

Abstract
Carboxylesterases are enzymes that hydrolyze a broad suite of endogenous and exogenous ester-containing compounds to the corresponding alcohol and carboxylic acid. These enzymes metabolize a number of therapeutics including the anti-tumor agent CPT-11, the anti-viral drug oseltamivir, and the anti-thrombogenic agent clopidogrel as well as many agrochemicals. In addition, carboxylesterases are involved in lipid homeostasis, including cholesterol metabolism and transport with a proposed role in the development of atherosclerosis. Several different scaffolds capable of inhibiting carboxylesterases have been reported, including organophosphates, carbamates, trifluoromethyl ketone-containing structures (TFKs), and aromatic ethane-1,2-diones. Of these varied groups, only the 1,2-diones evidence carboxylesterase isoform-selectivity, which is an important characteristic for therapeutic application and probing biological mechanisms. This study constructed a series of classical and 3D-QSAR models to examine the physiochemical parameters involved in the observed selectivity of three mammalian carboxylesterases: human intestinal carboxylesterase (hiCE), human carboxylesterase 1 (hCE1), and rabbit carboxylesterase (rCE). CoMFA-based models for the benzil-analogs described 88%, 95% and 76% of observed activity for hiCE, hCE1 and rCE, respectively. For TFK-containing compounds, two distinct models were constructed using either the ketone or gem-diol form of the inhibitor. For all three enzymes, the CoMFA ketone models comprised more biological activity than the corresponding gem-diol models; however the differences were small with described activity for all models ranging from 85–98%. A comprehensive model incorporating both benzil and TFK structures described 92%, 85% and 87% of observed activity for hiCE, hCE1 and rCE, respectively. Both classical and 3D-QSAR analysis showed that the observed isoform-selectivity with the benzil-analogs could be described by the volume parameter. This finding was successfully applied to examine substrate selectivity, demonstrating that the relative volumes of the alcohol and acid moieties of ester-containing substrates were predictive for whether hydrolysis was preferred by hiCE or hCE1. Based upon the integrated benzil and TFK model, the next generation inhibitors should combine the A-ring and the 1,2-dione of the benzil inhibitor with the long alkyl chain of the TFK-inhibitor in order to optimize selectivity and potency. These new inhibitors could be useful for elucidating the role of carboxylesterase activity in fatty acid homeostasis and the development of atherosclerosis as well as effecting the controlled activation of carboxylesterase-based prodrugs in situ.
Keywords: Carboxylesterase, Esterase, Benzil, Trifluoromethyl ketone, TFK, QSAR, CoMFA, Fatty acid metabolism, Cholesterol, Atherosclerosis
1. Introduction
Carboxylesterases (CaEs) are enzymes from the α/β hydrolase-fold that metabolize numerous endogenous and exogenous compounds by cleaving carboxylesters (RCOOR′) into the corresponding alcohol (R′OH) and the carboxylic acid (RCOOH).1–3 They have broad substrate specificity and play an important role in the detoxification of many insecticides4 and pharmaceuticals.5 It has also been suggested that CaE activity is important in cholesterol processing and fatty acid homeostasis;5,6 subsequently playing a role in cardiovascular disease.7 CaEs exert their hydrolytic capability via a two-step serine hydrolase mechanism that involves the formation of a covalent acyl-intermediate attached to the catalytic serine. The intermediate is then released via hydrolysis generating the corresponding alcohol and acid.4 CaEs can also perform transesterification reactions, such as the formation of cocaethylene from cocaine and alcohol8 and the production of fatty acid ethyl esters (FAEEs) from fatty acyl-Coenzyme A (CoA) and ethanol.9,10 Both of these reactions result in an increase in toxicity over the parent compounds, demonstrating that CaE activity is not always a detoxification process. In humans, two major CaEs have been identified, hCE1 and hiCE (hCE2), which exhibit isoform-specific hydrolysis activity. For example, the anti-thrombogenic agents aspirin and clopidogrel are hydrolyzed by CaEs and are coadministered to patients at risk of acute coronary syndromes,11 potentially resulting in drug:drug interactions that could affect the clinical efficacy. However, Tang and coworkers showed that aspirin is hydrolyzed predominately by hiCE and clopidogrel by hCE1.12 On the other hand, clopidogrel treatment has been shown to inhibit the hCE1-mediated activation of the anti-influenza prodrug oseltamivir, demonstrating that drug:drug interactions can occur in CaE-mediated metabolism.13
CaEs are ubiquitous enzymes and are found in a range of tissues including liver, kidney, small intestine and brain (see Ref. 14 and references therein). However, activity in plasma is species dependent and is low in humans.5 CaEs are typically expressed in the epithelial of most organs, suggesting that these enzymes serve in a protective role against xenobiotic exposure.14–16 Interest in this family of enzymes is steadily increasing and a number of recent advances have greatly expanded our understanding. The first mammalian crystal structure was published in 2002 and provided insight into the role that this enzyme plays in the hydrolysis of the chemotherapeutic agent CPT-11 (irinotecan-7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin).17 Since then, a number of follow-up structures have examined the interactions of hCE1 with a range of exogenous and endogenous substrates, including the Alzheimer’s drug tacrine,18 heroin and cocaine,19 the cholesterol-lowering drug mevastain20, the breast cancer drug tamoxifen20 and the chemical warfare agents soman and tabun.21 These papers have provided useful data concerning the nature of CaE-mediated hydrolysis and explained some of the structural requirements for both substrate hydrolysis and catalytic inhibition. Because CaEs metabolize a range of pharmaceuticals, the use of inhibitors could potentially be valuable in modulating the efficacy of these therapeutics.5 There is particular interest in the CaE-mediated activation of CPT-11, as hydrolysis of the parent compound in the small intestine to the active metabolite SN-38 (7-ethyl-10-hydroxycamptothecin) can lead to significant gastrotoxicity (e.g., mucosal damage resulting in diarrhea).22 It has been proposed that the development of CaE inhibitors selective for the intestinal form of the enzyme (hiCE) could potentially ameliorate this side-effect, yet not affect the activity of other CaEs necessary for CPT-11 activation in situ.23 The development of CaE-selective inhibitors would also be useful for studying the endogenous role(s) of the enzyme by examining the effects of inhibition upon downstream biological products.
There are a number of different structural motifs that have been developed to inhibit CaEs, with recent research efforts designed to increase the specificity of inhibition. Many of the early CaE inhibitors were related to organophosphates (OPs) and exerted their inhibitory effect by covalently modifying the enzyme.24 Another family of potent esterase inhibitors are the trifluoromethyl ketones (TFKs), which have been used to effectively inhibit a number of CaEs including juvenile hormone esterase,25 mouse and porcine liver CaEs,26 and human CaEs.27 TFKs are transition state analogs that form a reversible covalent bond with the enzyme, after undergoing nucleophilic attack by the catalytic serine.28 A key feature lacking in these different classes of inhibitors is specificity. Both OPs and TFKs inhibit a wide swathe of esterases and do not demonstrate selectivity, which is highly desired for potential therapeutic applications as discussed above. A series of isoform-selective inhibitors of mammalian CaEs has been reported that includes sulfonamides,23 aromatic ethane-1,2-diones based upon a benzil structure29–31 and a number of subsequent derivatives including indole-2,3-diones32 and fluorobenzils.33 The aromatic ethane-1,2-diones in particular demonstrated excellent selectivity among a rabbit CaE and two different human CaEs (hCE1 and hiCE).29
Given the high similarity among different CaEs, the selectivity exhibited by the aromatic ethane-1,2-diones is very interesting. hCE1 and hiCE share 47% amino acid sequence identity and rCE shares 81% and 46% sequence identity with hCE1 and hiCE, respectively. Initial 3D-QSAR analysis indicated that the selectivity afforded by these compounds was partly due to steric interactions and repulsions within the enzyme active sites.29 Further analysis demonstrated that when the carbonyl oxygen atoms of the 1,2-diones are cis-coplanar, the compounds demonstrate specificity for hCE1 and that conversely, when the dione oxygen atoms are not planar (or are trans-coplanar), the compounds are more potent hiCE inhibitors. 31 However, the full details of the selectivity are still unknown. Accordingly, to further examine this issue, this study combined a series of different aromatic ethane-1,2-diones to generate a CoMFA 3D-QSAR model in order to examine the electronic and steric parameters that contribute to the observed inhibition selectivity. A number of TFK-based CoMFA models were also generated, with specific emphasis on the geometry of the inhibitor, either ketone or gem-diol. The optimal TFK model was then combined with the model for the 1,2-dione motif in order to present a comprehensive CoMFA model of CaE inhibition. Together these results provide further information on the parameters necessary to selectivity inhibit mammalian CaEs and will be useful in the future design of second generation inhibitors.
2. Results
2.1. Classical QSAR analysis for the benzil-analogs
It has been previously demonstrated that the molecular hydrophobicity or the length of the alkyl chain of TFK compounds correlates positively with the potency of CaE inhibitors.28 Accordingly, the initial classical QSAR models for the benzil derivatives included a number of physicochemical properties centered on hydrophobicity and steric bulk using a Hansch–Fujita type analysis.34 L and B5 are the STERIMOL length and width parameter, respectively, in Ångströms from the axis, with L representing the length of the substituent along the axis connecting to the alpha atom of the substituent with the rest of the molecule, and B5 representing the maximum width from the L-axis.35 As shown in Table 1 the variability in steric bulk of the benzil derivatives was not as great as that for the TFK-containing compounds. The substituents on the benzene rings are relatively small compared to the alkyl groups of the TFK compounds in terms of the STERIMOL parameters: n-C4H9 (L/B5 = 6.17/4.54) — C12H25 (14.38/10.27) vs CH3 (2.87/2.04), halogens (2.65–3.82/1.35–1.95), COOH (3.91/2.66), NO2 (3.44/2.44), OCH3 (3.98/3.07), CH2Br (4.09/3.75).36 We evaluated a new steric parameter for the A-ring moiety (less substituted benzene ring, see Table 1 for a description of the A- and B-rings) that combined the steric effects of all substituents on the benzene ring. As described in Section 5, the two benzene rings are distinguished according to the substitution pattern, with the more substituted benzene ring termed the B-ring. Even though the QSAR correlations were poor, we were able to derive significant correlations using the volume parameter and log P as shown below. Inactive compounds were excluded from the analyses.
| (1) |
| (2) |
| (3) |
Table 1.
Structures of the benzil derivatives examined
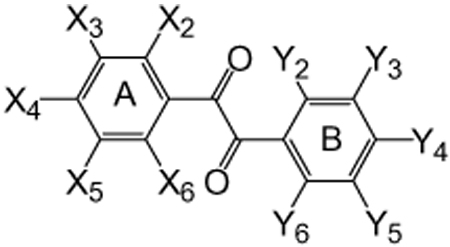 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | A-ring |
B-ring |
||||||||
| X2 | X3 | X4 | X5 | X6 | Y2 | Y3 | Y4 | Y5 | Y6 | |
| 1 | H | H | H | H | H | H | H | H | H | H |
| 2 | H | H | F | H | H | H | H | F | H | H |
| 3 | H | H | Cl | H | H | H | H | Cl | H | H |
| 4 | Cl | H | H | H | H | H | H | H | H | Cl |
| 5 | H | F | H | F | H | H | F | H | F | H |
| 6 | H | F | F | F | H | H | F | F | F | H |
| 7 | H | H | H | H | H | H | H | COOH | H | H |
| 8 | H | H | H | H | H | H | H | Cl | H | H |
| 9 | H | H | Cl | H | H | H | H | CH3 | H | H |
| 10 | H | H | H | H | H | H | H | CH3 | H | H |
| 11 | H | H | CH3 | H | H | H | H | CH3 | H | H |
| 12 | H | H | H | H | H | H | CH3 | CH3 | H | H |
| 13 | H | H | H | H | H | H | H | OCH3 | H | H |
| 14 | H | H | OCH3 | H | H | H | H | OCH3 | H | H |
| 15 | H | H | H | OCH3 | H | H | OCH3 | H | H | H |
| 16 | Cl | H | H | H | H | H | OCH3 | OCH3 | H | H |
| 17 | H | H | H | H | H | H | H | CH2Br | H | H |
| 18 | H | H | Br | NO2 | H | H | NO2 | Br | H | H |
| 19 | H | H | H | H | H | H | NO2 | CH3 | H | H |
| 20 | H | H | H | H | H | H | H | NO2 | H | H |
| 21 | H | H | H | H | H | H | H | NO2 | H | NO2 |
| 22 | H | NO2 | H | H | H | H | H | H | NO2 | H |
| 23 | OCH3 | H | Br | H | H | H | H | Br | H | OCH3 |
| 24 | F | F | F | F | F | Cl | Cl | Cl | Cl | Cl |
| 25 | H | H | H | H | H | H | H | COCOPh | H | H |
In these and the following equations, n is the number of compounds used for regression analyses, s is the standard deviation, r is the correlation coefficient, and F is the F-statistics value. The numbers in parentheses are 95% confidence intervals for each coefficient by t-test. The addition of the log P value increased the statistical significance of Eq. (2) (s = 0.661 and r2 = 0.142 without log P), but did not improve Eq. (1) or Eq. (3). For the inhibition of hiCE, an optimum volume (3.7) of the A-ring moiety was observed. Interestingly, the inhibition of hCE1 was negatively correlated with the volume while the inhibition of rCE was positively correlated. The correlation coefficients of these three equations, however, were insufficient to explain the observed SAR, with values ranging from 0.202–0.348. Even though attempts to formulate significant correlations using the Hammett sigma constant were unsuccessful, the electrostatic effects are most likely important in enzyme inhibition. Accordingly, we constructed CoMFA models to explicitly examine the electrostatic effects.
2.2. 3D-QSAR and CoMFA analysis for the benzil-analogs
CoMFA was performed with the combination of parameters including log P and volume (vol) for all active compounds shown in Table 1 and Figure 1. Compounds were superposed as described in Section 5 ( Fig. S1). Because the number of active compounds was ≤32 (depending upon the enzyme), the maximum component number (m) was set as 6 in the initial cross-validation test to identify the optimum number of components. All perturbations of log P, log P2, vol and vol2 were examined for the three enzymes and the corresponding statistics of the cross-validations are listed in Table 2. For the inhibition of hiCE and hCE1, correlations 7 and 16 were selected as the best CoMFAs in terms of q2 and spress values, in which log P, vol, and vol2 were used as external parameters. For the inhibition of rCE, correlation 24 was selected as the best, in which only the log P and vol parameter were used (the addition of vol2 was not significant). Attempts to use the CoMFA electrostatic term instead of the basic CoMFA (electrostatic + steric) term reduced the statistical significance. The conventional correlation equations for correlations 7, 16, and 24 (Table 1) are shown below.
| (4) |
| (5) |
| (6) |
Figure 1.

Structures of the dione-based carboxylesterase inhibitors examined in this study that are not displayed in Table 1.
Table 2.
CoMFA correlation statistics for carboxylesterase inhibition by benzil derivatives
| Parameters used for analysesb | Statistics for cross-validationa |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hiCE (n = 31)c |
hCE1 (n = 29) |
rCE (n = 32) |
|||||||||||||
| log P | log P2 | vol | vol2 | q2 | spress | m | Corr No.d | q2 | spress | m | Corr No. | q2 | spress | m | Corr No. |
| — | — | — | — | 0.539 | 0.582 | 4 | 1 | 0.015 | 0.722 | 2 | 10 | 0.296 | 0.771 | 2 | 19 |
| ○ | — | — | — | 0.538 | 0.594 | 5 | 2 | 0.548 | 0.532 | 6 | 11 | 0.341 | 0.76 | 3 | 20 |
| ○ | ○ | — | — | 0.521 | 0.618 | 6 | 3 | 0.485 | 0.568 | 6 | 12 | 0.157 | 0.875 | 4 | 21 |
| — | — | ○ | ○ | 0.508 | 0.601 | 4 | 4 | 0.079 | 0.712 | 3 | 13 | 0.284 | 0.792 | 3 | 22 |
| — | — | ○ | ○ | 0.554 | 0.572 | 4 | 5 | 0.083 | 0.711 | 3 | 14 | 0.280 | 0.794 | 3 | 23 |
| ○ | — | ○ | — | 0.511 | 0.611 | 5 | 6 | 0.594 | 0.504 | 6 | 15 | 0.374 | 0.754 | 4 | 24 |
| ○ | — | ○ | ○ | 0.557 | 0.582 | 5 | 7 | 0.603 | 0.498 | 6 | 16 | 0.325 | 0.783 | 5 | 25 |
| ○ | ○ | ○ | — | 0.541 | 0.604 | 6 | 8 | 0.545 | 0.533 | 6 | 17 | 0.106 | 0.918 | 5 | 26 |
| ○ | ○ | ○ | ○ | 0.548 | 0.600 | 6 | 9 | 0.546 | 0.533 | 6 | 18 | 0.206 | 0.865 | 5 | 27 |
The statistical quality of the models is described in terms of: q = leave-one-out cross-validated correlation coefficient, spress = cross-validated standard error, m = number of components. The maximum component number (m) in the cross-validation test was set as 6 for all CoMFA analyses. Correlations 7, 16 and 24 were selected as the optimal equations for hiCE, hCE1 and rCE, respectively. The final statistics for these equations were: Corr 7, SE = 0.307, r2 = 0.876; Corr 16, SE = 0.176, r2 = 0.950; Corr 24 SE = 0.466, r2 = 0.760.
The parameters employed in the CoMFA analysis are indicated as included (○), or not included (—) in the generation of the final equation.
The number of compounds used in the CoMFA analysis based upon the activity data in Table 3.
Correlation number.
As shown above, the vol2 term was significant in Eq. (4) and Eq.(5), but not in Eq. (6), indicating that there is an optimum volume for inhibition of hiCE (the optimum volume for inhibition of hCE1 is out of range). These results are consistent with those of Eq. (1) and the CoMFA contour views are shown in Figure 2. Overall, the three different enzymes displayed distinct CoMFA maps, with very few fields in common. Generally, it appears that for all enzymes there is a small region of favorable positive electronic potential around the 5- and 6-positions of the A-ring, while hCE1 and rCE share a favorable negative potential region around the 4-position of the B-ring. In addition, there is a general trend of unfavorable steric bulk around the 3- and 4-position of the A-ring. A negative potential region is also found at the 4-position of the A-ring for hCE1. Because the external volume parameter was used for the A-ring (smaller ring) moiety, the steric effect of the CoMFA analysis was not as evident as that for the B-ring moiety. This effect is especially pronounced for hiCE, which displays a large sterically unfavorable region surrounding the A-ring. hiCE evidenced significant favorable positive potential in the space bridging the A- and B-rings, while this field was negative for hCE1 and weakly mixed for rCE. hCE1 displayed a significant favorable positive field around the 3-position of the B-ring, which was negative for rCE and mixed for hiCE. There is a sterically unfavorable region at the 4- and 5-positions of the B-ring for rCE and a small favorable region appears at the 4-position of the B-ring for hiCE. The observed and predicted inhibition constants for the three enzymes using the optimal classical QSAR and CoMFA equations are provided in Table 3.
Figure 2.

CoMFA maps for benzil-analogs for the three carboxylesterases examined. (A) hiCE (Eq. (4)), (B) hCE1 (Eq. (5)) and (C) rCE (Eq. (6)). The template molecule is 1-(3,4-dimethylphenyl)-2-phenylethane-1,2-dione (12). The contours are shown to surround regions where a higher steric bulk increases (green) or decreases (yellow) the inhibitory activity and a negative (red) or positive electronic potential (blue) amplifies biological activity.
Table 3.
Observed and predicted values for benzil-analog mediated carboxylesterase inhibition using classical QSAR and CoMFA analysesa
| No. | pKi |
log P | vol | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| hiCE |
hCE1 |
rCE |
|||||||||
| Obsdb | Eq. (1) | Eq. (4) | Obsd | Eq. (2) | Eq. (5) | Obsd | Eq. (3) | Eq. (6) | |||
| 1 | 7.83 | 7.20 | 6.97 | 7.35 | 6.68 | 6.96 | 6.99 | 6.96 | 6.73 | 3.38 | 3.33 |
| 2 | 6.78 | 7.36 | 7.06 | 6.64 | 6.61 | 6.69 | 6.40 | 7.11 | 7.12 | 3.76 | 3.49 |
| 3 | 6.97 | 7.44 | 7.30 | 6.74 | 6.62 | 6.79 | 8.02 | 7.30 | 7.64 | 4.90 | 3.69 |
| 4 | 6.45 | 7.42 | 6.70 | 5.78 | 6.69 | 5.79 | 6.66 | 7.22 | 6.34 | 4.90 | 3.61 |
| 5 | 7.63 | 7.43 | 7.26 | 7.13 | 6.53 | 6.99 | 7.76 | 7.24 | 7.47 | 4.06 | 3.63 |
| 6 | 6.59 | 7.43 | 7.11 | 6.43 | 6.41 | 6.53 | 7.32 | 7.37 | 7.66 | 4.07 | 3.77 |
| 7 | 7.15 | 7.10 | 7.15 | 6.28 | 6.75 | 6.28 | 7.19 | 6.89 | 7.04 | 3.39 | 3.26 |
| 8 | 7.74 | 7.20 | 7.35 | 7.32 | 6.82 | 7.14 | 7.62 | 6.96 | 7.10 | 4.17 | 3.33 |
| 9 | 7.64 | 7.44 | 7.64 | 6.80 | 6.56 | 6.73 | 7.79 | 7.33 | 7.41 | 4.67 | 3.72 |
| 10 | 7.48 | 7.19 | 7.71 | 6.90 | 6.78 | 7.03 | 6.92 | 6.95 | 7.03 | 3.88 | 3.32 |
| 11 | 7.22 | 7.42 | 7.37 | 6.27 | 6.45 | 6.41 | 7.30 | 7.39 | 7.41 | 4.38 | 3.79 |
| 12 | 8.39 | 7.19 | 8.22 | 7.00 | 6.85 | 7.13 | 6.97 | 6.95 | 7.29 | 4.33 | 3.32 |
| 13 | 7.99 | 7.21 | 7.76 | 6.76 | 6.71 | 6.64 | 6.70 | 6.97 | 6.94 | 3.60 | 3.34 |
| 14 | 7.15 | 7.17 | 7.06 | 5.47 | 6.06 | 5.33 | 6.24 | 7.67 | 7.35 | 3.61 | 4.08 |
| 15 | 6.86 | 7.21 | 7.16 | 5.81 | 6.09 | 5.79 | 7.14 | 7.64 | 7.41 | 3.61 | 4.05 |
| 16 | 8.05 | 7.44 | 8.30 | 5.48 | 6.46 | 5.46 | 7.44 | 7.32 | 7.46 | 4.04 | 3.71 |
| 17 | 7.67 | 7.16 | 7.92 | 7.11 | 6.84 | 7.26 | 7.82 | 6.93 | 7.45 | 4.16 | 3.30 |
| 18 | — | 6.57 | 7.32 | — | 4.95 | 5.87 | 8.06 | 7.96 | 8.41 | −1.38 | 4.39 |
| 19 | 8.10 | 7.19 | 8.11 | 6.53 | 6.23 | 6.56 | 7.92 | 6.95 | 7.77 | 0.59 | 3.32 |
| 20 | 7.51 | 7.17 | 7.52 | 6.67 | 6.15 | 6.68 | 7.66 | 6.94 | 7.69 | 0.09 | 3.31 |
| 21 | 6.68 | 7.13 | 6.58 | — | 5.63 | 5.41 | 6.28 | 6.91 | 6.39 | −3.20 | 3.28 |
| 22 | 6.40 | 7.27 | 6.37 | 4.74 | 4.99 | 4.72 | 6.57 | 7.59 | 5.85 | −3.20 | 4.00 |
| 23 | 7.14 | 7.25 | 7.14 | — | 6.29 | 6.57 | 7.27 | 7.61 | 7.30 | 4.68 | 4.02 |
| 24 | 6.42 | 7.31 | 6.23 | 5.77 | 6.77 | 5.87 | 6.08 | 7.55 | 6.27 | 7.20 | 3.96 |
| 25 | 8.25 | 7.14 | 8.25 | 8.10 | 6.97 | 8.07 | 8.21 | 6.92 | 8.63 | 4.89 | 3.29 |
| 26 | 5.98 | 6.36 | 6.03 | 6.67 | 6.96 | 6.79 | 6.06 | 6.57 | 6.55 | 2.84 | 2.92 |
| 27 | 5.18 | 6.19 | 5.18 | 6.44 | 7.01 | 6.38 | 5.62 | 6.52 | 5.88 | 2.84 | 2.86 |
| 28 | 7.24 | 7.35 | 6.84 | 7.52 | 6.77 | 7.34 | 8.66 | 7.09 | 7.76 | 4.61 | 3.47 |
| 29 | 6.63 | 7.33 | 6.76 | 6.68 | 6.78 | 6.57 | 8.15 | 7.07 | 7.33 | 4.61 | 3.45 |
| 30 | 5.10 | 5.52 | 5.49 | 5.93 | 6.92 | 5.96 | 5.07 | 6.33 | 5.37 | 1.24 | 2.66 |
| 31 | 6.82 | 6.94 | 6.67 | 6.90 | 6.41 | 7.09 | 6.32 | 6.81 | 6.31 | 0.90 | 3.17 |
| 32 | 7.07 | 6.00 | 6.88 | 6.59 | 5.96 | 6.67 | 8.98 | 8.15 | 8.86 | 5.73 | 4.59 |
Experimental data are taken from Wadkins et al.29
Obsd = observed or experimental pKi values.
2.3. Classical QSAR analysis for the TFK compounds
The inhibition of CaE activity by TFK-containing compounds was previously quantitatively analyzed using another 3D-QSAR procedure (Quasar 5.0) by Wadkins.27 They demonstrated that the activity correlated with log P or length of the alkyl group for a set of sulfur-containing compounds with a terminal –COCF3 group. However, in their discussion, sulfoxide and sulfonyl-type compounds with –COCF3 (keto form) as well as the corresponding gem-diol form were not included. In this study, the TFK QSAR models were generated by first analyzing all compounds using the classical Hansch–Fujita QSAR method.34 The steric parameters STERIMOL L and B5 were used for the alkyl chain after the oxygen atom and the sulfur atom (as well as its oxidized analogs). For compound 39, octyl was used as the alkyl chain.
The results for all three enzymes in the benzil-based CoMFA studies showed that log P was an important parameter in describing inhibitor potency (Table 2), which agreed with earlier TFK-based CoMFA studies.37 Accordingly, log P was used to begin the construction of the TFK-based models. Initially, inhibitor geometry was chosen after those published by Wadkins,27 where the hydration state of the ketone was based upon 1H NMR observations. The data reported by Wadkins27 determined Ki values for 5 min and 24 h incubation of enzyme and inhibitor. For the development of the initial classical QSAR models, the 5 min data were employed. This approach generated statistically significant equations for all three enzymes that described the majority of biological activity (Eq. (7)–Eq. (9)).
| (7) |
| (8) |
| (9) |
However, studies by Wheelock37 suggested that the geometry of the hydration state can significantly affect the outcome of QSAR studies. The TFK studies were therefore divided into two distinct data sets of either all gem-diol (Eq. (7a), Eq. (8a), and Eq. (9a)) or all ketone (Eq. (7b), Eq. (8b), and Eq. (9b)) compounds and re-analyzed. In these equations, the Δ term indicates the corresponding parameter value for the H atom.
| (7a) |
| (7b) |
| (8a) |
| (8b) |
| (9a) |
| (9b) |
Results showed no significant differences between the QSAR equations for mixed geometries or selected geometries. Interestingly, the largest log P value for the TFKs analyzed in this study was for compound 33, which had values of 6.07 for the gem-diol and 6.63 for the ketone. However, both values are lower than the predicted optimum values in Eq. (9a) and Eq. (9b) (6.77 and 7.61, respectively). However, without the squared log P term, the correlations were worse (s = 0.429; r2 = 0.748) than those of (9a) and (9b) as shown below.
| (9c) |
| (9d) |
For hiCE and hCE1, the addition of the squared log P term improved the correlation, but the log P2 and B5 parameter became insignificant (hiCE: B5 > 74.5%, log P2 > 87.4%; hCE1: log P > 82.7%). Accordingly, as these values were less than the >95% cut-off normally employed, the resulting equations are not shown. Interestingly, the inhibition of all three CaEs correlated strongly with hydrophobicity (the correlation coefficients, r2, for log P only were: hiCE = 0.85 and 0.83 for the gem-diol and ketone, respectively; hCE1 = 0.52 and 0.49 for the gem-diol and ketone, respectively; and rCE = 0.61 and 0.58 for the gem-diol and ketone, respectively). The correlations with the squared log P term were also strong, but consistently weaker than for the log P term, whereas the steric parameters evidenced weak or no correlations (the correlation coefficients for steric parameters were: hiCE = 0.71 and 0.69 for L and B5, respectively; hCE1 = 0.30 and 0.25 for L and B5, respectively; and rCE = 0.41 and 0.40 for L and B5, respectively). The inhibition of hCE1 was enhanced with the molecular hydrophobicity, but decreased with the maximum width (B5) of alkyl groups. The observed and predicted Ki values for the TFK-inhibitors are shown in Table 4.
Table 4.
Observed and predicted values for TFK-mediated carboxylesterase inhibition using classical QSAR and CoMFA analyses
| No. | TFK structureb | pKia |
log Pc | L | B5 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hiCE |
hCE1 |
rCE |
||||||||||||||||||||
| Obsd 5 min | Obsd 24 h | Eq. (7a) | Eq. (7b) | Eq. (10a) | Eq. (10b) | Obsd 5 min | Obsd 24 h | Eq. (8a) | Eq. (8b) | Eq. (11a) | Eq. (11b) | Obsd 5 min | Obsd 24 h | Eq. (9a) | Eq. (9b) | Eq. (12a) | Eq. (12b) | |||||
| 33 | 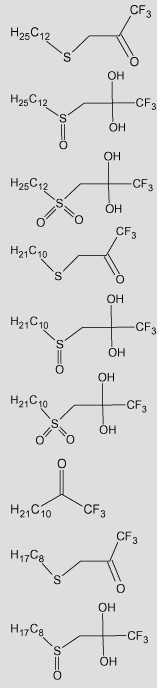 |
7.82 | 8.27 | 8.24 | 8.24 | 7.71 | 7.75 | 7.15 | 8.13 | 7.23 | 7.26 | 7.11 | 7.03 | 6.80 | 8.48 | 6.73 | 6.76 | 6.84 | 6.81 | 6.63 (6.07) | 14.38 | 10.27 |
| 34 | 7.27 | 9.40 | 7.07 | 7.14 | 6.77 | 6.86 | 5.72 | 9.40 | 5.87 | 5.89 | 5.65 | 5.64 | 5.96 | 8.85 | 6.24 | 6.23 | 5.73 | 5.84 | 5.12 (4.43) | 14.38 | 10.27 | |
| 35 | 7.49 | 9.00 | 7.01 | 7.08 | 7.65 | 7.70 | 6.13 | 9.40 | 5.80 | 5.81 | 6.33 | 6.30 | 6.82 | 9.15 | 6.20 | 6.19 | 6.95 | 6.88 | 5.04 (4.34) | 14.38 | 10.27 | |
| 36 | 6.82 | 8.34 | 7.49 | 7.47 | 7.08 | 7.13 | 6.89 | 8.40 | 7.07 | 7.11 | 7.02 | 6.89 | 6.80 | 8.55 | 6.96 | 7.00 | 6.88 | 6.80 | 5.57 (5.02) | 12.33 | 8.33 | |
| 37 | 5.72 | 9.30 | 6.32 | 6.37 | 6.00 | 5.90 | 5.28 | 9.30 | 5.71 | 5.74 | 5.53 | 5.46 | 5.60 | 8.52 | 6.13 | 6.15 | 5.57 | 5.66 | 4.06 (3.37) | 12.33 | 8.33 | |
| 38 | 7.10 | 8.96 | 6.25 | 6.32 | 6.82 | 6.82 | 6.10 | 9.52 | 5.64 | 5.67 | 6.05 | 6.04 | 6.66 | 8.80 | 6.07 | 6.09 | 6.55 | 6.41 | 3.98 (3.28) | 12.33 | 8.33 | |
| 39 | 7.66 | 5.69 | 7.31 | 7.14 | 7.94 | 7.59 | 6.82 | 8.64 | 7.20 | 7.09 | 6.63 | 6.93 | 6.85 | 5.49 | 7.10 | 7.06 | 6.85 | 6.97 | 5.11 (4.76) | 10.27 | 7.39 | |
| 40 | 6.66 | 8.46 | 6.74 | 6.70 | 6.64 | 6.67 | 6.72 | 8.38 | 6.55 | 6.54 | 6.87 | 6.75 | 6.82 | 8.11 | 6.72 | 6.72 | 6.85 | 6.75 | 4.51 (3.96) | 10.27 | 7.39 | |
| 41 | 5.62 | 9.30 | 5.57 | 5.60 | 5.47 | 5.49 | 4.83 | 9.22 | 5.18 | 5.17 | 4.45 | 4.66 | 5.31 | 8.19 | 5.56 | 5.56 | 5.25 | 5.26 | 3.00 (2.31) | 10.27 | 7.39 | |
| 42 | 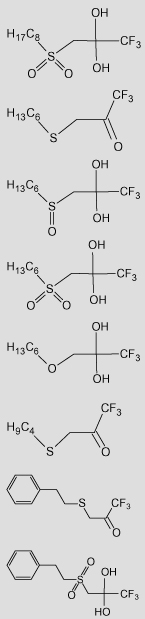 |
5.33 | 8.24 | 5.51 | 5.55 | 5.64 | 5.62 | 5.82 | 9.22 | 5.12 | 5.10 | 5.62 | 5.65 | 5.32 | 7.43 | 5.49 | 5.48 | 5.67 | 5.62 | 2.92 (2.23) | 10.27 | 7.39 |
| 43 | 6.25 | 8.19 | 5.99 | 5.94 | 6.03 | 6.30 | 6.48 | 8.28 | 6.20 | 6.19 | 6.39 | 6.58 | 6.60 | 8.00 | 6.39 | 6.37 | 6.45 | 6.62 | 3.46 (2.90) | 8.22 | 5.96 | |
| 44 | 4.17 | 9.30 | 4.82 | 4.84 | 4.51 | 4.43 | 4.10 | 8.48 | 4.85 | 4.82 | 4.18 | 4.24 | 4.64 | 7.66 | 4.90 | 4.89 | 4.71 | 4.75 | 1.95 (1.26) | 8.22 | 5.96 | |
| 45 | 4.56 | 8.43 | 4.75 | 4.78 | 4.68 | 4.56 | 5.01 | 9.15 | 4.77 | 4.74 | 5.16 | 5.09 | 4.82 | 7.46 | 4.80 | 4.80 | 4.98 | 4.95 | 1.87 (1.17) | 8.22 | 5.96 | |
| 46 | 5.04 | 8.92 | 5.45 | 5.43 | 5.38 | 4.99 | 5.48 | 9.00 | 5.57 | 5.55 | 5.87 | 5.37 | 5.57 | 8.77 | 5.76 | 5.74 | 5.79 | 5.51 | 2.76 (2.14) | 8.22 | 5.96 | |
| 47 | 5.77 | 7.78 | 5.23 | 5.17 | 5.44 | 5.64 | 6.10 | 8.27 | 5.85 | 5.82 | 6.02 | 6.16 | 6.38 | 8.11 | 5.84 | 5.79 | 6.07 | 6.21 | 2.40 (1.84) | 6.17 | 4.54 | |
| 48 | 6.12 | 8.32 | 5.59 | 5.54 | 5.89 | 6.19 | 6.92 | 8.19 | 6.62 | 6.69 | 6.51 | 6.79 | 6.39 | 8.26 | 6.54 | 6.57 | 6.37 | 6.50 | 2.91 (2.35) | 8.33 | 3.58 | |
| 49 | 4.29 | 6.93 | 4.37 | 4.39 | 4.07 | 4.06 | 4.86 | 8.22 | 5.19 | 5.24 | 5.02 | 4.84 | 4.87 | 6.97 | 4.77 | 4.82 | 4.71 | 4.66 | 1.32 (0.62) | 8.33 | 3.58 | |
The experimental Ki values (Obsd) are from Wadkins.27 Predicted Ki values for the classical QSAR are based upon Eq. (7a),Eq. (8a), and Eq. (9a) for gem-diol geometry and Eq. (7b),Eq. (8b), and Eq. (9b) for ketone geometry. Predicted Ki values for the 3D-QSAR are based upon Eq. (10a),Eq. (11a), and Eq. (12a) for gem-diol geometry and Eq. (10b),Eq. (11b), and Eq. (12b) for ketone geometry.
TFK structures are drawn as shown in Wadkins27, in which the hydration of the gem-diol/ketone is shown based upon 1H NMR observations. However, the compounds exist in a dynamic equilibrium between ketone and gem-diol in solution.
The log P value for the ketone is the first value and the gem-diol is given in parentheses.
2.4. 3D-QSAR analysis for the TFK compounds
Because earlier work had demonstrated that the ketone form of TFK-inhibitors performed better in CoMFA analyses, the geometries (ketone vs gem-diol) were also separated in this study.37 The incubation time of inhibitor and enzyme has also been shown to significantly affect the observed inhibition potency, with earlier studies evidencing distinct differences following a 5 min or 24 h inhibitor/enzyme incubation.27 Accordingly, multiple CoMFA models were built using these four different conditions in order to determine which combination provided the most statistically significant model. The TFK-based inhibitors were superposed in both the gem-diol and ketone forms (Fig. S2). The different correlations and parameters employed are shown in Table 5 for the gem-diol and Table 6 for the ketone. Results showed that the CoMFA statistics for the ketone geometry consistently were slightly improved over that for the gem-diol forms and that the 5 min incubation was significantly improved over the 24 h incubation. The best equations for the gem-diol geometry for all three enzymes are given below as Eq. (10a), Eq. (11a), and Eq. (12a) (correlations 30a, 33a and 35a, respectively; Table 5), and the equivalent ketone equations are Eq. (10b), Eq. (11b), and Eq. (12b) (correlations 39a, 42a and 44a, respectively; Table 6).
| (10a) |
| (11a) |
| (12a) |
| (10b) |
| (11b) |
| (12b) |
Table 5.
CoMFA statistics for TFK-inhibitors in gem-diol conformationa
| Enzyme | q2 | spress | m | r2 | SE | log P | log P2 | Corr No. |
|---|---|---|---|---|---|---|---|---|
| Data for 5 min Ki incubation | ||||||||
| hiCEb | 0.275 | 1.156 | 4 | 0.966 | 0.251 | — | — | 28a |
| 0.767 | 0.586 | 1 | 0.845 | 0.478 | ○ | — | 29a | |
| 0.771 | 0.625 | 3 | 0.945 | 0.305 | ○ | ○ | 30a | |
| hCE1c | 0.474 | 0.709 | 3 | 0.922 | 0.273 | — | — | 31a |
| 0.719 | 0.518 | 3 | 0.925 | 0.267 | ○ | — | 32a | |
| 0.732 | 0.506 | 3 | 0.939 | 0.241 | ○ | ○ | 33a | |
| rCEd | 0.563 | 0.611 | 4 | 0.960 | 0.184 | — | — | 34a |
| 0.794 | 0.419 | 4 | 0.959 | 0.188 | ○ | — | 35a | |
| 0.787 | 0.427 | 4 | 0.957 | 0.191 | ○ | ○ | 36a | |
| Data for 24 h Ki incubation | ||||||||
| hiCE | 0.120 | 1.026 | 4 | 0.979 | 0.160 | — | — | 28b |
| −0.091 | 1.143 | 4 | 0.962 | 0.215 | ○ | — | 29b | |
| −0.075 | 1.135 | 4 | 0.957 | 0.227 | ○ | ○ | 30b | |
| hCE1 | 0.442 | 0.423 | 3 | 0.921 | 0.159 | — | — | 31b |
| 0.463 | 0.400 | 2 | 0.867 | 0.199 | ○ | — | 32b | |
| 0.503 | 0.416 | 4 | 0.934 | 0.152 | ○ | ○ | 33b | |
| rCE | 0.050 | 0.947 | 3 | 0.936 | 0.246 | — | — | 34b |
| −0.169 | 0.978 | 1 | 0.256 | 0.780 | ○ | — | 35b | |
| −0.167 | 0.977 | 1 | 0.155 | 0.831 | ○ | ○ | 36b | |
The parameters employed in the CoMFA analysis are indicated as included (○), or not included (—) in the generation of the final equation. The statistical quality of the models is described in terms of: q = leave-one-out cross-validated correlation coefficient, spress = cross-validated standard error, m = number of components, r = conventional correlation coefficient, SE = conventional standard error.
Correlation 30a was selected as the best correlation and is shown as Eq. (10a) and displayed in Figure 3.
Table 6.
CoMFA statistics for TFK-inhibitors in ketone conformationa
| Enzyme | q2 | spress | m | r2 | SE | log P | log P2 | Corr No. |
|---|---|---|---|---|---|---|---|---|
| Data for 5 min Ki incubation | ||||||||
| hiCEb | 0.447 | 1.010 | 4 | 0.952 | 0.299 | — | — | 37a |
| 0.789 | 0.624 | 4 | 0.969 | 0.241 | ○ | — | 38a | |
| 0.803 | 0.603 | 4 | 0.969 | 0.237 | ○ | ○ | 39a | |
| hCE1c | 0.638 | 0.612 | 4 | 0.968 | 0.181 | — | — | 40a |
| 0.822 | 0.429 | 4 | 0.979 | 0.146 | ○ | — | 41a | |
| 0.825 | 0.425 | 4 | 0.982 | 0.137 | ○ | ○ | 42a | |
| rCEd | 0.673 | 0.528 | 4 | 0.957 | 0.191 | — | — | 43a |
| 0.861 | 0.345 | 4 | 0.966 | 0.171 | ○ | — | 44a | |
| 0.859 | 0.347 | 4 | 0.969 | 0.161 | ○ | ○ | 45a | |
| Data for 24 h Ki incubation | ||||||||
| hiCE | 0.296 | 0.918 | 4 | 0.986 | 0.128 | — | — | 37b |
| 0.115 | 1.029 | 4 | 0.976 | 0.169 | ○ | — | 38b | |
| 0.112 | 1.031 | 4 | 0.975 | 0.171 | ○ | ○ | 39b | |
| hCE1 | 0.563 | 0.374 | 3 | 0.912 | 0.168 | — | — | 40b |
| 0.606 | 0.342 | 2 | 0.849 | 0.212 | ○ | — | 41b | |
| 0.623 | 0.362 | 4 | 0.950 | 0.132 | ○ | ○ | 42b | |
| rCE | 0.099 | 0.959 | 3 | 0.968 | 0.180 | — | — | 43b |
| −0.135 | 0.963 | 1 | 0.230 | 0.793 | ○ | — | 44b | |
| −0.134 | 0.963 | 1 | 0.146 | 0.836 | ○ | ○ | 45b | |
The parameters employed in the CoMFA analysis are indicated as included (○), or not included (—) in the generation of the final equation. The statistical quality of the models is described in terms of: q = leave-one-out cross-validated correlation coefficient, spress = cross-validated standard error, m = number of components, r = conventional correlation coefficient, SE = conventional standard error.
Correlation 39a was selected as the best correlation and is shown as Eq. (10b) and displayed in Figure 3.
As shown in Table 5 and Table 6, the addition of log P improved the correlation for all enzymes, which is consistent with what is known about TFK QSAR. The addition of the squared log P term improved the correlations for the human enzymes in the ketone geometry (Corr 38 vs 39 and 41 vs 42), but did not improve the correlation for rCE (Corr 44 vs 45). The CoMFA maps for both the gem-diol and the ketone are shown in Figure 3. The maps are extremely similar for all three enzymes, with only slight variations in the field magnitude. In addition, the gem-diol and ketone exhibited very similar maps, with the ketone fields consistently displaying larger fields relative to the gem-diols. Key areas of biological interaction were centered around the sulfur moiety beta to the ketone/diol and then a number of steric interactions were observed along the carbon backbone of the aliphatic chain. For hiCE, a region where positive electronic potential increases inhibition potency was observed surrounding the beta/gamma position to the ketone/diol. This activity was most pronounced for thioether-containing compounds (33, 36, 40, 43, 47 and 48). An area of beneficial steric bulk was observed for long chain sulfone-containing compounds (35 and 38) at the end of the alkyl chain template. For hCE1, the positive electronic potential surrounding the beta/gamma position to the ketone/diol only increased the inhibition potency for the non-sulfur substituted compound (39). Two regions of beneficial steric bulk were observed, one for the thioether-containing compounds (33, 36, 40, 43, 47 and 48) in the middle of the alkyl chain template and one for sulfone- containing compounds (35, 38 and 42) at the end of the template chain. A region of unfavorable steric bulk was observed for the sulfoxide-containing compounds at the end of the alky chain template (34 and 37). The results for rCE were similar to hCE1, with the main difference being the absence of the sterically favorable region in the middle of the alkyl chain for the thioether-containing compounds. Figures S3–S5 describe the contour maps for all three enzymes in more detail.
Figure 3.

CoMFA maps for the TFK-containing inhibitors examined in this study for the three carboxylesterases examined. Maps are shown for both the gem-diol and ketone geometries using the Ki data for 5 min incubation of inhibitor and enzyme. For hiCE the displayed maps are (A) gem-diol (Eq. (7a)) and (B) ketone (Eq. (7b)), for hCE1 the displayed maps are (C) gem-diol (Eq. (8a)) and (D) ketone (Eq. (8b)), for rCE the displayed maps are (E) gem-diol (Eq. (9a)) and (F) ketone (Eq. (9b)). The template molecule is 1,1,1-trifluoro-3-dodecylsulfanyl-propan-2,2-diol or 1,1,1-trifluoro-3-dodecylsulfanyl-propan-2-one (33). The contours are shown to surround regions where a higher steric bulk increases (green) or decreases (yellow) the inhibitory activity and a negative (red) or positive electronic potential (blue) amplifies biological activity.
2.5. 3D-QSAR analysis for the benzil-analogs and TFK compounds combined
In order to develop a comprehensive model of CaE inhibition, the TFK and benzil models were combined. Because the ketone geometry with a 5 min inhibitor/enzyme incubation had been identified as the best model, those parameters were used in the combined TFK and benzil model. The resulting correlations are provided in Table 7, with the three best equations shown below as (13), (14) and (15).
| (13) |
| (14) |
| (15) |
Table 7.
CoMFA statistics for combined TFK and benzil CoMFA modela
| Enzyme | q2 | spress | m | r2 | SE | log P | log P2 | vol | vol2 | Corr No. |
|---|---|---|---|---|---|---|---|---|---|---|
| hiCEb | 0.593 | 0.712 | 5 | 0.922 | 0.310 | — | — | — | — | 46 |
| 0.627 | 0.689 | 6 | 0.934 | 0.290 | ○ | — | — | — | 47 | |
| 0.641 | 0.676 | 6 | 0.927 | 0.304 | ○ | ○ | — | — | 48 | |
| 0.603 | 0.703 | 5 | 0.920 | 0.315 | — | — | ○ | — | 49 | |
| 0.596 | 0.708 | 5 | 0.919 | 0.318 | — | — | ○ | ○ | 50 | |
| 0.648 | 0.669 | 6 | 0.932 | 0.295 | ○ | — | ○ | — | 51 | |
| 0.584 | 0.719 | 5 | 0.871 | 0.401 | ○ | — | ○ | ○ | 52 | |
| 0.641 | 0.677 | 6 | 0.923 | 0.313 | ○ | ○ | ○ | — | 53 | |
| 0.681 | 0.645 | 7 | 0.932 | 0.298 | ○ | ○ | ○ | ○ | 54 | |
| hCE1c | 0.334 | 0.688 | 2 | 0.632 | 0.511 | — | — | — | — | 55 |
| 0.517 | 0.600 | 4 | 0.843 | 0.342 | ○ | — | — | — | 56 | |
| 0.535 | 0.589 | 4 | 0.840 | 0.346 | ○ | ○ | — | — | 57 | |
| 0.400 | 0.661 | 3 | 0.712 | 0.458 | — | — | ○ | — | 58 | |
| 0.359 | 0.675 | 2 | 0.591 | 0.539 | — | — | ○ | ○ | 59 | |
| 0.621 | 0.532 | 4 | 0.838 | 0.348 | ○ | — | ○ | — | 60 | |
| 0.623 | 0.531 | 4 | 0.850 | 0.334 | ○ | — | ○ | ○ | 61 | |
| 0.596 | 0.549 | 4 | 0.818 | 0.369 | ○ | ○ | ○ | — | 62 | |
| 0.593 | 0.551 | 4 | 0.811 | 0.376 | ○ | ○ | ○ | ○ | 63 | |
| rCEd | 0.600 | 0.663 | 3 | 0.807 | 0.461 | — | — | — | — | 64 |
| 0.689 | 0.592 | 4 | 0.874 | 0.376 | ○ | — | — | — | 65 | |
| 0.634 | 0.642 | 4 | 0.863 | 0.393 | ○ | ○ | — | — | 66 | |
| 0.534 | 0.715 | 3 | 0.823 | 0.441 | — | — | ○ | — | 67 | |
| 0.530 | 0.718 | 3 | 0.827 | 0.436 | — | — | ○ | ○ | 68 | |
| 0.632 | 0.643 | 4 | 0.852 | 0.408 | ○ | — | ○ | — | 69 | |
| 0.630 | 0.645 | 4 | 0.841 | 0.423 | ○ | — | ○ | ○ | 70 | |
| 0.587 | 0.681 | 4 | 0.829 | 0.438 | ○ | ○ | ○ | — | 71 | |
| 0.582 | 0.686 | 4 | 0.819 | 0.451 | ○ | ○ | ○ | ○ | 72 |
The parameters employed in the CoMFA analysis are indicated as included (○), or not included (—) in the generation of the final equation. The TFK data are for the ketone geometry with Ki values for a 5 min incubation. The statistical quality of the models is described in terms of: q = leave-one-out cross-validated correlation coefficient, spress = cross-validated standard error, m = number of components, r = conventional correlation coefficient, SE = conventional standard error.
Correlation 49 was selected as the best correlation and is shown as Eq. (13) and displayed in Figure 5.
As described above, the QSAR models differed between the benzil-analog and TFK compounds, which is expected based upon their structural differences. Accordingly, combining both groups of compounds into a single model should provide a greater indication of the physicochemical parameters involved in CaE inhibitor binding and potency. The benzil-analogs and TFK compounds were superposed using four common atoms –CC(O)C– to execute the CoMFA analysis; however significant results were not expected due to the poor superposition between the benzil and TFK compounds (Fig. 4). Surprisingly, the statistical quality of the models (Table 8) show that the combined models effectively described a significant portion of observed biological activity, with model quality varying on an enzyme-specific basis. The mixed model for hiCE was superior to the benzil-only model (r2 = 0.92 relative to 0.88) and almost as good as the TFK-only model (r2 = 0.97), whereas the hCE1 mixed model (r2 = 0.85) evidenced lower correlations than either the benzil- or TFK-only models (r2 = 0.97and 0.98, respectively) and the rCE mixed model (r2 = 0.87) gave varied results with the benzil-only model being lower (r2 = 0.76), but the TFK model greater (r2 = 0.97). The important observation from these results is that a mixed model of CaE inhibition based upon structurally divergent compounds can describe from 85% to 92% of observed biological activity.
Figure 4.

Superposition for all 49 compounds used to generate the mixed TFK and benzil-analog CoMFA analysis.
Table 8.
Squared correlation coefficients for all QSAR modelsa
| Inhibitor | hiCE | hCE1 | rCE | |||
|---|---|---|---|---|---|---|
| Benzil | Eq. (1)b | 0.33 | Eq. (2) | 0.35 | Eq. (3) | 0.20 |
| Eq. (4) | 0.88 | Eq. (5) | 0.97 | Eq. (6) | 0.76 | |
| Eq. (7a) | 0.85 | Eq. (8a) | 0.81 | Eq. (9a) | 0.83 | |
| TFK | Eq. (7b) | 0.84 | Eq. (8b) | 0.82 | Eq. (9b) | 0.82 |
| Eq. (10a) | 0.95 | Eq. (11a) | 0.94 | Eq. (12a) | 0.96 | |
| Mixed | Eq. (10b) | 0.97 | Eq. (11b) | 0.98 | Eq. (12b) | 0.97 |
| Eq. (13) | 0.92 | Eq. (14) | 0.85 | Eq. (15) | 0.87 |
The squared Pearson’s correlation coefficients (r2) were determined for experimental and predicted Ki values for each QSAR equation. The individual experimental and predicted Ki values for the benzil-analogs are given in Table 3, for the TFK compounds in Table 4 and for the mixed TFK and benzil model in Supplementary Table S1.
Eq. (1)–Eq. (3) are for the classical QSAR analysis for the benzil-analogs (n ≤ 32). Eq. (4)–Eq. (6) are for 3D-QSAR analysis with CoMFA for the benzil-analogs (n ≤ 32). Eq. (7a), Eq. (8a), and Eq. (9a) are for the classical QSAR analysis for the TFK-inhibitors with the gem-diol geometry (n = 17). Eq. (7b), Eq. (8b), and Eq. (9b) are for the classical QSAR analysis for the TFK-inhibitors with the ketone geometry (n = 17). Eq. (10a), Eq. (11a), and Eq. (12a) are for the 3D-QSAR analysis with CoMFA for the TFK-inhibitors with the gem-diol geometry (n = 17). Eq. (10b), Eq. (11b), and Eq. (12b) are for the 3DQSAR analysis with CoMFA for the TFK-inhibitors with the ketone geometry (n = 17). Eq. (13)–Eq. (15) are for the 3D-QSAR analysis with CoMFA for the mixture of benzil-analogs and TFK-inhibitors with the ketone geometry (n ≤ 49).
The resulting CoMFA maps for the mixed models are displayed in Figure 5. The main differences in the combined models relative to the initial benzil-analog models involve an increase in the description of the steric fields in the active site. For all three enzymes, large regions of unfavorable steric activity are observed surrounding the upper portion of the B-ring, whereas steric bulk is favorable in the lower portion of the B-ring. In addition, the extent of the steric fields surrounding the A-ring were reduced in the combined model, with a small sterically favorable field around the 3-, 4- and 5-positions of hCE1 and rCE and an unfavorable field around the 4- and 5-postions in hiCE. Notably, the inhibition of hCE1 correlated negatively with the steric effects, suggesting that the active site gorge of hCE1 is smaller than that of hiCE and rCE. However, it is possible that if the alkyl group ‘tails’ of the TFK-inhibitors were not in extended conformations that a better superposition and subsequently improved model may have been generated.
Figure 5.

CoMFA maps for the mixed TFK and benzil-analog models for the three carboxylesterases examined. (A) hiCE (Eq. (13)), (B) hCE1 (Eq. (14)) and (C) rCE (Eq. (15)). The template molecule is 1-(3,4-dimethylphenyl)-2-phenylethane-1,2-dione (12). The contours are shown to surround regions where a higher steric bulk increases (green) or decreases (yellow) the inhibitory activity and a negative (red) or positive electronic potential (blue) amplifies biological activity.
To construct the mixed model, the volume parameters were calculated for the benzil-analog A-ring and the CF3 moiety of the TFK-containing inhibitors. The volume parameters were previously identified as significant for the benzil-analog model, but insignificant for the TFK-based model. Results for the mixed model showed that the vol2 term was only significant for hCE1. This finding is due to the contributions of the TFK-containing inhibitors, which all have identical volumes and is consistent with the TFK-only CoMFA. This result demonstrates an important caution when generating combined models from compounds with significant structural divergence. In this case, the lack of variability in the volume of the TFK-containing inhibitors reduces the overall statistical significance of this parameter in the final model, thereby masking this important parameter. Overall, the combined model suggests that maximum inhibitor potency and selectivity can be achieved through a combination of the two scaffolds, with the benzil-analog A-ring conveying selectivity, the 1,2-dione moiety necessary for biological activity, and the TFK alkyl chain providing potency.
3. Discussion
Previous work with benzil-analogs identified the necessary physiochemical properties for efficient selective inhibition of mammalian CaEs: (1) the 1,2-dione moiety, (2) aromatic (or potentially highly hydrophobic) domains adjacent to the dione and (3) substitutions on the aromatic rings that do not impede inhibitor access to the active site.29 The results of the QSAR analyses performed in this study were consistent with these properties. In particular, the need for highly hydrophobic domains adjacent to the dione was observed. This point is further strengthened by the benzil-TFK mixed model, which suggested that the hydrophobic aliphatic chain of the TFK-inhibitors would enhance benzil-analog inhibition potency. In addition, the substitutions on the A-ring were found to be extremely important for inhibitor selectivity. The models were not able to evaluate the necessity of including the 1,2-dione, as this moiety was defined by the user to be the basis of the compound alignment for the CoMFA analysis. However, the importance of the electron-deficient ketone in the inhibitory mechanism of nucleophilic catalytic mechanisms has been well-established in the literature.29,31,38
A number of previous studies have examined the physicochemical parameters surrounding TFK-mediated CaE inhibition using both classical and 3D-QSAR.27,28,37–39 However, the majority of these studies focused on the insect ortholog (juvenile hormone esterase, JHE). This study is the first to our knowledge to build either classical or CoMFA-based models to examine TFK-mediated inhibition of mammalian esterases. Because previous studies have shown that the geometry of the inhibitor can be important for inhibition potency and model construction, we built a series of models where all inhibitors were in either the ketone or the gem-diol conformation. One of the remaining questions regarding TFK-mediated esterase inhibition involves the active form of the inhibitor geometry during the inhibition process. Previous CoMFA-based studies with JHE suggested that the ketone was the active form based upon superior model statistics relative to the gem-diol-based models.37 These results were further confirmed in this study with mammalian CaEs in that for all three enzymes examined, the ketone-based models evidenced improved statistical power relative to the corresponding gem-diol models. The magnitude of the difference was small, but the consistency of the trend is further evidence to support the hypothesis that the ketone geometry is the ‘active’ form of the inhibitor. However, this hypothesis still needs to be confirmed with experimental evidence. Interestingly, for the classical QSAR model, no significant differences were observed between the ketone and gem-diol geometries. Earlier work with JHE showed significant differences between the ketone and gem-diol models (r2 = 0.72 and 0.69, respectively).37 Accordingly, the evidence to date indicates that the ketone is the active form of the inhibitor, but that the energy difference is extremely small. This would suggest that the interconversion between ketone and gem-diol is facile and does not represent a limiting factor in the inhibition process.
Aliphatic TFK-containing inhibitors tend to exhibit a strong positive correlation with lipophilicity;28 however, the inhibition potency can be affected by specific structural substitutions that do not affect the overall hydrophobicity or volume of the molecule. This correlation tends to be weaker for cyclic or branched chain aliphatic inhibitors. TFKs are considered to be slow tight-binding inhibitors, with binding equilibrium reached on the order of hours to days.38,40 Accordingly, published data has examined equilibrium times of 5 min to 24 h.27 Based upon these previously published works, we hypothesized that the models generated from data sets of increased equilibrium times would provide statistically improved results as the system had been given sufficient time to reach ‘true equilibrium’. However, the opposite effect was observed in that the models generated with 5 min Ki data were vastly superior. The exact reasons behind these observations are unfortunately unclear. It is possible, that a 24 h enzyme:inhibitor incubation resulted in loss of enzyme activity, thus affecting the observed inhibitor potency. However, similar studies with benzil-analogs did not observe significant differences in inhibition potency between a 1 h and a 24 h enzyme:inhibitor incubation.29 Previous work examining the effect of extended enzyme:inhibitor incubations using porcine liver CaE with TFKs showed significant loses in enzyme activity by 24 h (data not shown). It may therefore be necessary to have stabilizing cofactors present in the assay system for extended incubations. Ideally, a series of time-points should be examined, with a structural range of compounds examined for enzyme inhibition at time-points ranging from 5 min to 24 h.
By combining the individual CoMFA models for benzil-analogs and TFK compounds, we were able to construct a more inclusive model of CaE inhibition. Both classes of compounds inhibit CaE activity through a similar mechanism that involves nucleophilic attack by a catalytic serine residue. However, benzil-mediated inhibition involves a cycling reaction in which the serine residue attacks one of the carbonyl groups to form a reversible covalent intermediate that can revert to benzil and the free enzyme. Benzil can also be hydrolyzed to the two benzyl ring products benzaldehyde and benzoic acid.20 TFK-mediated CaE inhibition involves a direct nucleophilic attack on the carbon atom of the ketone (or potentially the gem-diol) to form the transition state analog complex. This process is reversible; however, hydrolysis products have not been observed. Accordingly, the inhibition mechanisms are sufficiently similar that a unified CoMFA model could be constructed. A major caveat though is the fact that the superposition of the compounds is performed manually, with the superposed atoms defined by the user, thereby introducing significant bias into the system. For this study, it has been established that the ketone moiety is vital for CaE inhibition with either benzil-analogs or TFKs. Accordingly, both atoms of the carbonyl moiety as well as an adjacent carbon atom on each side of the carbonyl were used as the template for the superposition (Fig. 4).
According to Bencharit,41 the opening of the hCE1 active site gorge is at the top of the enzyme and the catalytic amino acids reside at the bottom. The active sites of hCE1 exist as relatively hydrophobic deep gorges within the enzymes. It is likely that the gorge size varies among the different CaEs. As shown in Eq. (1)–Eq. (3), the gorge of the inhibitor binding cite appears to be larger in rCE compared to that of the human enzymes. Interestingly, there is also a difference in gorge size between hiCE and hCE1, with hiCE appearing to be larger than hCE1. As shown above, the vol2 term was significant in Eq. (4) and Eq. (5), but not in Eq. (6), indicating that there is an optimum volume for inhibition of the human enzymes [calculated to be 3.4 for hiCE (Eq. (4)) and 0.69 for hCE1 (Eq. (5))]. The volume range of the benzil-analogs is 2.66–4.59 (Table 3). Accordingly, the optimal volume of hCE1 means that inhibition increases with decreasing volume, while the volume parameter for rCE positively correlated with inhibition, and the optimal volume for hiCE was in the range of the benzil-analogs (3.4). These results demonstrate that the volume parameter plays a role in CaE selectivity. It is important to stress that these results do not contradict that of the TFK-containing compounds, which did not evidence selectivity (the volume range was 2.01–2.02). Because all TFK-containing inhibitors have the same volume for the CF3 group, there is no volume variability and subsequently the parameter is not significant in model construction. This fact heavily influenced the 3D-QSAR analysis for the combined TFK/benzil model resulting in a significant reduction of the significance of the volume in generation of the model.
The models developed in the current study support previously published work reporting that the volume of the esterase active site is important for isoform-selectivity.12,17,41 Towards this end, the volumes of the acid and alcohol moieties of a number of CaE substrates were calculated to further explore isoform-selectivity (Table 9). CaEs are generally reported as exhibiting broad substrate selectivity; however, studies have shown that there are distinct isoform-specific preferences. Tang reported that hCE1 demonstrates selectivity for esters containing a large acid moiety (small alcohol), whereas the opposite is true with hiCE.12 They applied an alcohol:acid size-based ratio to explain the observed substrate selectivity. hCE1, but not hiCE, was shown to hydrolyze clopidogrel (alcohol:acid volume = 1.78:7.21), whereas aspirin (alcohol:acid volume = 4.12:1.42) was preferably hydrolyzed by hiCE. This trend has also been observed in the hydrolysis of cocaine, which contains two esters. hCE1 hydrolyzes cocaine to benzoylecgonine (alcohol: acid volume = 1.72:7.22), whereas hiCE hydrolyzes the benzoyl group (alcohol:acid volume = 5.70:3.13) to give the ecgonine methyl ester.8 CPT-11 is selectively hydrolyzed by hiCE (alcohol: acid volume = 10.22:5.96)42, whereas oseltamivir is preferred by hCE1 (alcohol:acid volume = 2.33:7.58).13 However, the pyrethroid permethrin (alcohol:acid volume = 6.10:4.81) appears to be an exception, with hydrolysis ~3-fold more rapid by hCE1 than hiCE, with exact rates dependent upon the stereochemistry.43 A comparison of the standard substrates p-nitrophenyl acetate (alcohol: acid volume = 4.07:1.40), o-nitrophenyl acetate (alcohol:acid volume = 4.20:1.48) and 4-methylumbelliferone acetate (alcohol: acid volume = 5.28:1.48) show that hiCE-mediated hydrolysis is ~2-fold,43 1.4-fold,44 and 30-fold45 greater than hCE1, respectively. Accordingly, these limited data suggest that an alcohol volume of ~3 is the approximate cut-off between selectivity between hCE1 and hiCE. These observations are supported by Eq. (1) and Eq. (2) for hiCE and hCE1, respectively. These two equations were graphed in Figure S6, intersecting at a volume of ~3 and ~6. Accordingly, these data agree with the above results for the alcohol: acid volume, suggesting that alcohol volumes >3 shift the isoform preference from hCE1 to hiCE and that alcohol volumes >6 shift the preference back to hCE1 from hiCE.
Table 9.
Volume and ClogP parameters for carboxylesterase substrates
| Structure | Name | Vola | ClogPb |
|---|---|---|---|
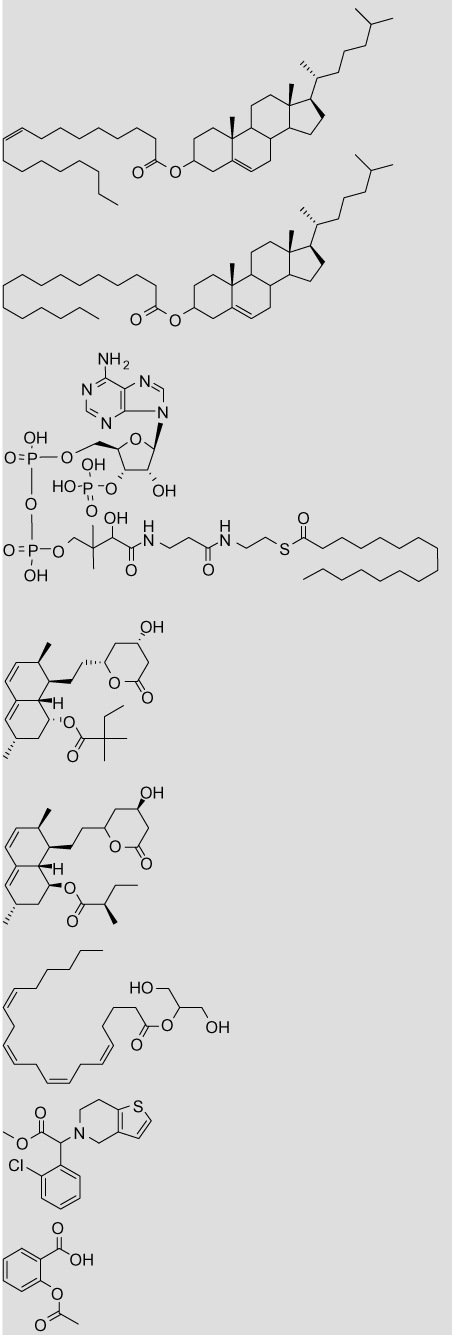 |
Cholesteryl oleate | 9.42 (11.81) | 18.45 |
| Cholesteryl palmitate | 9.81 (11.81) | 17.83 | |
| Palmityl-CoA | 9.11 (18.35) | 3.46 | |
| Simvastatin | 3.34 (9.27) | 4.68 (4.70) | |
| Lovastatin | 3.09 (9.43) | 4.26 (4.30) | |
| 2-Arachidonoyl glycerol | 10.49 (3.43) | 6.32 | |
| Clopidogrel | 7.21 (1.78) | 4.21 | |
| Aspirin | 1.42 (4.12) | 1.12 (1.02) | |
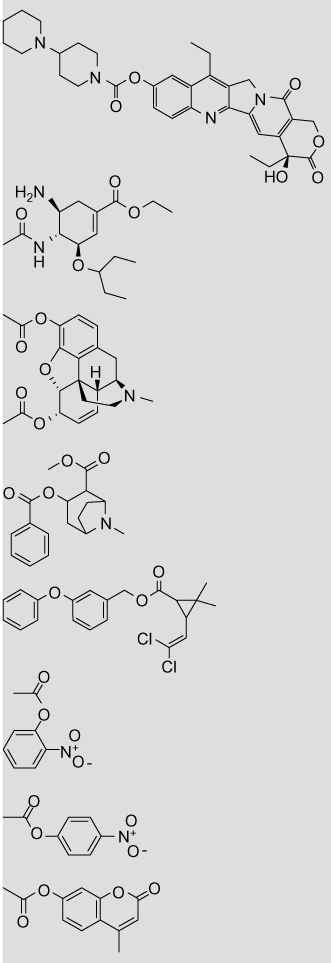 |
CPT-11 | 5.96 (10.22) | 2.72 |
| Oseltamivir | 7.58 (2.33) | 2.33 | |
| Heroin | 1.48 (8.54), 1.48 (8.68) | 1.58 (1.50) | |
| Cocaine | 7.22 (1.72), 3.13 (5.70) | 2.30 (2.57) | |
| Permethrin | 4.81 (6.10) | 6.50 (7.38) | |
| o-Nitrophenyl acetate | 1.48 (4.20) | 1.55 (1.50) | |
| p-Nitrophenyl acetate | 1.40 (4.07) | 1.50 (1.53) | |
| 4-Methyl umbelliferone acetate | 1.48 (5.28) | 1.90 (2.11) |
Volumes are given for the substrate hydrolysis products: the acid and the alcohol, with the alcohol value in parenthesis. The carbonyl group was excluded from all calculations. In cases where the substrate contains multiple ester moieties (cocaine and heroin), the volumes are given for the hydrolysis products of each ester. Volumes are not given for lactone hydrolysis in the statins (simvastatin and lovastatin) as the acid and alcohol moieties are still linked following hydrolysis.
The log P values were calculated with the program ClogP as described in Section 5. Values in parentheses are measured log P taken from literature sources.48
Based upon the volume, it would be predicted that the fatty acid cholesteryl esters and palmityl-CoA would be predominantly metabolized by hCE1, whereas the cholesterol-lowering therapeutics simvastatin and lovastatin would be hydrolyzed by hiCE. However, this prediction is complicated because the statins contain multiple esters consisting of different sized alcohols and acids. Work by Fleming on mevastatin reported no observable hCE1-mediated hydrolysis, suggesting that it is a hiCE-mediated process; however in vitro studies showed that mevastatin is a weak inhibitor of hCE1.20 There are of course multiple physiochemical interactions that are important for substrate binding and subsequent hydrolysis; however this simple volume parameter appears to be useful predictor. This information provides a potentially useful ‘rule-of-thumb’ to estimate which isoform will hydrolyze a given substrate and accordingly where in the body the substrate is likely to be metabolized. An important caveat is that it has been reported that the acyl- and alcohol-binding pockets of hCE1 can switch depending on which substrate is bound.19 It is therefore possible that this alcohol:acid volume-associated trend does not hold constant. For example, 2-arachidonyl glycerol consists of a small alcohol (volume = 3.43) and a large acid (volume = 10.49), suggesting that it would be hydrolyzed by hiCE. However, measurement of 2-arachidonyl glycerol hydrolysis by p-nitrobenzyl esterase from Bacillus subtilis suggested that the purported binding mechanism was more similar to hCE1 than hiCE.46
Based upon the models designed in the current study, isoform-selective inhibition appears to be volume dependent. Accordingly, the variability in the A-ring of the benzil-analogs affords the observed selectivity, whereas the TFK-containing inhibitors do not vary in their volume on the ‘A-ring’ (the CF3 moiety). Therefore next generation inhibitors should combine these two different scaffolds, employing the A-ring from the benzyl analogs with the long aliphatic chain of the TFK-inhibitors. A potential scaffold would be similar to 1-phenylpentadecane-1,2-dione, in which the A-ring substitution could be varied to control isoform-selectivity. This general scaffold is hypothesized to provide optimized selectivity and inhibition potency. The length of the alkyl chain could be varied to test inhibition potency. It would also be interesting to introduce various degrees of unsaturation into the alkyl chain to ascertain the ideal geometry.
4. Summary
This study has developed classical and 3D-QSAR models to describe the isoform-selectivity of mammalian CaEs. In addition, the geometry of the ‘active’ form of TFK-containing inhibitors was further examined, with results supporting that of previous studies suggesting that the ketone is the active form. However, the statistical differences between ketone and gem-diol based models was minimal, indicating that additional experimental investigations are required to determine the active inhibitor form. According to both the classical and 3D-QSAR models, the volume parameter was central in determining CaE-isoform-selectivity. This finding was successfully used to explain isoform-selective substrate hydrolysis, with the volume of the acid moiety of ester substrates predictive of isoform preference. This information could be useful in designing the next generation of isoform-selective CaE inhibitors. Towards this end, a general scaffold centered around a 1-phenylpentadecane-1,2-dione structure was suggested as a logical basis for future structural explorations. New isoform-selective inhibitors could be useful tools in elucidating the role of CaEs in fatty acid homeostasis, cholesterol trafficking and the development of cardiovascular disease.
5. Experimental
5.1. QSAR model development for the benzil-analogs
All structures used in this study are listed in Table 1 and Figure 1. Molecule construction was performed using the SYBYL building module (ver 6.91; Tripos Co., St. Louis, MO, USA). All conformations were optimized using the semi-empirical molecular orbital method PM3 and atomic charges were calculated with AM1, another semi-empirical molecular orbital method, using MOPAC5.0. The coordinates of the crystal structure of benzil (1) were downloaded from the Cambridge database and saved as a mol2 file.20 Compound 1 was used as a template to construct the other substituted compounds. Although benzil is a symmetrical molecule, substitutions on the benzene ring can render the compound unsymmetrical. There are other complications in that the benzil bridge structure is symmetrical and there are three bonds with free rotations. Therefore, the conformation of each compound was fixed prior to superposition. Since two ortho (2) and (6) and two meta-positions (3 and 3′) were distinguishable for some structures, we attempted to assign these positions. Subsequently, substitution positions close to the carbonyl oxygens were defined as 2 and 2′ as shown in Figure S7. In this study, the B-ring generally contained a higher degree of substitution than the A-ring.
In the first step, we constructed four conformers (2,2′, 2,6′, 6,2′, 6,6′) for o,o′-dichlorobenzil as shown in Figure S7. In order to identify the most stable conformer, each conformer was optimized using PM3, even though two conformers (2,6′ vs 6,2′) were not distinguishable in their ordinal planes. As a result of the calculations, the 2,2′-conformer (Pattern 1) was selected as the template for further calculations, even though the difference between Pattern 1 and 3 was only 4 kcal/mol with respect to the heat of formation energy.
Para-substituted compounds were constructed by adding the appropriate substituent to the benzil structure at either the 4 or 4′ position. For compounds with meta-substitution, there are two potential configurations (3,3′ and 5,5′). However, in this study we placed all meta-groups at the 5- (and 5′-) position and not at 3-(and 3′-) position, except for compounds containing a nitro group at the meta-position. It was found that this procedure resulted in improved results. The position of nitro-groups at the meta-position was fixed, on an enzyme dependent basis, in order to improve the CoMFA results. The 3,3′-nitrobenzil substitution was selected for hiCE and rCE, and the 5,5′-nitrobenzil substitution was used for hCE1. The six common atoms –C–C(=O)–C(=O)–C– were used in the lattice space (40.0 Å × 40.0 Å × 40.0 Å) to superpose compounds. The superposition of all 32 compounds is shown in Figure S1.
5.2. QSAR model development for the TFK compounds
All conformations of TFK compounds were calculated according to previously published procedures.37 The four common atoms –C–C(=O)–C– were used in the lattice space (28.5 Å × 19.0 Å × 25.0 Å) to superpose gem-diol-type compounds and (26.9 Å × 23.6 Å × 24.8 Å) to superpose ketone-type compounds.
5.3. QSAR model development for the mixed benzil and TFK model
The four common atoms –C–C(=O)–C– were used in the lattice space (29.3 Å × 23.5 Å × 24.7 Å) to superpose the benzil-analogs and ketone-type TFK compounds.
5.4. Determination of external QSAR parameters
Log P values were calculated using the program ClogP47 (Windows Version 4.0, BioByte Corp; CA, USA) and substituent volumes were calculated using the SYBYL module, Molprop volume, and 1/100 calculated values were used as the volume parameter. The L and B5 parameters were taken from literature sources.48
Supplementary Material
Acknowledgments
This research was supported by the Åke Wibergs Stiftelse, the Fredrik and Ingrid Thurings Stiftelse and The Royal Swedish Academy of Sciences. C.E.W. was supported by a Center for Allergy Research Fellowship. P.M.P. was supported by NIH grants CA108775, a Cancer Center Core Grant P30 CA21765, and the American Lebanese Syrian Associated Charities.
Footnotes
Supplementary data
Supplementary data includes figures of the superposition of the benzil-analogs for the CoMFA models (Fig. S1), superposition of the TFK CoMFA models ( Fig. S2), and detailed descriptions of the CoMFA contour maps for the ketone TFK models of hiCE (Fig. S3), hCE1 (Fig. S4), and rCE (Fig. S5). Figure S6 provides a graph of the classical QSAR equations for the benzil-analogs displaying the volume dependence of hCE1 and hiCE. The four conformer patterns tested to determine the most stable conformation of the benzil-analogs for construction of the superposition for the CoMFA models are shown in Figure S7. Supplementary Table S1 provides predicted pKi values as well as the associated physicochemical parameters for all compounds examined in this study using the mixed benzil-analog and TFK-containing inhibitor CoMFA analysis. Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2008.11.008.
References and notes
- 1.Ollis DL, et al. Protein Eng. 1992;5:197. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 2.Cygler M, et al. Protein Sci. 1993;2:366. doi: 10.1002/pro.5560020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heikinheimo P, et al. Structure Fold Des. 1999;7:R141. doi: 10.1016/s0969-2126(99)80079-3. [DOI] [PubMed] [Google Scholar]
- 4.Wheelock CE, et al. J. Pestic. Sci. 2005;30:75. [Google Scholar]
- 5.Redinbo MR, et al. Drug Discov. Today. 2005;10:313. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- 6.Redinbo MR, et al. Biochem. Soc. Trans. 2003;31:620. doi: 10.1042/bst0310620. [DOI] [PubMed] [Google Scholar]
- 7.Crow JA, et al. Biochim. Biophys. Acta. 2008;643:1781. doi: 10.1016/j.bbalip.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dean RA, et al. FASEB J. 1991;5:2735. doi: 10.1096/fasebj.5.12.1916095. [DOI] [PubMed] [Google Scholar]
- 9.Bora PS, et al. J. Mol. Cell. Cardiol. 1996;28:2027. doi: 10.1006/jmcc.1996.0195. [DOI] [PubMed] [Google Scholar]
- 10.Diczfalusy MA, et al. J. Lipid Res. 2001;42:1025. [PubMed] [Google Scholar]
- 11.Cooke GE, et al. J. Am. Coll. Cardiol. 2006;47:541. doi: 10.1016/j.jacc.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 12.Tang M, et al. J. Pharmacol. Exp. Ther. 2006;319:1467. doi: 10.1124/jpet.106.110577. [DOI] [PubMed] [Google Scholar]
- 13.Shi D, et al. J. Pharmacol. Exp. Ther. 2006;319:1477. doi: 10.1124/jpet.106.111807. [DOI] [PubMed] [Google Scholar]
- 14.Satoh T, et al. Annu. Rev. Pharmacol. Toxicol. 1998;38:257. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 15.Stadnyk AW, et al. J. Histochem. Cytochem. 1990;38:1. doi: 10.1177/38.1.1688447. [DOI] [PubMed] [Google Scholar]
- 16.Munger JS, et al. J. Biol. Chem. 1991;266:18832. [PubMed] [Google Scholar]
- 17.Bencharit S, et al. Nat. Struct. Biol. 2002;9:337. doi: 10.1038/nsb790. [DOI] [PubMed] [Google Scholar]
- 18.Bencharit S, et al. Chem. Biol. 2003;10:341. doi: 10.1016/s1074-5521(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 19.Bencharit S, et al. Nat. Struct. Biol. 2003;10:349. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 20.Fleming CD, et al. J. Mol. Biol. 2005;352:165. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Fleming CD, et al. Biochemistry. 2007;46:5063. doi: 10.1021/bi700246n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potter PM, et al. Curr. Med. Chem. 2006;13:1045. doi: 10.2174/092986706776360969. [DOI] [PubMed] [Google Scholar]
- 23.Wadkins RM, et al. Mol. Pharmacol. 2004;65:1336. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]
- 24.Casida JE, et al. Chem. Res. Toxicol. 2004;17:983. doi: 10.1021/tx0499259. [DOI] [PubMed] [Google Scholar]
- 25.Abdel-Aal YAI, et al. Science. 1986;233:1073. doi: 10.1126/science.3738525. [DOI] [PubMed] [Google Scholar]
- 26.Ashour MBA, et al. Biochem. Pharmacol. 1987;36:1869. doi: 10.1016/0006-2952(87)90483-7. [DOI] [PubMed] [Google Scholar]
- 27.Wadkins RM, et al. Mol. Pharmacol. 2007;71:713. doi: 10.1124/mol.105.021683. [DOI] [PubMed] [Google Scholar]
- 28.Székács A, et al. In: Rational Approaches to Structure–Activity and Ecotoxicology of Agrochemicals. Draber W, Fujita T, et al., editors. Boca Raton, FL: CRC Press; 1992. p. 219. [Google Scholar]
- 29.Wadkins RM, et al. J. Med. Chem. 2005;48:2906. doi: 10.1021/jm049011j. [DOI] [PubMed] [Google Scholar]
- 30.Hyatt JL, et al. J. Med. Chem. 2005;48:5543. doi: 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- 31.Hyatt JL, et al. J. Med. Chem. 2007;50:5727. doi: 10.1021/jm0706867. [DOI] [PubMed] [Google Scholar]
- 32.Hyatt JL, et al. J. Med. Chem. 2007;50:1876. doi: 10.1021/jm061471k. [DOI] [PubMed] [Google Scholar]
- 33.Hicks LD, et al. Bioorg. Med. Chem. 2007;15:3801. doi: 10.1016/j.bmc.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansch C, et al. J. Am. Chem. Soc. 1964;86:1616. [Google Scholar]
- 35.Verloop A. In: Pesticide, Chemistry, Human Welfare and Environment. Miyamoto J, Kearney P, editors. Oxford: Pergamon Press; 1983. p. 339. [Google Scholar]
- 36.Hansch C, et al. Exploring QSAR-Hydrophobic Electronic and Steric Constants. Washington, DC: American Chemical Society; 1995. [Google Scholar]
- 37.Wheelock CE, et al. Bioorg. Med. Chem. 2003;11:5101. doi: 10.1016/j.bmc.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 38.Wheelock CE, et al. J. Med. Chem. 2002;45:5576. doi: 10.1021/jm020072w. [DOI] [PubMed] [Google Scholar]
- 39.Wogulis M, et al. Biochemistry. 2006;45:4045. doi: 10.1021/bi0521644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wheelock CE, et al. Bioorg. Med. Chem. 2008;16:2114. doi: 10.1016/j.bmc.2007.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bencharit S, et al. J. Mol. Biol. 2006;363:201. doi: 10.1016/j.jmb.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wadkins RM, et al. Mol. Pharmacol. 2001;60:355. doi: 10.1124/mol.60.2.355. [DOI] [PubMed] [Google Scholar]
- 43.Ross MK, et al. Biochem. Pharmacol. 2006;71:657. doi: 10.1016/j.bcp.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 44.Hatfield MJ, et al. Personal communication. 2008.
- 45.Pindel EV, et al. J. Biol. Chem. 1997;272:14769. doi: 10.1074/jbc.272.23.14769. [DOI] [PubMed] [Google Scholar]
- 46.Streit TM, et al. Biol. Chem. 2008;389:149. doi: 10.1515/BC.2008.017. [DOI] [PubMed] [Google Scholar]
- 47.Leo AJ. Chem. Rev. 1993;93:1281. [Google Scholar]
- 48.Hansch C, et al. Exploring QSAR. Washington, DC: American Chemical Society; 1995. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.