstrating the specific role of lymphocytic Wnt10b, iPTH has no anabolic activity in mice lacking T cell produced Wnt10b. Therefore, T cell mediated activation of Wnt signaling in osteoblastic cells plays a key permissive role in the mechanism by which iPTH increases bone strength, suggesting that T cell osteoblast cross-talk pathways may provide pharmacological targets for bone anabolism.
Introduction
Sustained overproduction or in vivo continuous delivery of Parathyroid hormone (PTH) is a cause of bone loss (Grey et al., 1996; Potts, 1998). However when injected daily, a regimen known as intermittent PTH treatment, the hormone markedly stimulates trabecular and cortical bone formation. Although this bone forming activity is antagonized, in part, by a stimulation of bone resorption, the net effect of intermittent PTH treatment is an improvement in bone microarchitecture and increased strength (Qin et al., 2004; Zaidi, 2007). As a result, intermittent treatment with the 1-34 fragment of PTH is an FDA approved treatment modality for postmenopausal osteoporosis (Neer et al., 2001).
PTH promotes bone formation by increasing the number of osteoblasts (OBs) through multiple effects, including activation of quiescent lining cells (Dobnig and Turner, 1995), increased OB proliferation (Nishida et al., 1994; Pettway et al., 2007) and differentiation (Nishida et al., 1994; Schmidt et al., 1995), and attenuation of pre-OB and OB apoptosis (Almeida et al., 2005; Bellido et al., 2003; Jilka et al., 1999). However, the specific contribution of each of these effects to the overall anabolic activity of PTH remains controversial.
PTH binds to the PTH/PTH-related protein (PTHrP) receptor (PPR or PTH-1R), which is expressed on OBs, osteocytes and bone marrow (BM) stromal cells (SCs) (Calvi et al., 2001; Qin et al., 2004). Ligand binding to PPR activates the cyclic AMP–dependent protein kinase A, and calcium-dependent protein kinase C signaling pathways (Gensure et al., 2005; Jilka, 2007).
PTH signaling has been observed to intersect Wnt pathways in OBs. Activation of Wnt signaling induces OB proliferation (Kato et al., 2002) and differentiation (Bodine and Komm, 2006), prevents OB apoptosis (Almeida et al., 2005; Bodine et al., 2005), and augments OB production of OPG (Glass et al., 2005), a soluble decoy receptor for the osteoclastogenic cytokine RANKL. Wnt proteins initiate a canonical signaling cascade by binding to receptors of the Frizzled family together with coreceptors, members of the low-density lipoprotein receptor-related protein (LRP) family, LRP5 and LRP6, which results in the stabilization of cytosolic β-catenin. Interaction of β-catenin with transcription factors of the lymphoid enhancer-binding factor/T cell factor family in the nucleus subsequently regulate the transcription of Wnt target genes (Behrens et al., 1996). Wnt signaling plays a critical role in bone formation as inactivating mutations of LRP5 cause osteoporosis in humans (Behrens et al., 1996), and low bone mass in mice (Kato et al., 2002).
PTH is a canonical Wnt signaling agonist which increases β catenin levels in osteoblastic cells (Kulkarni et al., 2005), an effect which occurs through modulation of both the protein kinase A and protein kinase C pathways (Tobimatsu et al., 2006). PTH, once bound to PPR, is also capable of forming a complex with LRP6 which results in LRP6 signaling and β catenin activation (Wan et al., 2008). Thus PTH activates Wnt signaling in osteoblastic cells trough both Wnt ligands dependent and independent mechanisms. Moreover, PTH down regulates the production of sclerostin, an osteocyte derived Wnt signaling antagonist (Bellido et al., 2005), Dickkopf-1, a soluble LRP5 and LRP6 signaling inhibitor (Kulkarni et al., 2005) and Sfrp-4, a factor which binds Wnt proteins thus antagonizing both canonical and non-canonical Wnt signaling (Qin et al., 2003).
These reports suggest that Wnt signaling in cells of the osteoblastic lineage plays a role in PTH induced anabolism. However, uncertainty remains with regard to the relevance of canonical Wnt signaling in the mode of action of PTH, as the silencing of LRP5 does not abrogate trabecular bone anabolism (Iwaniec et al., 2007; Sawakami et al., 2006). Furthermore, the identity and the source of Wnt ligands which activate Wnt signaling in response to PTH treatment are not completely known.
While SCs, OBs and osteocytes represent the major targets of PTH in bone, accessory cells that express PPR may play a contributory or permissive role. Among them are T lymphocytes, a lineage responds to PTH (Geffner et al., 1995; Stojceva-Taneva et al., 1993), stimulates OB differentiation (Rifas et al., 2003) and express Wnt10b (Hardiman et al., 1996; Ouji et al., 2006), a Wnt ligand produced in the BM that inhibits adipogenesis and stimulates osteoblastogenesis (Bennett et al., 2005). We have reported that a continuous infusion of PTH fails to induce bone resorption and cortical bone loss in mice lacking T cells (Gao et al., 2008). This is because T cells increase the capacity of SCs to support PTH induced osteoclastogenesis through CD40L, a surface molecule of activated T cells that induces CD40 signaling in osteoblastic cells (Gao et al., 2008). While in that study T cells were found not to be required for continuous PTH treatment to stimulate bone formation, little information is available on whether T cells contribute to the anabolic response to intermittent PTH.
In the present studies we report that intermittent PTH treatment increases the production of Wnt10b by BM CD8+ cells and that deletion of T cells, or T cell expressed Wnt10b, results in blunted trabecular bone anabolism.
Results
T cell deficient mice exhibit a blunted anabolic response to iPTH
To determine whether T lymphocytes are required for intermittent PTH to exert its effects on bone, 5 week old wild type (WT) and congenic TCRβ -/- mice, a strain completely devoid of αβ T cells, were injected daily with vehicle or 80 μg/kg of hPTH 1-34 for 4 weeks, a treatment modality referred to hereafter as iPTH. To control for strain dependent confounders, the study included TCRβ -/- mice subjected to adoptive transfer of WT T cells 1 week before initiation of iPTH treatment, a procedure which is followed by the engraftment and homeostatic expansion of the donor T cells (Gao et al., 2007; Roggia et al., 2001). Dual X-ray absorptiometry (DXA) was utilized to measure in vivo total body and femoral bone mineral density (BMD). At baseline WT and TCRβ -/- mice had similar BMD values (Supplemental Fig. 1). Due to growth, BMD increased in all vehicle treated groups during the 4 weeks of the experiment (Fig.1a). In WT and T cell reconstituted TCRβ -/- mice iPTH induced a ∼ 55-85 % greater increase in the total body BMD and a ∼70-80 % greater gain in femur BMD, as compared to vehicle. Conversely, in T cell deficient TCRβ -/- mice iPTH augmented total body and femur BMD only ∼30% and ∼25 % more than vehicle, respectively.
Figure 1.
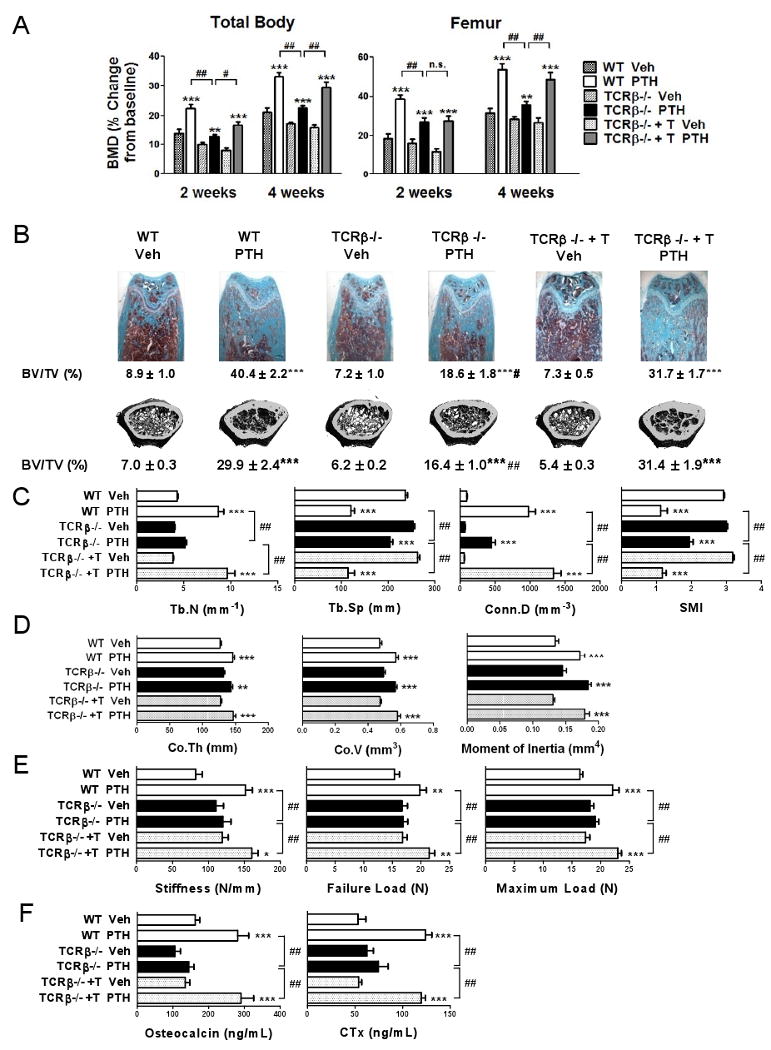
Analysis of the effects (mean ± SEM) of iPTH treatment in WT mice, TCRβ -/- mice, and TCRβ -/- mice previously subjected to adoptive transfer of WT T cells. (n = 9-11 mice per group). A In vivo total body and femoral BMD measurements by DXA at 2 and 4 weeks of treatment. B Femoral histology and ex vivo μCT analysis of femoral trabecular bone. Representative longitudinal histological sections stained by Goldner's Trichrome stain, cross sectional μCT reconstructions, and trabecular bone volume (BV/TV) measured by bone histomorphometry and μCT are shown. C Measurements of trabecular structural indices by μCT. Trabecular number (Tb.N), Trabecular space (Tb.Sp), Connectivity density (Conn.D), and Structure model index (SMI) are shown. D Ex vivo μCT analysis of femoral cortical bone. E Mechanical testing of the femur by 4-point bending tests. F Serum levels of osteocalcin, a marker of formation, and CTx, a marker of resorption. * = p<0.05, ** = p<0.01 and *** = p<0.001 compared to baseline and/or to the corresponding vehicle treated group. # = p < 0.05 and ## = p < 0.001 compared to T cell replete iPTH treated mice.
Cancellous bone was analyzed by histology and micro-computed tomography (μCT) utilizing femurs harvested at sacrifice. Histomorphometric measurements of trabecular bone volume (BV/TV) revealed a less pronounced anabolic response to PTH in TCRβ -/- mice than in controls (Fig.1b). μCT analysis showed that iPTH increased in all groups. However, BV/TV was ∼ 46-48 % lower in iPTH treated TCRβ -/- mice, as compared to iPTH treated WT and T cell reconstituted controls. Parameters of trabecular structure (Fig.1c) were also differentially affected in T cell deficient and T cell replete mice, as trabecular number (Tb.N), trabecular separation (Tb.Sp), connectivity density (Conn.D), and structure model index (SMI) were more substantially improved in WT and reconstituted mice than in TCRβ -/- mice.
By contrast, μCT analysis of cortical bone showed that iPTH induced similar increases in cortical thickness (Co.Th), cortical volume (Co.V), and moment of inertia in all groups of mice (Fig.1d), thus demonstrating that T cells specifically augment the capacity of iPTH to improve architecture in trabecular bone.
Analysis of femoral structural properties by 4-point bending tests revealed that iPTH increased stiffness, failure load, and maximum load in WT mice and T cell reconstituted TCRβ -/- mice, but not in T cell deficient TCRβ -/- mice (Fig. 1e). As structural properties reflect the strength of the whole bone, these findings demonstrate that lack of T cells blunts iPTH induced trabecular anabolism by a degree sufficient to hamper the improvement in overall bone strength caused by iPTH.
Measurements of serum osteocalcin, a marker of bone formation, suggested that iPTH increased bone accretion in T cell replete but not in T cell deficient mice (Fig.1f). Similarly, assessment of the serum levels of C-terminal telopeptide of collagen (CTx), a biochemical marker of resorption revealed that iPTH increased bone resorption in WT mice and T cell reconstituted TCRβ -/- mice, but not in T cell deficient mice. Analysis of the secondary spongiosa by bone histomorphometry confirmed that iPTH increased trabecular bone formation in T cell replete mice but not in TCRβ -/- mice (Table 1), as assessed by measurements of mineral apposition rate (MAR), and bone formation rate (BFR). In contrast, in all groups iPTH failed to cause a significant increase in endocortical MAR (Ec.MAR). iPTH had no significant effects on indices of trabecular bone resorption in T cell deficient mice while it caused an increase in total OC surface (Oc.S) in WT and T cell reconstituted mice. In response to iPTH, T cell replete mice also exhibited a decrease in both the number of OCs per bone surface (N.Oc/BS) and the percentage of surfaces covered by OCs (Oc.S/BS), a phenomenon explained by the fact that in control mice iPTH increased bone surfaces more markedly than OC number and OC surfaces.
Table 1.
Effect (mean ± SEM) of iPTH treatment on histomorphometric indices of trabecular and cortical bone turnover in WT mice, TCRβ -/- mice, and TCRβ -/- mice subjected to adoptive transfer of WT T cells 1 week before the initiation of iPTH treatment. n = 9-11 mice per group.
| WT Veh |
WT PTH |
TCRβ-/- Veh |
TCRβ-/- PTH |
TCRβ-/- + T Veh |
TCRβ-/- + T PTH |
|
|---|---|---|---|---|---|---|
| MAR (μm/day) |
1.74 ± 0.05 | 2.06 ± 0.07* | 1.89 ± 0.07 | 2.02 ± 0.08 | 1.75 ± 0.10 | 2.06 ± 0.09* |
| BFR/BS (μm3/ μm2/day) |
0.62 ± 0.06 | 1.36 ± 0.05*** | 0.59 ± 0.10 | 0.88 ± 0.08 | 0.57 ± 0.06 | 1.37 ± 0.10*** |
| Ob.S (mm) |
2.2 ± 0.3 | 12.0 ± 0.7*** | 2.4 ± 0.2 | 5.4 ± 0.5*# | 2.4 ± 0.2 | 12.8 ± 0.7*** |
| Oc.S (mm) |
1.0 ± 0.1 | 1.7 ± 0.2* | 0.7 ± 0.1 | 1.0 ± 0.2 | 1.0 ± 0.1 | 1.8 ± 0.2* |
| N.Oc/BS (mm-1) |
3.1 ± 0.1 | 1.9 ± 0.3** | 2.9 ± 0.3 | 2.5 ± 0.1 | 3.0 ± 0.3 | 1.8 ± 0.1*** |
| Oc.S/BS (%) |
10.1 ± 0.4 | 6.5 ± 0.9** | 9.1 ± 0.7 | 8.0 ± 0.5 | 9.6 ± 0.9 | 6.2 ± 0.4*** |
| Ec.MAR (μm/day) |
2.15 ± 0.2 | 2.27 ± 0.10 | 2.10 ± 0.1 | 2.30 ± 0.20 | 2.27 ± 0.20 | 2.33 ± 0.20 |
MAR (Mineral apposition Rate) and BFR (Bone Formation rate) are dynamic indices of trabecular bone formation. Ob.S (the length of bone surface occupied by osteoblasts) is a static index of trabecular bone formation, Oc.S (the length of bone surface occupied by OCs), N.OC/BS (the number of osteoclasts per mm2 bone surface), and OcS/BS (the percentage of bone surface occupied by osteoclasts), are indices of trabecular bone resorption. Ec.MAR is an index of endocortical bone formation.
= p<0.05,
= p<0.01 and
= p<0.001 compared to the corresponding vehicle treated group.
= p < 0.05 compared to T cell replete iPTH treated mice.
The finding that iPTH fails to stimulate in vivo bone resorption in TCRβ -/- mice may suggest that iPTH induces T cell production of osteoclastogenic cytokines. Since T cells secrete large amounts of soluble RANKL (Kong et al., 1999) we measured the production of RANKL by BM and spleen CD4+ and CD8+T cells harvested from WT mice treated with iPTH or vehicle for 4 weeks. These studies revealed that iPTH increased the production of RANKL by BM CD8+ cells by 2-3 folds, while it had no effects on BM CD4+ cells (Supplemental Fig. 2a,b) suggesting that RANKL production by BM CD8+ cells may play a role in the stimulation of bone resorption induced by iPTH. Additional experiments disclosed that iPTH increased by ∼30-35 % the fraction of TNFα+ CD8+ cells and IFNγ+ CD8+ cells (Supplemental Fig. 3a), although this difference was not statistically significant. The stimulation of cytokine production by CD8+ cells caused by PTH was associated with a non significant, increase in the expression of the activation marker CD69, and a minimal stimulation of CD25 expression (Supplemental Fig. 3b). iPTH had negligible effects on the expression of both markers on CD4+ cells.
To investigate the effects of iPTH in additional strains of T cell deficient mice, iPTH was injected for 4 weeks in nude mice, a strain with an severe deficiency of αβ T cells, and Rag2 -/- mice, a strain which lacks both T and B cells. μCT analysis revealed (Fig. 2a) that iPTH caused a smaller increase in BV/TV in Rag2 -/- mice and no increase in BV/TV in nude mice, as compared to WT controls. Measurements of serum osteocalcin showed that iPTH failed to stimulate bone formation in both nude and Rag2 -/- mice, while it caused a significant increase in osteocalcin levels in WT mice. Together, these findings not only confirm that T cells potentiate the anabolic activity of iPTH by stimulating bone formation, but suggest that while in some strains T cells play an amplificatory role, in others the presence of T cells is an essential requirement for iPTH mediated trabecular bone anabolism.
Figure 2.
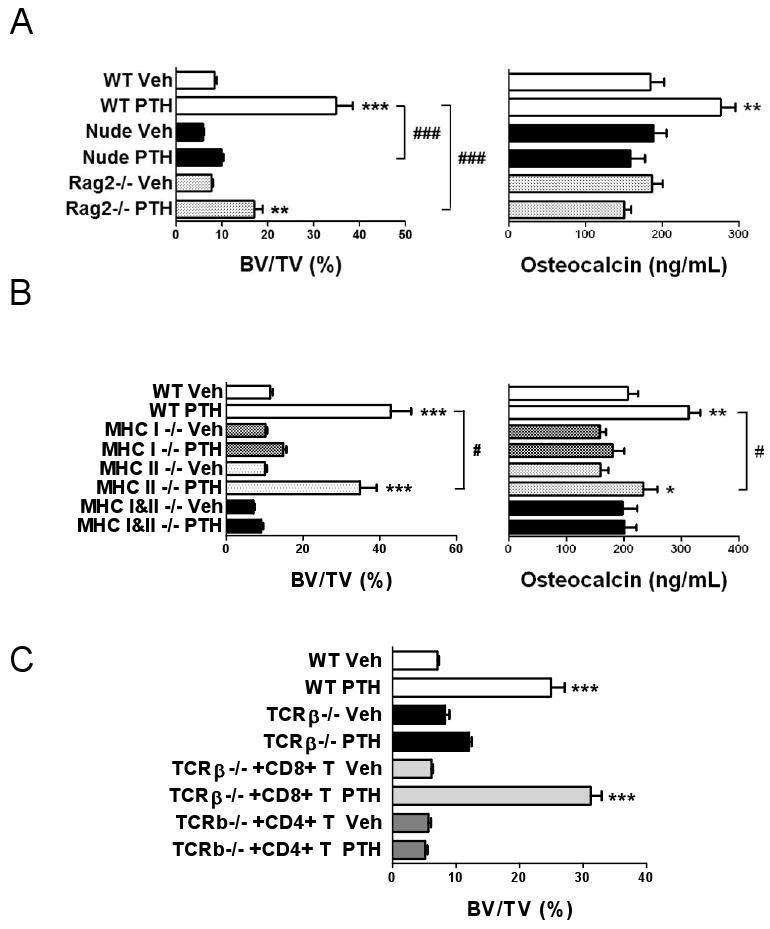
Analysis of the effects (mean ± SEM) of iPTH treatment in additional strains of T cell deficient mice. A Trabecular BV/TV and serum osteocalcin levels in nude and RAG2 -/- mice. n = 11-13 mice per group. B Trabecular BV/TV and serum osteocalcin levels in class II MHC -/- mice which lack CD4 T cells, class I MHC -/- mice which lack CD8 T cells, and double MHC -/- mice which lack both populations. (n = 6-8 mice per group). C Trabecular BV/TV in TCRβ -/- mice subjected to adoptive transfer of either CD4+ or CD8+ T cells 1 week before initiation of iPTH. (n = 5-10 mice per group) *** = p<0.001 compared to the corresponding vehicle treated group. * = p<0.05, ** = p<0.01 and *** = p<0.001 compared to the corresponding vehicle treated group. # = p < 0.05, and ### = p < 0.001 compared to the indicated groups.
To characterize the subset of T cells required for iPTH to promote bone anabolism, iPTH was injected in class II MHC -/- (Abb -/-) mice which lack CD4 T cells, class I MHC -/- (β2m -/-) mice which lack CD8 T cells, and double MHC -/- (Abb/β2m) mice which lack both populations. iPTH markedly increased BV/TV and osteocalcin in WT mice, caused a slightly blunted response in class II MHC -/- mice, and induced no changes in class I MHC -/- and double MHC -/- mice(Fig. 2b). These findings suggest that iPTH promotes bone anabolism mostly through CD8+ T cells, while CD4+ cells play a small contributory role.
The pivotal role of CD8+ cells was confirmed by adoptively transferring CD4+ or CD8+ cells into TCRβ -/- mice. Measurements of BV/TV by μCT revealed (Fig. 2c) that iPTH increased BV/TV in WT mice and TCRβ -/- animals reconstituted with CD8+ T cells, while it had no effects in T cell deficient mice TCRβ -/- mice and TCRβ -/- animals reconstituted with CD4+ T cells.
T cell deficient mice exhibit a blunted osteoblastogenic response to iPTH
BM from WT, TCRβ -/- mice, and T cell reconstituted TCRβ -/- mice treated with iPTH, was utilized to assess the formation of alkaline phosphatase (ALP) positive colony forming unit-fibroblast (CFU-F), herein defined CFU-ALP, an index of SC commitment to the osteoblastic lineage. iPTH treatment increased by ∼2 fold CFU-ALP formation in the BM of T cell replete mice while it had no effect in that of TCRβ -/- mice (Fig. 3a), thus indicating that T cells potentiate the capacity of iPTH to increase the number of SCs with osteogenic potential.
Figure 3.
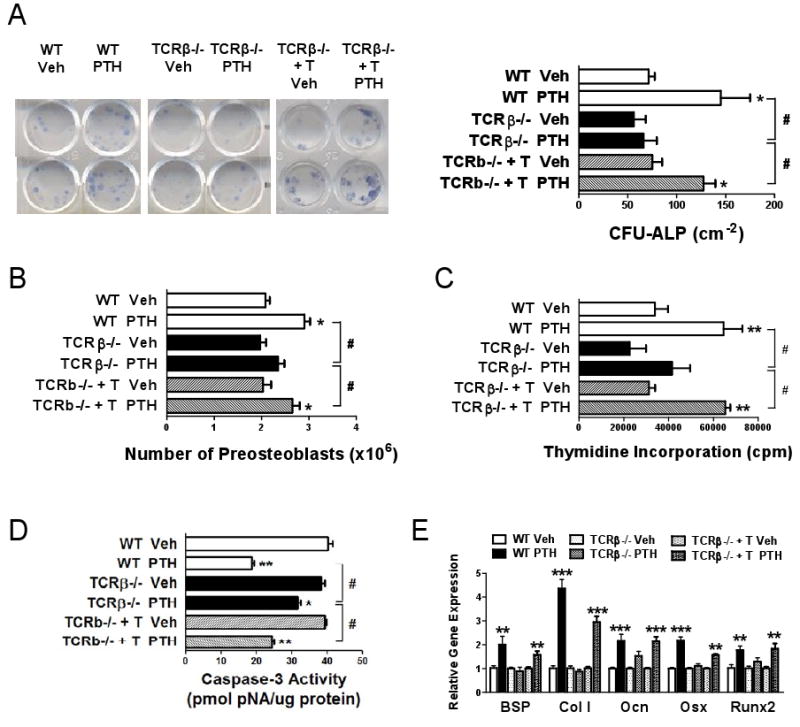
Analysis of the effects (mean ± SEM) of iPTH treatment on osteoblastogenesis and on pre-OB apoptosis in WT, TCRβ -/- mice, and TCRβ -/- mice previously subjected to adoptive transfer of WT T cells. A Whole BM was cultured for 7 days to assess the formation CFU-ALP. The left panel shows representative duplicate wells per group. The right panel shows the average of the colonies counted in 6 wells. B BM harvested at sacrifice was cultured for 1 week and pre-OBs purified and counted. C Pre-OBs were purified from BM cultured for 1 week, seeded in equal number and pulsed with [3H]-thymidine for 18 hours, to assess their proliferation. Data are expressed in CPM. D Pre-OBs were purified from BM cultured for 1 week and the rate of apoptosis quantified by determinations of caspase3 activity. E Pre-OBs were purified from BM cultured for 1 week and the level of OB marker gene mRNAs, Bone sialoprotein (BSP), type I collagen (Col1a1), Osteocalcin (Ocn), Osterix (Osx), and Runt Related Transcription factor 2 (Runx2) analyzed by RT-PCR. n = 4-5 per group. * = p<0.05, ** = p<0.01 and *** = p<0.001 compared to the corresponding vehicle treated group. # = p < 0.001 compared to the indicated group.
To directly evaluate the role of T cells in iPTH induced osteoblastogenesis, BM from iPTH treated T cell replete and TCRβ -/- mice was cultured for 1 week to allow SCs to proliferate and differentiate into pre-OBs. Pre-OBs were then purified and counted. This analysis revealed (Fig. 3b) that in vivo iPTH treatment increases the number of pre-OBs by ∼ 50 % in samples from WT and T cell reconstituted TCRβ -/- mice. In contrast, iPTH had no effects on the number of pre-OBs from TCRβ -/- mice. To investigate the mechanism involved, BM was cultured for 1 week, and pre-OBs purified and used to determine their rate of proliferation and apoptosis. These experiments revealed that iPTH increases significantly the proliferation of WT and T cell reconstituted TCRβ -/- mice pre-OBs, but not those from TCRβ -/- mice (Fig. 3c). Moreover, iPTH decreased the rate of pre-OB apoptosis in all groups of mice. However, the anti-apoptotic effect of iPTH was smaller in pre-OBs from TCRβ -/- mice than in those from WT and T cell reconstituted TCRβ -/- controls (Fig. 3d). Analysis of the expression levels of osteoblastic genes in pre-OBs revealed that iPTH increases the expression of type 1 collagen mRNA by ∼ 5 fold and that of Runx2, Osterix, Bone Sialoprotein, and Osteocalcin mRNAs by ∼ 2 fold in pre-OBs from WT mice and T cell reconstituted TCRβ -/- mice (Fig. 3e). In contrast, iPTH had no effects on the expression of OB-related genes in cells from TCRβ -/- mice, thus demonstrating that T cells promote the capacity of iPTH to expand the osteoblastic pool and cell differentiation along the osteoblastic lineage.
T cells regulate osteoblastogenesis through Wnt10b
To determine whether T cells are required for iPTH to activate Wnt signaling in osteoblastic cells, BM from iPTH treated WT and TCRβ -/- mice was cultured for 1 week and pre-OBs purified. Pre-OB expression of mRNA for genes known to be upregulated by Wnt signaling was then assessed by real time RT-PCR. The analyzed genes were chosen based on known expression patterns during differentiation of primitive mesenchymal cells to the osteoblast phenotype. Some of the selected genes are known to play a direct role in regulating OB differentiation (Vaes et al., 2005) while others are not involved in OB differentiation but are sensitive markers of Wnt activation (Jackson et al., 2005). These analysis revealed (Fig. 4a) that iPTH activates Wnt signaling in pre-OBs through T cells, as pre-OB levels of mRNA for 9 of the 12 tested genes were all increased by iPTH in pre-OBs from WT but not in those from T cell deficient TCRβ -/- mice, while the mRNA levels of the remaining 3 genes were not increased by iPTH in both WT and TCRβ -/- mice. The 9 genes which were stimulated by iPTH were: aryl-hydrocarbon receptor (Ahr), axin2, cystein rich protein 61 (Cyr61), naked cuticle 2 homolog (Nkd2), transgelin (tagln), transforming growth factor β3 (TGFβ3), thrombospondin 1 (Thbs1), Twist gene homolog 1 (Twst1) and Wnt1 inducible signaling pathway protein 1 (Wisp1). The 3 genes which were not stimulated by iPTH were cyclin D1 (Ccnd1), insulin-like growth factor binding protein 2 (Igfbp2), and osteomodulin (Omd).
Figure 4.
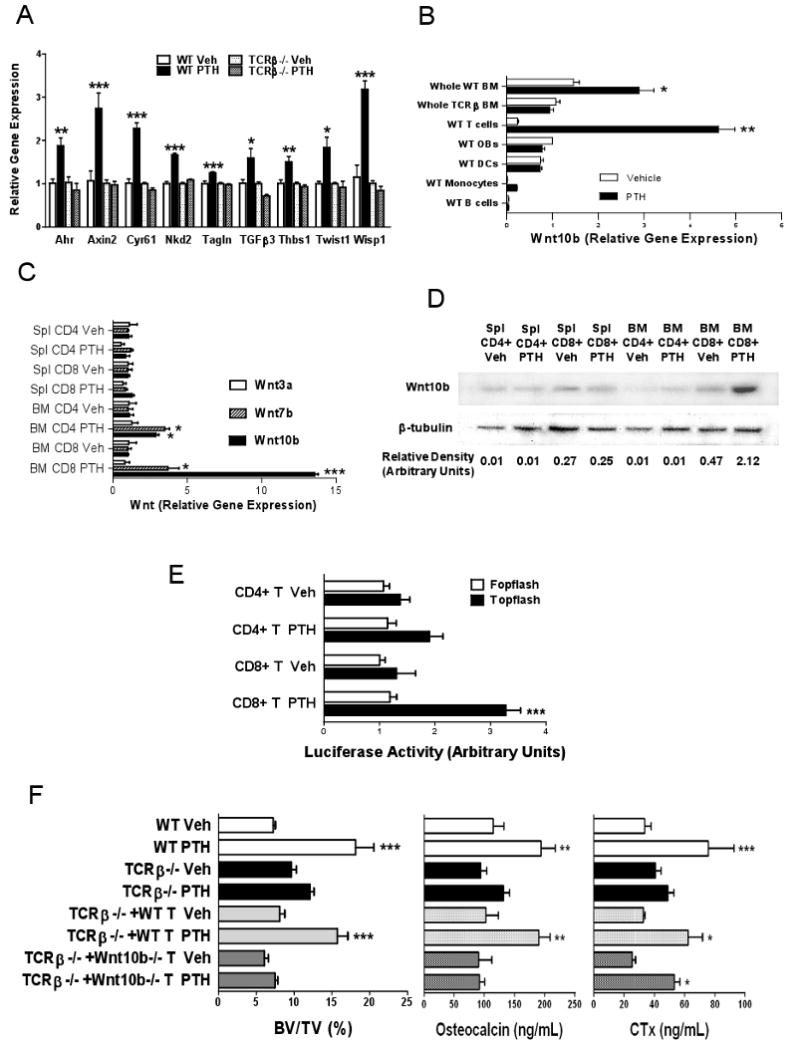
Analysis of the role of Wnt10b and Wnt signaling in iPTH induced anabolism. All data are expressed as mean ± SEM. * = p<0.05, ** = p<0.01 and *** = p<0.001 compared to WT vehicle. A Effect of iPTH on the expression of mRNA of genes known to be upregulated by Wnt signaling, aryl-hydrocarbon receptor (Ahr), axin2, cystein rich protein 61 (Cyr61), naked cuticle 2 homolog (Nkd2), transgelin (tagln), transforming growth factor beta 3 (TGFβ3), thrombospondin 1 (Thbs1), Twist gene homolog 1 (Twst1) and Wnt1 inducible signaling pathway protein 1 (Wisp1). BM was harvested at sacrifice from WT and TCRβ -/- treated with vehicle of iPTH. BM was cultured for 1 week, pre-OBs purified, and mRNA levels determined by RT-PCR. n=4 mice pre group. B Effect of iPTH on the expression of Wnt10b mRNA in whole BM and BM T cells, dendritic cells, monocytes, B cells and pre-OBs purified from BM samples obtained at sacrifice. Data are expressed relative to pre-OBs from vehicle treated mice. n = 10 mice per group. C Effect of iPTH on the expression of Wnt3a, Wnt7b and Wnt10b mRNA in spleen and BM CD4+ and CD8+ T cells purified from samples harvested at sacrifice. n = 10 mice per group. D Measurement of Wnt10b protein levels by Western blotting in spleen and BM CD4+ and CD8+ T cells purified from samples harvested at sacrifice. The bottom panel shows the densitometric quantification of the data shown in the top panel. E Effect of T cells from iPTH treated mice on Wnt signaling in the MC3T3-E1 osteoblastic cell line. MC3T3-E1 cells were transiently transfected with the TCF-luciferase reporter (TOPFLASH) or a control vector lacking the TCF binding site (FOPFLASH) and cocultured with either CD4+ or CD8+ T cells derived from the BM of WT mice treated with iPTH. Data are expressed as the mean of triplicate determinations of luciferase activity normalized for pRL-TK activity. n=4 mice per group. F Analysis of the effects of iPTH treatment in WT mice, TCRβ -/- mice, and TCRβ -/- mice subjected to adoptive transfer of T cells derived from WT mice and Wnt10b -/- mice 1 week before initiation of iPTH. The panels show trabecular BV/TV as measured by μCT at sacrifice, and serum osteocalcin and CTx at 4 weeks. n = 8 mice per group * = p<0.05, ** = p<0.01 and *** = p<0.001 compared to the corresponding vehicle treated group.
Having established that T cells are required for iPTH to activate Wnt signaling in OBs, we sought to identify the ligands involved. Wnt10b is a candidate factor produced by mesenchymal stem cells and T lymphocytes (Hardiman et al., 1996; Ouji et al., 2006) which promotes OB differentiation and increases bone density (Bennett et al., 2005). To determine whether iPTH increases Wnt10b expression and the responsible cell lineages, BM was harvested from WT and TCRβ -/- mice treated with iPTH or vehicle for 4 weeks and assayed for Wnt10b mRNA levels. BM from WT mice was also utilized to purify pre-OBs, dendritic cells (DCs), T cells, monocytes and B cells. Analysis by real time RT-PCR disclosed that iPTH increases the whole BM expression of Wnt10b mRNA by 2 fold in WT but not in TCRβ -/- mice. Among individual lineages of WT BM, T lymphocytes were the only cells that responded to iPTH with a significant increase (∼5 fold) in Wnt10b expression (Fig. 4b).
To characterize the PTH responsive T cell subset CD4+ and CD8+ cells were purified from the spleen and the BM of iPTH treated WT mice and analyzed by real time RT-PCR. These studies disclosed that iPTH treatment increases the levels of Wnt10b mRNA in BM CD4+ and CD8+ T cells by ∼ 4 and ∼13 fold, respectively (Fig 4c). In contrast, iPTH had no effects on Wnt10b mRNA levels in spleen CD4+ and CD8+ T cells. Analysis of Wnt7a and Wnt3a, two ligands which also promote bone anabolism disclosed that iPTH caused a ∼ 4 fold increase in CD4+ cell production of Wnt7b mRNA, but failed to alter the mRNA levels of Wnt3a in all populations of T cells. Further analysis by western blotting confirmed that iPTH upregulates the production of Wnt10b by BM CD8+ T cells by ∼4.5 fold (Fig 4d). In contrast, Wnt7b protein was not detected in spleen and BM CD4+ and CD8+ T cells (Supplemental Fig. 4a), indicating T cell produced Wnt7b does not contribute to activate Wnt signaling in OBs.
To determine whether T cell produced Wnt10b has the capacity to activate canonical Wnt signaling in OBs, CD4+ and CD8+ T cells purified from the BM of iPTH treated WT mice were cocultured with the osteoblastic cell line MC3T3-E1, transiently transfected with the β-catenin/ TCF-luciferase reporter plasmid TOPFLASH, or with the negative control plasmid FOPFLASH (Almeida et al., 2005). Analysis of luciferase activity in transfected MC3T3-E1 cells revealed that while the addition of CD8+ T cells from iPTH treated mice activates TCF-mediated transcription, the addition of CD4+ T cells does not (Fig 4e). Together, these findings demonstrate that iPTH upregulates the CD8+ T cell production of Wnt10b and that T cell produced Wnt10b activates canonical Wnt signaling in osteoblastic cells.
WT and Wnt10b -/- donor mice were similar with respect to CD4+/CD8+ ratio (Supplemental Fig. 5a) and numbers of naïve and memory BM T cells (Supplemental Fig. 5b). Furthermore WT and Wnt10b -/- T cells exhibit similar rates of proliferation (Supplemental Fig. 5c) and expression of activation markers in response to T cell receptor cross-linking (Supplemental Fig. 5d). However, mice lacking Wnt10b -/- have a markedly decreased bone volume and bone formation (Bennett et al., 2005), a phenotype that prevents establishing the role of Wnt10b in PTH induced bone anabolism by comparing the response of WT and Wnt10b -/- mice to iPTH. Therefore, to demonstrate the specific relevance of T cell produced Wnt10b in vivo, equal numbers of splenic T cells from WT and Wnt10b -/- mice were adoptively transferred into 6 week old TCRβ -/- mice. After 1 week, reconstituted mice were treated with vehicle or iPTH for 4 weeks. FACS analysis of splenocytes harvested at sacrifice from mice subjected to adoptive transfer of T cells revealed that mice reconstituted with WT T cells and Wnt10b -/- T cells had a similar numbers of CD4+ and CD8+ cells (Supplemental Fig. 6).
Measurements of femoral BV/TV by μCT and of serum indices of turnover at 4 weeks revealed (Fig. 4g) that iPTH increased BV/TV and osteocalcin in WT mice and TCRβ -/- animals reconstituted with WT T cells, while it had no effects in both T cell deficient mice and TCRβ -/- mice reconstituted Wnt10b -/- T cells. In contrast, iPTH failed to augment CTx levels in T cell deficient mice, but increased CTx levels in TCRβ -/- mice reconstituted with WT or Wnt10b -/- T cells. These findings demonstrate that iPTH stimulates bone formation and promotes bone anabolism through T cell produced Wnt10b.
In vitro PTH treatment stimulates T cell Wnt10b production
To investigate whether T cells have the capacity to directly respond to PTH, 99 % pure splenic CD4+ and CD8+ T cells were prepared from untreated WT mice by immunomagnetic cell sorting (Supplemental Fig. 7). The PTH receptor PPR was found to be expressed in both CD4+ and CD8+ cells, although the levels of the receptor were higher in CD8+ cells (Fig. 5a). Stimulation of T cells by PTH induced a significant increase in the production of cAMP in both CD4+ and CD8+ cells within 10 minutes (Fig. 5b), demonstrating that both T cell lineages are responsive to PTH. In vitro treatment with PTH for 3 hours increased the expression of Wnt10b mRNA by non-activated CD8+ T cells cultured with and without the phosphodiesterase inhibitor 3-isobutyl-1-methyl-xanthine (IBMX) (Fig. 5c), although the stimulatory effect of PTH was significantly higher in IBMX pretreated cells. PTH did not induce a higher expression of Wnt10b in activated CD8+ cells, indicating that T cell activation is not required for PTH induction of Wnt10b production. Pretreatment with H89, a specific PKA inhibitor, abolished PTH increase in Wnt10b mRNA expression in resting CD8+ T cells (Fig. 5d), suggesting that PTH increases Wnt10b mRNA expression primarily through the Gs/cAMP/PKA pathway. PTH also increased Wnt10b mRNA production by IBMX treated human CD8+ T cells (Fig. 5e), while it did not alter Wnt10b mRNA levels in murine pre OBs (Fig. 5f). These findings demonstrate that PTH has the capacity to directly stimulates T cell production of Wnt10b in vitro.
Figure 5.
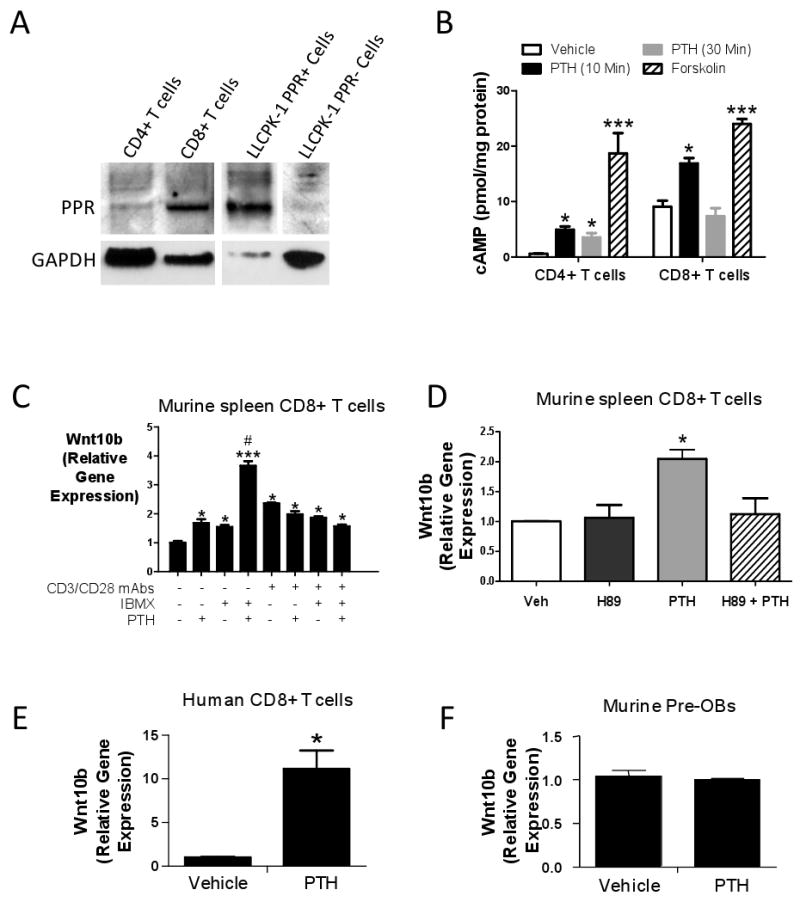
A Measurement of PPR expression by Western blotting in spleen CD4+ and CD8+ T cells from intact mice. PPR-negative LLCPK-1 cells stably transfected with empty vector (LLCPK-1 PPR-) or PPR expression vector (LLCPK-1 PPR+) are negative and positive controls, respectively. CD4+, CD8+, LLCPK-1 PPR- and LLCPK-1 PPR+ lanes were run in the same gel but were non contiguous. B Effect (mean ± SEM) of in vitro PTH treatment (50 nM) on the production of cAMP by spleen CD4+ and CD8+ T cells from intact mice. Forskolin was used as a positive control. * = p<0.05, *** = p<0.001 as compared to the corresponding vehicle. C Effect (mean ± SEM) of in vitro PTH treatment on the expression of Wnt10b mRNA by murine CD8+ T cells. Splenic CD8+ T cells were purified, stimulated with or without plate bound anti-CD3 plus anti-CD28 mAbs for 24 hours, and cultured with PTH (50 nM) for 3 hours. The phosphodiesterase inhibitor IBMX (100 μM) or vehicle was added in the indicated samples 1 hour before PTH. * = p<0.05 and *** = p<0.001 compared to mAbs-, IBMX- PTH- samples. # = p<0.05 compared to the other groups. D Effect (mean ± SEM) of the PKA inhibitor H89 on the PTH induced production of Wnt10b by unstimulated splenic CD8+ T cells. Purified cells were cultured with PTH (50 nM) and H89 (25 μM) for 3 hours. * = p<0.05 compared to the other groups. E (mean ± SEM) Effect of in vitro PTH treatment on the expression of Wnt10b mRNA by ex vivo expanded human CD8+ T cells. Data are from one of three representative experiments. * = p<0.05 compared to vehicle. F Effect (mean ± SEM) of In vitro PTH treatment on the osteoblastic expression of Wnt10b. Pre-OBs were purified from the BM of untreated WT mice and cultured with PTH 50 nM) for 3 hours.
Discussion
We report that iPTH markedly increases the production of Wnt10b by bone marrow CD8+ T cells, and that the activation of Wnt signaling in osteoblastic cells by T cell produced Wnt10b plays a relevant role in the mechanism by which iPTH increases bone strength. This insight supports a role for immune cell-bone cell cross talk in the mechanism of action of PTH.
T cells were found to augment the capacity of iPTH to improve architecture in trabecular but not in cortical bone. Although the reason of this selectivity is unknown, a lack of access of T cells to cortical surfaces is not a likely explanation, as T cells reach endosteal and periosteal bone surface through blood vessels and recirculate in and out of the BM (Di Rosa, 2008).
The relevance of T cells in vivo was demonstrated in TCRβ -/- mice and confirmed in additional T cell deficient strains, RAG2 -/-, nude, and MHC -/- mice. Our findings extend earlier observations by Pettway et al, who reported that daily injections of PTH for up to 7 weeks did not induce vertebral bone growth in nude mice (Pettway et al., 2005). In MHC -/- and nude mice the anabolic activity of iPTH was completely absent, while in the others it was markedly but not completely silenced. Moreover, in TCRβ -/- mice the anabolic activity of iPTH was more attenuated in animals treated from 7 to 11 weeks of age than in those started on iPTH at 5 weeks.
Osteoblastic cells produce several bone anabolic Wnt ligands including Wnt10b, Wnt7a and Wnt3b (Luo et al., 2004; Rawadi et al., 2003). These factors are likely to contribute to the T cell independent anabolic activity of iPTH, and quantitative differences in their production may account, in part, to the strain and age dependent variability in the response to iPTH observed herein. Furthermore, the magnitude of the anabolic response to iPTH in T cell null mice may be related to a strain and age dependent capacity of iPTH to inhibit the bone cells production of Wnt inhibitors such as sclerostin, (Bellido et al., 2005; Keller and Kneissel, 2005), Dickkopf-1 (Kulkarni et al., 2005) and Sfrp-4 (Qin et al., 2003). These factors have been shown to contribute to the anabolic activity of iPTH through T cell independent mechanisms. Since B cells are regulated by PTH (Alexiewicz et al., 1990), the response of RAG2 -/- mice to iPTH might also have been determined by the lack of B cells which is a feature of Rag2 -/- mice.
The enhancement of bone formation induced by iPTH is accompanied by a stimulation of bone resorption which is driven by increased production of RANKL and decreased release of OPG in the bone microenvironment (Ma et al., 2001). The direct effects of PTH on RANKL/OPG production are mitigated, in part, by the iPTH induced activation of β catenin in OBs, as this transcriptional regulator stimulates their production of OPG (Glass et al., 2005) and represses that of RANKL (Spencer et al., 2006). The latter is one of the mechanisms which prevent bone resorption from offsetting the anabolic activity of iPTH. Accordingly, we found that iPTH increased CTx levels and total OC surfaces in T cell replete mice. In agreement with studies by others (Iida-Klein et al., 2002), the stimulation of bone resorption induced by iPTH was not reflected by measurements of the number of OCs and OC surfaces per unit of trabecular bone surface. This was mainly because in control mice iPTH increased bone surfaces more markedly than OC number and OC surfaces. As a result, percent of OC surfaces were observed to be slightly decreased. By contrast, neither CTx nor histomorphometric indices of bone resorption were significantly affected by iPTH in T cell deficient mice. In a previous study continuous PTH treatment was similarly found unable to stimulate bone resorption in T cell depleted mice (Gao et al., 2008). The capacity of iPTH to elicit a resorptive response in T cell replete mice but not in T cell deficient animals may be explained, in part, by the increased production of RANKL by BM CD8+ T cells induced by iPTH. Another likely contributory mechanism is the capacity of T cells to lead to the generation of BM SCs which respond to PTH by producing higher levels of RANKL and lower levels of OPG, as compared to the SCs which differentiate in the BM of T cell deficient mice (Gao et al., 2008).
We show herein that osteoblastic cells from WT mice treated with iPTH in vivo exhibited increased commitment to the osteoblastic lineage, proliferation, differentiation and life span in vitro, as compared to the corresponding cells from T cell deficient mice. Thus, T cells, like PTH, affect all aspects of OB life cycle. Remarkably, these differences were demonstrated in OBs purified from BM cultured for 7 days without the addition of PTH, suggesting that in vivo the hormone regulates early commitment steps of SCs and their osteoblastic progeny through T cell produced Wnt10b, and that these steps are not reversed by the absence of PTH and T cells in vitro. This model is consistent with the capacity of Wnt signaling to guide cell fate determination (Moon et al., 2002). A similar paradigm has been described in ovariectomized mice, a model where estrogen withdrawal in vivo leads to the formation of SCs which exhibit an increased osteoclastogenic activity which persists in vitro for 4 weeks (Kimble et al., 1996).
Activation of the Wnt signaling pathway in cells of the osteoblastic lineage plays a relevant role in iPTH induced anabolism (Bodine and Komm, 2006; Kulkarni et al., 2005), but the specific signaling molecules involved remains uncertain. LRP5 is now recognized to stimulate bone formation by inhibiting serotonin synthesis in the duodenum, rather than functioning as a Wnt coreceptor in OBs (Yadav et al., 2008). Accordingly, LRP5 -/- mice are fully responsive to iPTH (Iwaniec et al., 2007; Sawakami et al., 2006). iPTH may promote Wnt signaling by activating the closely related receptor LRP6, which is also expressed in cells of the osteoblastic lineage (Jilka, 2007). Alternatively, iPTH could activate non-canonical Wnt signaling pathways that are independent of LRP5 and LRP6. Our data show that T cells activate canonical Wnt signaling in pre-OBs. However, we have not determined whether T cells activate LRP5 or LRP6 signaling in pre-OBs, nor the effects of T cells on non-canonical Wnt signaling.
The specific Wnt ligands responsible for the iPTH induced activation of Wnt signaling are also unknown. We found that iPTH markedly upregulates the production of Wnt10b by BM CD8+ T cells, and that the capacity of activating Wnt signaling in an osteoblastic line is a feature of CD8+ T cells. The relevance of CD8+ cells was demonstrated by the inability of iPTH to promote bone anabolism in class I MHC -/- mice, while the pivotal role of T cell produced Wnt10b was revealed by the hampered effect of iPTH on BV/TV and bone turnover in TCRβ -/- mice reconstituted with T cells from Wnt10b -/- mice. iPTH also caused a small increase in the production of Wnt10b by BM CD4+ cells which was associated with a slightly diminished anabolic response in class II MHC -/- mice, suggesting that production of Wnt10b by CD4+ cells contributes, in small part, to the anabolic activity of iPTH.
It is likely that iPTH directly targets CD8+ T cells and stimulates their production of Wnt10b. This hypothesis is supported by the strong expression of PPR in CD8+ T cells, and the capacity of in vitro PTH treatment to promote cAMP production and Wnt10b expression in CD8+ murine and human lymphocytes. However, we cannot conclusively exclude that in vivo iPTH treatment might induce CD8+ T cell production of Wnt10b indirectly.
While in vitro PTH treatment increased Wnt10b mRNA expression in splenic T cells, iPTH upregulated Wnt10b production only by BM T cells. This diversity might be explained by the different dose and time of exposure to PTH. However, since adoptive transfer of spleen T cells into TCRβ -/- mice was followed by a restoration of a full responsiveness to iPTH, the data suggest that the capacity of T cells to upregulate their production of Wnt10b in response to iPTH is not an intrinsic feature of T cells, but rather is induced by environmental cues.
In summary, this study indicates that T cells represent a regulatory component of the BM microenvironment involved in the anabolic response to iPTH. Bone anabolism is induced by PPR signaling in SCs and their osteoblastic progeny, but T cells play a permissive role by producing Wnt10b in response to stimulation by iPTH. Understanding the cross-talk between T cells and osteoblastic cells may thus yield novel therapeutic strategies for potentiating bone anabolic agents.
Experimental Procedures
Animals
The animal procedures were approved by the Institutional Animal Care and Use Committee of Emory University. Additional information is provided as on line Supplemental information.
Intermittent administration of PTH
80 μg/kg/day of human PTH (1-34) (Bachem California, Inc., Torrance, CA) or vehicle were injected daily subcutaneously for 4 weeks in 5-6 weeks old mice.
Purification of T and B cells, monocytes and dendritic cells, and T cell Transfer
Information is provided as on line Supplemental information.
Flow cytometry
Splenocytes and BM cells were stained with APC anti-mouse CD4 (BioLegend, San Diego, CA), PerCP anti-mouse CD8, FITC Anti-mouse MHCII, PE Anti-mouse CD20, PE Anti-mouse Cd11b, PE anti-mouse CD69 and FITC anti-mouse CD25 (BD Biosciences, San Jose, CA) mAbs and propidium Iodide solution. Samples were analyzed by flow cytometry on a FACSort flow cytometer (BD Biosciences).
In vivo BMD measurements
Total body and femoral BMD were measured in anesthetized mice using a PIXImus2 bone densitometer (GE Medical System, Lunar, Madison, WI) as described (Cenci et al., 2003).
μCT measurements
μCT scanning and analysis was performed as reported previously (Gao et al., 2007), using a Scanco μCT-40 scanner (Scanco Medical, Bassersdorf, Switzerland). Cortical bone volume and cortical thickness were determined by analyzing 80 slices at the mid –diaphysis of the femurs.
Bone histology and quantitative bone histomorphometry
Histology of the distal metaphysis of the left femora was performed by the Histomorphometry and Molecular Analysis Core Laboratory of the Center for Metabolic Bone Disease, University of Alabama at Birmingham as described (Gao et al., 2008). Additional information is provided as on line Supplemental information.
Mechanical testing
The mechanical properties of the right femora were analyzed via four-point bending as described elsewhere (Robertson et al., 2006). Each femur was tested to failure at a displacement rate of 0.05 mm/s, with the supports centered on the mid-diaphysis and the anterior side of the bone in tension. The resultant force displacement curves were analyzed to obtain force and deflection data.
Markers of bone turnover
Serum osteocalcin was measured using Rat-MID™ Osteocalcin ELISA kit (Immunodiagnostic Systems Inc., Fountain Hills, AZ). Serum CTx, was measured using the RatLaps™ ELISA kit (Immunodiagnostic Systems Inc., Fountain Hills, AZ).
CFU-ALP assay
Colony forming assays were carried out as described (Gao et al., 2008).
Real-time RT-PCR, thymidine incorporation assay, apoptosis assay, luciferase assay, cAMP assay, Western blots and Statistical Analysis
Information is provided as on line Supplemental information.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health (AR54625 and AG28278). We are grateful to Laurie McCauley (University of Michigan) for their review of the manuscript.
Abbreviations
- BM
Bone marrow
- BMM
BM macrophage
- BMD
Bone mineral density
- CTx
C-terminal telopeptides
- DXA
Dual X-ray Absorptiometry
- IBMX
3-isobutyl-1-methyl-xanthine
- μCT
micro computerized tomography
- OB
osteoblast
- OC
osteoclast
- OPG
osteoprotegerin
- RANKL
receptor activator of nuclear factor-κB ligand
- Sfrp-4
secreted frizzled-related protein-4
- SCs
stromal cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexiewicz JM, Klinger M, Pitts TO, Gaciong Z, Linker-Israeli M, Massry SG. Parathyroid hormone inhibits B cell proliferation: implications in chronic renal failure. J Am Soc Nephrol. 1990;1:236–244. doi: 10.1681/ASN.V13236. [DOI] [PubMed] [Google Scholar]
- Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. The Journal of biological chemistry. 2005;280:41342–41351. doi: 10.1074/jbc.M502168200. [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O'Brien CA, Manolagas SC, Jilka RL. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–4583. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- Bellido T, Ali AA, Plotkin LI, Fu Q, Gubrij I, Roberson PK, Weinstein RS, O'Brien CA, Manolagas SC, Jilka RL. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. The Journal of biological chemistry. 2003;278:50259–50272. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102:3324–3329. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine PV, Billiard J, Moran RA, Ponce-de-Leon H, McLarney S, Mangine A, Scrimo MJ, Bhat RA, Stauffer B, Green J, Stein GS, Lian JB, Komm BS. The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. Journal of cellular biochemistry. 2005;96:1212–1230. doi: 10.1002/jcb.20599. [DOI] [PubMed] [Google Scholar]
- Bodine PV, Komm BS. Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:33–39. doi: 10.1007/s11154-006-9002-4. [DOI] [PubMed] [Google Scholar]
- Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–286. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, Sierra O, Pacifici R. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci U S A. 2003;100:10405–10410. doi: 10.1073/pnas.1533207100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rosa F. T-lymphocyte interaction with stromal, bone and hematopoietic cells in the bone marrow. Immunol Cell Biol. 2008;87:20–29. doi: 10.1038/icb.2008.84. [DOI] [PubMed] [Google Scholar]
- Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136:3632–3638. doi: 10.1210/endo.136.8.7628403. [DOI] [PubMed] [Google Scholar]
- Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, Weitzmann MN, Pacifici R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117:122–132. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Wu X, Terauchi M, Li JY, Grassi F, Galley S, Yang X, Weitzmann MN, Pacifici R. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell metabolism. 2008;8:132–145. doi: 10.1016/j.cmet.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geffner ME, Bersch N, Cortez AB, Bailey RC, Golde DW. Growth-promoting actions of parathyroid hormone, adrenocorticotrophic hormone, and thyroid-stimulating hormone: in vitro studies in normal and pygmy T-lymphoblast cell lines. Pediatr Res. 1995;37:507–511. doi: 10.1203/00006450-199504000-00021. [DOI] [PubMed] [Google Scholar]
- Gensure RC, Gardella TJ, Juppner H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem Biophys Res Commun. 2005;328:666–678. doi: 10.1016/j.bbrc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Developmental cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Grey AB, Stapleton JP, Evans MC, Reid IR. Accelerated bone loss in post-menopausal women with mild primary hyperparathyroidism. Clin Endocrinol (Oxf) 1996;44:697–702. doi: 10.1046/j.1365-2265.1996.744565.x. [DOI] [PubMed] [Google Scholar]
- Hardiman G, Albright S, Tsunoda J, McClanahan T, Lee F. The mouse Wnt-10B gene isolated from helper T cells is widely expressed and a possible oncogene in BR6 mouse mammary tumorigenesis. Gene. 1996;172:199–205. doi: 10.1016/0378-1119(96)00109-6. [DOI] [PubMed] [Google Scholar]
- Iida-Klein A, Zhou H, Lu SS, Levine LR, Ducayen-Knowles M, Dempster DW, Nieves J, Lindsay R. Anabolic action of parathyroid hormone is skeletal site specific at the tissue and cellular levels in mice. J Bone Miner Res. 2002;17:808–816. doi: 10.1359/jbmr.2002.17.5.808. [DOI] [PubMed] [Google Scholar]
- Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, Brommage R. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22:394–402. doi: 10.1359/jbmr.061118. [DOI] [PubMed] [Google Scholar]
- Jackson A, Vayssiere B, Garcia T, Newell W, Baron R, Roman-Roman S, Rawadi G. Gene array analysis of Wnt-regulated genes in C3H10T1/2 cells. Bone. 2005;36:585–598. doi: 10.1016/j.bone.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40:1434–1446. doi: 10.1016/j.bone.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–446. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Chan L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–158. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Kimble RB, Srivastava S, Ross FP, Matayoshi A, Pacifici R. Estrogen deficiency increases the ability of stromal cells to support murine osteoclastogenesis via an interleukin-1and tumor necrosis factor- mediated stimulation of macrophage colony-stimulating factor production. The Journal of biological chemistry. 1996;271:28890–28897. doi: 10.1074/jbc.271.46.28890. [DOI] [PubMed] [Google Scholar]
- Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ, Martin TJ, Gillespie MT, Onyia JE. Effects of parathyroid hormone on Wnt signaling pathway in bone. Journal of cellular biochemistry. 2005;95:1178–1190. doi: 10.1002/jcb.20506. [DOI] [PubMed] [Google Scholar]
- Luo Q, Kang Q, Si W, Jiang W, Park JK, Peng Y, Li X, Luu HH, Luo J, Montag AG, Haydon RC, He TC. Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. The Journal of biological chemistry. 2004;279:55958–55968. doi: 10.1074/jbc.M407810200. [DOI] [PubMed] [Google Scholar]
- Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, Chandrasekhar S, Martin TJ, Onyia JE. Catabolic effects of continuous human PTH (1--38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology. 2001;142:4047–4054. doi: 10.1210/endo.142.9.8356. [DOI] [PubMed] [Google Scholar]
- Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- Nishida S, Yamaguchi A, Tanizawa T, Endo N, Mashiba T, Uchiyama Y, Suda T, Yoshiki S, Takahashi HE. Increased bone formation by intermittent parathyroid hormone administration is due to the stimulation of proliferation and differentiation of osteoprogenitor cells in bone marrow. Bone. 1994;15:717–723. doi: 10.1016/8756-3282(94)90322-0. [DOI] [PubMed] [Google Scholar]
- Ouji Y, Yoshikawa M, Shiroi A, Ishizaka S. Wnt-10b secreted from lymphocytes promotes differentiation of skin epithelial cells. Biochem Biophys Res Commun. 2006;342:1063–1069. doi: 10.1016/j.bbrc.2006.02.028. [DOI] [PubMed] [Google Scholar]
- Pettway GJ, Meganck JA, Koh AJ, Keller ET, Goldstein SA, McCauley LK. Parathyroid hormone mediates bone growth through the regulation of osteoblast proliferation and differentiation. Bone. 2007 doi: 10.1016/j.bone.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettway GJ, Schneider A, Koh AJ, Widjaja E, Morris MD, Meganck JA, Goldstein SA, McCauley LK. Anabolic actions of PTH (1-34): use of a novel tissue engineering model to investigate temporal effects on bone. Bone. 2005;36:959–970. doi: 10.1016/j.bone.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Potts J. Primary hyperparathyroidism. In: A LV, Krane S, editors. Metabolic Bone Diseases. San Diego: Academic Press; 1998. pp. 411–442. [Google Scholar]
- Qin L, Qiu P, Wang L, Li X, Swarthout JT, Soteropoulos P, Tolias P, Partridge NC. Gene expression profiles and transcription factors involved in parathyroid hormone signaling in osteoblasts revealed by microarray and bioinformatics. The Journal of biological chemistry. 2003;278:19723–19731. doi: 10.1074/jbc.M212226200. [DOI] [PubMed] [Google Scholar]
- Qin L, Raggatt LJ, Partridge NC. Parathyroid hormone: a double-edged sword for bone metabolism. Trends Endocrinol Metab. 2004;15:60–65. doi: 10.1016/j.tem.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res. 2003;18:1842–1853. doi: 10.1359/jbmr.2003.18.10.1842. [DOI] [PubMed] [Google Scholar]
- Rifas L, Arackal S, Weitzmann MN. Inflammatory T cells rapidly induce differentiation of human bone marrow stromal cells into mature osteoblasts. Journal of cellular biochemistry. 2003;88:650–659. doi: 10.1002/jcb.10436. [DOI] [PubMed] [Google Scholar]
- Robertson G, Xie C, Chen D, Awad H, Schwarz EM, O'Keefe RJ, Guldberg RE, Zhang X. Alteration of femoral bone morphology and density in COX-2-/- mice. Bone. 2006;39:767–772. doi: 10.1016/j.bone.2006.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, Pacifici R. Up-regulation of TNF-producing T cells in the bone marrow: A key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U S A. 2001;98:13960–13965. doi: 10.1073/pnas.251534698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. The Journal of biological chemistry. 2006;281:23698–23711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- Schmidt IU, Dobnig H, Turner RT. Intermittent parathyroid hormone treatment increases osteoblast number, steady state messenger ribonucleic acid levels for osteocalcin, and bone formation in tibial metaphysis of hypophysectomized female rats. Endocrinology. 1995;136:5127–5134. doi: 10.1210/endo.136.11.7588250. [DOI] [PubMed] [Google Scholar]
- Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG. Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. Journal of cell science. 2006;119:1283–1296. doi: 10.1242/jcs.02883. [DOI] [PubMed] [Google Scholar]
- Stojceva-Taneva O, Fadda GZ, Smogorzewski M, Massry SG. Parathyroid hormone increases cytosolic calcium of thymocytes. Nephron. 1993;64:592–599. doi: 10.1159/000187406. [DOI] [PubMed] [Google Scholar]
- Tobimatsu T, Kaji H, Sowa H, Naito J, Canaff L, Hendy GN, Sugimoto T, Chihara K. Parathyroid hormone increases beta-catenin levels through Smad3 in mouse osteoblastic cells. Endocrinology. 2006;147:2583–2590. doi: 10.1210/en.2005-1627. [DOI] [PubMed] [Google Scholar]
- Vaes BL, Dechering KJ, van Someren EP, Hendriks JM, van de Ven CJ, Feijen A, Mummery CL, Reinders MJ, Olijve W, van Zoelen EJ, Steegenga WT. Microarray analysis reveals expression regulation of Wnt antagonists in differentiating osteoblasts. Bone. 2005;36:803–811. doi: 10.1016/j.bone.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008;22:2968–2979. doi: 10.1101/gad.1702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–837. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.