

Abstract
Development of a functional organ requires the establishment of its proper size as well as the establishment of the relative proportions of its individual components. In the zebrafish heart, organ size and proportion depend heavily on the number of cells in each of its two major chambers, the ventricle and the atrium. Heart size and chamber proportionality are both affected in zebrafish fgf8 mutants. To determine when and how FGF signaling influences these characteristics, we examined the effect of temporally controlled pathway inhibition. During cardiac specification, reduction of FGF signaling inhibits formation of both ventricular and atrial cardiomyocytes, with a stronger impact on ventricular cells. After cardiomyocyte differentiation begins, reduction of FGF signaling can still result in a deficiency of ventricular cardiomyocytes. Consistent with two temporally distinct roles for FGF, we find that increased FGF signaling induces a cardiomyocyte surplus only before cardiac differentiation begins. Thus, FGF signaling first regulates heart size and chamber proportionality during cardiac specification and later refines ventricular proportion by regulating cell number after the onset of differentiation. Together, our data demonstrate that a single signaling pathway can act reiteratively to coordinate organ size and proportion.
Keywords: zebrafish, organogenesis, heart development, chamber formation, ventricle, atrium, FGF, Fgf8, acerebellar
Introduction
Organ size is highly consistent among individuals of the same species. Similarly, the distinct subunits of an organ typically develop with the same relative proportions in each individual (Cook and Tyers, 2007; Hidalgo and ffrench-Constant, 2003). Comparison between different species suggests that organ size and proportion are highly dependent on cell number, although cell size can also influence organ dimensions (Auman and Yelon, 2004; Hidalgo and ffrench-Constant, 2003; Nijhout, 2003; Potter and Xu, 2001; Raff, 1996; Trumpp et al., 2001). Since organ size and proportion are critical for normal organ function, it is important to elucidate the mechanisms that regulate these parameters.
In the heart, each chamber serves as a distinct functional subunit, and it has long been recognized that the relative proportions of the cardiac chambers are constant under normal physiological conditions. All normal adult human hearts have essentially the same number of myocardial nuclei (Linzbach, 1960), suggesting that the cell number in each chamber does not vary either. However, little is known about how heart size and relative chamber proportion are coordinated during embryonic development. Specifically, it is unclear when these characteristics are acquired and if they are established simultaneously or separately during development. It also remains unknown whether heart size and chamber proportionality are established by the same signaling pathways or require different sets of signals.
Fibroblast growth factors (FGFs) are appealing candidates for signals that regulate the establishment of cardiomyocyte number. FGFs comprise a large family of secreted polypeptides thought to signal in a dose-dependent manner through receptor tyrosine kinases (Böttcher and Niehrs, 2005). Studies in a number of model organisms have implicated FGF signaling in cardiac specification (Zaffran and Frasch, 2002). In Drosophila, heartless (Fgfr) mutants completely lack the dorsal vessel (Beiman et al., 1996); this phenotype reflects roles of Fgfr both in mesoderm migration and in cardiac fate assignment (Michelson et al., 1998). FGF signaling is also required to establish cardiac identity in Ciona, and activation of FGF transcriptional targets causes the transformation of anterior tail muscle cells into heart cells (Davidson et al., 2006). In chick, FGF8 from the anterior endoderm seems to contribute to its ability to induce the expression of cardiac genes such as NKX2-5 and MEF2c (Alsan and Schultheiss, 2002; Zhu et al., 1999). Fgf8 is also required to initiate nkx2.5 expression in zebrafish (Reifers et al, 2000): acerebellar (ace, fgf8) mutants exhibit weak nkx2.5 expression at early stages, although expression recovers as development proceeds. In mouse, it has been difficult to address the role of FGF signaling in cardiac specification, since Fgf8−/− (Sun et al., 1999) and Fgfr1−/− (Deng et al., 1994) mutant mice fail to complete gastrulation. However, consistent with the roles of FGFs in other species, in vitro studies have shown that Fgfr1−/− embryoid bodies fail to express cardiac genes and do not form contractile foci (Dell’Era et al., 2003).
FGFs may also contribute to the establishment of chamber proportion. Prior studies have pointed out that loss of FGF signaling causes more prominent defects in ventricles than it does in atria. In zebrafish, fgf8 mutants exhibit small hearts with particularly notable reductions of the ventricle (Reifers et al., 2000). Additionally, mouse embryos hypomorphic for Fgf8 (compound heterozygous Fgf8neo/−) display a hypoplastic right ventricle and outflow tract along with a complex series of other cardiovascular abnormalities (Abu-Issa et al., 2002), as do tissue-specific knockout mice in which Fgf8 is deleted from the anterior heart field, a territory giving rise to the ventricles and outflow tract (Ilagan et al., 2006; Park et al., 2006). Consistent with a differential effect of FGF signaling on ventricular and atrial cells, application of exogenous FGF2 or FGF4 in chick embryos promotes ventricular myosin heavy chain 1 (VMHC1) gene expression and decreases atrial myosin heavy chain (AMHC1) expression (Lopez-Sanchez et al., 2002). It is not yet clear whether this series of observations reflects a role of FGF signaling in setting the proper ratio of ventricular and atrial cell numbers.
Here, we test the hypothesis that FGF signaling influences both heart size and chamber proportionality through the establishment of the proper numbers of cardiomyocytes. Using the zebrafish heart as a model, we systematically evaluate the impact of FGF signaling on the number of ventricular and atrial cardiomyocytes by comparing the roles of FGF at early stages, during cardiac specification, and at later stages, following myocardial differentiation. We find that inhibition of FGF signaling during cardiac specification reduces the numbers of both types of cardiomyocytes, with the ventricular lineage exhibiting greater sensitivity over a longer period of time. Even after myocardial differentiation is underway, FGF inhibition remains able to affect the number of ventricular cardiomyocytes. Furthermore, we find that increased FGF signaling can induce excessive cardiomyocyte formation, but only prior to myocardial differentiation. Taken together, our results indicate that FGF signaling has reiterative roles in regulating heart size and chamber proportionality: early in development, FGF signaling helps to establish properly sized and proportioned cardiac progenitor pools, and, at later stages, FGF signaling continues to contribute to the regulation of ventricular cell number, potentially by controlling population maintenance or growth.
Materials and Methods
Zebrafish
Zebrafish and embryos were maintained at 28.5°C in standard laboratory conditions. In addition to wild-type fish, we used carriers of the aceti282a mutation (Reifers et al., 1998), carriers of the transgene Tg(cmlc2:DsRed2-nuc) (Mably et al., 2003), and carriers of the transgene Tg(hsp70:ca-fgfr1). The Tg(hsp70:ca-fgfr1) construct allows heat shock-inducible expression of a constitutively active form of Xenopus Fgfr1. A point mutation in the Fgfr1 kinase domain (K562E) results in autophosphorylation of the receptor in the absence of bound ligand (Neilson and Friesel, 1996). A transgenic line was generated by injecting 40 ng/mL of linearized, purified construct into zebrafish embryos at the 1-cell stage (Higashijima et al., 1997). We raised 200 injected embryos, screened the resulting fish for germline integration of the transgene, and established lines from successful founders. The Tg(hsp70:ca-fgfr1) construct also contains a cassette in which a 0.8 kb dsred coding sequence (plus SV40 polyA; BD Biosciences) is driven by the 0.7 kb zebrafish α-crystallin promoter (Kurita et al., 2003); this generates strong red fluorescence in the lens that is recognizable by 48 hours post fertilization (hpf) and persists throughout adulthood, thereby facilitating identification of transgenic fish in the F1 and subsequent generations.
Immunofluorescence and cell counting
To count cardiomyocytes in embryos carrying the transgene Tg(cmlc2:DsRed2-nuc), we used immunofluorescence to detect DsRed in cardiomyocyte nuclei and atrial myosin heavy chain (Amhc) in atrial cells, as described previously (Schoenebeck et al., 2007). Embryos were compressed under a cover slip and photographed with a Zeiss Axiocam on Zeiss M2Bio and Axioplan microscopes. Zeiss AxioVision 3.0.6 software and Adobe Photoshop Creative Suite were used to process images before counting fluorescent nuclei in each chamber. When comparing sets of cell number data, Student’s t-test (homocedastic, two-tail distribution) was used to determine if the differences between the means of data sets were statistically significant.
In situ hybridization
Anti-sense amhc, vmhc, cmlc2, pea3 and tbx5 probes were used for in situ hybridization as previously described (Berdougo et al., 2003; Brown et al., 1998; Yelon et al., 1999; Yelon et al., 2000). Embryos were examined on a Zeiss Axioplan microscope and photographed with a Zeiss Axiocam. Zeiss AxioVision 3.0.6 software and Adobe Photoshop Creative Suite were used to process images. Areas of gene expression were measured using Image J (http://rsb.info.nih.gov/ij/index.html). When comparing expression area data, Student’s t-test (homocedastic, two-tail distribution) was used to determine if the differences between the means of data sets were statistically significant.
SU5402 treatments
A 1mM stock of SU5402 (Calbiochem) in DMSO was diluted to a working concentration of 12.5 μM in E3 medium (Nusslein-Volhard and Dahm, 2002), a concentration of SU5402 that causes a strong reduction but does not completely abolish FGF signaling. Additional experiments utilized a working concentration of 9 μM SU5402, a concentration sufficient to induce circulation defects and pericardial edema. Up to 10 embryos were treated for discrete periods of time in glass vials in a final volume of 1 mL. Vials were kept in a nutator in the dark at 28.5°C. After treatment, embryos were washed in 50 mL of E3 medium and placed in new glass vials with fresh E3 medium. Control embryos were treated with a corresponding dilution of DMSO in E3 medium.
In order to address the effectiveness of SU5402, we analyzed the expression of the Fgf signaling pathway target gene polyoma enhancer activator 3 (pea3), which is known to be abolished with higher doses of SU5402 (Roehl and Nüsslein-Volhard, 2001). The concentration of SU5402 used in our experiments strongly reduces pea3 expression as soon as 30 minutes after exposure, and pea3 expression is restored 2 hours after SU5402 removal (SRM and DY, unpublished data). The length of this recovery period is similar to that previously reported for SU5402 by others (Crump et al., 2004; Maroon et al., 2002; Nechiporuk et al., 2005).
Heat shock conditions
Embryos from outcrosses of fish heterozygous for Tg(hsp70:ca-fgfr1) were kept at 28.5°C and heat shocked at desired stages. Embryos were placed in 40 mL of embryo medium in a Petri dish on top of a covered heat block for 20 minutes at 37°C. Following heat shock, transgenic embryos were identified by their elongated body morphology at the 19-somite or 21-somite stages or by the expression of the α-crystallin:dsred cassette in the lens at 48 hpf. Heat shocked non-transgenic sibling embryos served as controls.
Results
acerebellar mutants have small hearts with disproportionately reduced ventricles
Prior studies have shown that the zebrafish mutation acerebellar (aceti282a), a loss-of-function allele of fgf8, causes formation of undersized hearts with a particular reduction of the ventricle (Reifers et al., 1998; Reifers et al., 2000). Wild-type hearts display characteristically curved and expanded chambers (Fig. 1A), whereas ace mutant hearts resemble immature, linear tubes (Fig. 1B). Cardiomyocyte cell shape abnormalities contribute to the dysmorphic nature of the ace mutant heart (SRM and DY, unpublished data): in particular, ventricular cardiomyocytes in ace mutants do not undergo the regionalized processes of cellular enlargement and elongation that usually accompany chamber emergence (Auman et al., 2007). Since normal chamber emergence requires blood flow (Auman et al., 2007), this phenotype is consistent with the failure of ace mutants to establish circulation (Reifers et al., 2000). However, it is unclear whether the chamber morphology defects in ace mutants simply reflect errors in morphogenesis or may also be due to anomalies in cell number.
Figure 1.
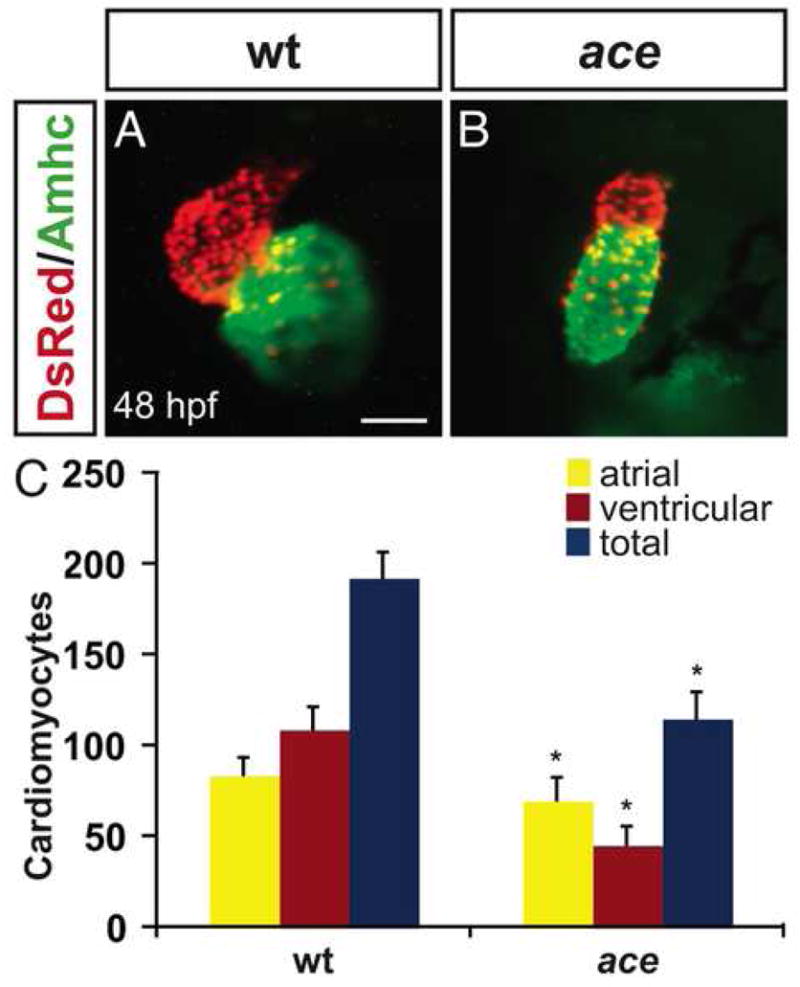
ace mutant embryos have reduced numbers of both ventricular and atrial cardiomyocytes. (A,B) Frontal views of hearts from wild-type (A) and ace mutant (B) embryos at 48 hpf; immunofluorescence detects DsRed (red) in all cardiomyocyte nuclei and atrial myosin heavy chain (Amhc; green) in atrial cells. (A) In a wild-type heart, the ventricle (red) and atrium (green) exhibit typical looping and expansion. (B) In an ace mutant heart, the chambers are unlooped and small, with a particularly apparent reduction of the ventricle. Scale bar represents 50 μm; both images are shown at the same magnification. (C) Quantification of cardiomyocytes at 48 hpf reveals that the numbers of both ventricular and atrial cells are significantly decreased in ace mutants, with ventricular cell number being more affected then atrial cell number. Graph indicates mean and standard deviation for each data set; asterisks indicate statistically significant differences relative to wild-type (p<0.005, Student’s t-test). n=13 for wild-type, and n=19 for ace mutants; see also Supplemental Table 1.
To determine whether the small dimensions of ace mutant hearts are related to reduced cell numbers, we counted ventricular and atrial cardiomyocytes in wild-type and ace mutant embryos. At 48 hours post fertilization (hpf), wild-type hearts typically contain approximately 30% more ventricular cells than atrial cells (Fig. 1C; Supplemental Table 1). In ace mutant hearts, total cell number is significantly reduced (Fig. 1C; Supplemental Table 1). Both ventricular and atrial cardiomyocyte populations are affected, but the ventricular cell loss is more dramatic than the atrial cell loss (Fig. 1C; Supplemental Table 1). As a result, ace mutant hearts typically contain approximately 50% more atrial cells than ventricular cells (Fig. 1C; Supplemental Table 1). Thus, in ace mutants, cell number deficiencies underlie abnormalities in both organ size and chamber proportionality.
Chamber disproportionality is evident prior to heart tube assembly in ace mutants
The overall reduction in cardiomyocyte number observed in ace mutants is likely to reflect early defects in cardiac specification, since expression of several pre-cardiac markers fails to initiate properly in the ace mutant lateral plate mesoderm (LPM) (Reifers et al., 2000). However, these data do not resolve whether Fgf8 affects ventricular and atrial lineages simultaneously. To evaluate ventricular and atrial cardiomyocyte populations in the LPM, we examined expression of the earliest known chamber-specific genes ventricular myosin heavy chain (vmhc) and atrial myosin heavy chain (amhc) at the 21-somite stage. At this timepoint, the two bilateral cardiac fields have migrated toward the midline where they fuse to form a ring of cardiomyocytes (Berdougo et al., 2003; Yelon et al., 1999). Even at this early stage, it is clear that ace mutants exhibit diminished populations of both ventricular cells (Fig. 2A,B) and atrial cells (Fig. 2C,D). Furthermore, quantification of the areas of vmhc and amhc expression reveals that the effects on the ventricular population are more striking than the effects on the atrial population (Fig. 2E,F; Supplemental Table 2). Since the size and organization of individual vmhc-expressing and amhc-expressing cells do not appear to be altered in ace mutants at this stage, the areas of vmhc and amhc expression suggest that ace mutants have only one-third of the number of vmhc-expressing cells and two-thirds of the number of amhc-expressing cells seen in wild-type embryos (Fig. 2F). Together, our data reveal that the influence of fgf8 on both ventricular and atrial lineages begins at early stages, prior to heart tube assembly.
Figure 2.
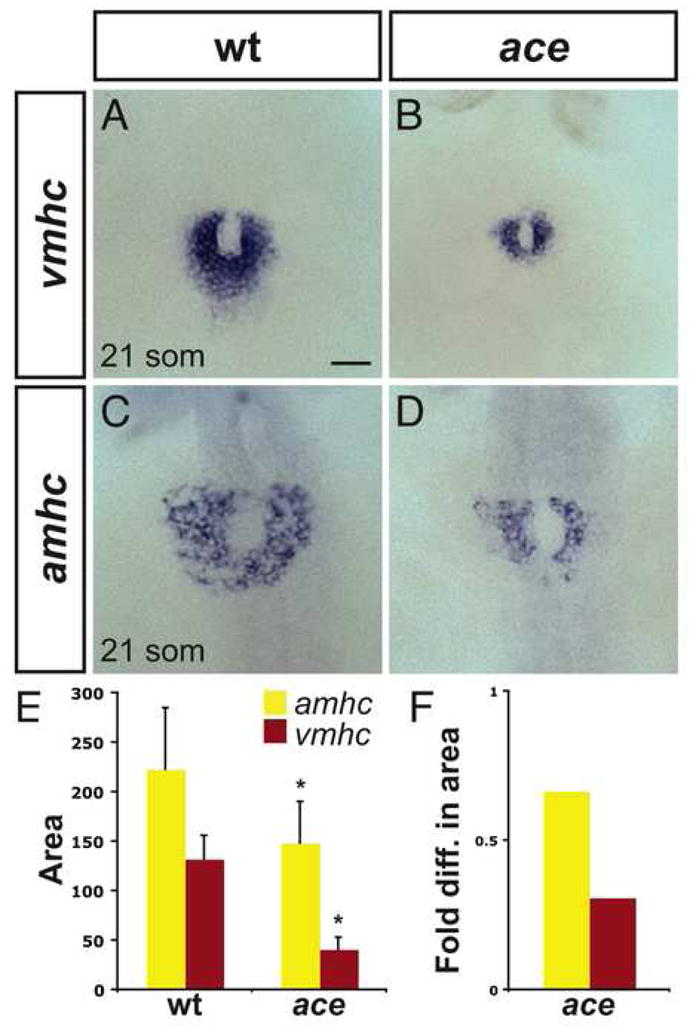
Chamber disproportionality is evident prior to heart tube assembly in ace mutants. (A–D) In situ hybridization depicts expression of vmhc (A, B) and amhc (C, D) at the 21-somite stage; dorsal views, anterior to the top. Scale bar represents 50 μm; all images are shown at the same magnification. (A) In wild-type embryos, vmhc is expressed in a ring of ventricular cardiomyocytes just prior to heart tube extension. (B) In ace mutant embryos, the population of vmhc-expressing cells is clearly reduced (n=14/15). (C) amhc is expressed in a ring of atrial cardiomyocytes, surrounding the ventricular cardiomyocytes. (D) The population of amhc-expressing cells is also reduced in ace mutant embryos (n=11/13). (E) Graph indicates mean and standard deviation of areas of expression (in μm2) of amhc and vmhc in wild-type and ace mutant embryos. Asterisks indicate statistically significant differences relative to wild-type (p<0.005, Student’s t-test). n≥10 for all data sets; see also Supplemental Table 2. (F) Graph indicates fold difference in mean areas of gene expression relative to wild-type.
FGF signaling influences both the ventricular and atrial lineages during gastrulation stages
We wondered whether the impact of Fgf8 on cardiomyocyte numbers could begin as early as gastrulation stages, when fgf8 transcripts accumulate in a dorsoventral gradient at the margin of the embryo, with the highest levels of fgf8 expression positioned dorsally (Fürthauer et al., 1997; Reifers et al., 1998). Fate maps of the zebrafish blastula indicate that myocardial progenitors reside in a region of the lateral margin that overlaps with the fgf8 expression domain (Keegan et al., 2004; Stainier et al., 1993). Furthermore, ventricular and atrial progenitors are spatially organized prior to gastrulation, with ventricular progenitors located more dorsally and atrial progenitors located more ventrally (Keegan et al., 2004). Thus, myocardial progenitors, particularly ventricular progenitors, are likely to be exposed to Fgf8 during gastrulation.
To test whether FGF signaling during gastrulation is required for production of the proper number of cardiomyocytes, we treated embryos with 12.5 μM SU5402, a specific inhibitor of FGFR tyrosine kinase activity (Mohammadi et al., 1997). Since the effects of SU5402 are rapid and reversible (see Materials and Methods), adding and removing the compound at different stages allowed us to create a transient period of attenuated FGF signaling and evaluate its impact on cardiomyocyte number at 48 hpf (Fig. 3A). We compared embryos exposed to SU5402 continuously (from 30% epiboly to 48 hpf; Fig. 3A,D,E) to embryos exposed only during gastrulation (from 30% epiboly to tailbud stage; Fig. 3A,F,G). Embryos continuously exposed to SU5402 morphologically resemble ace mutants (Fig. 3D; SRM and DY, unpublished data): they lack the midbrain-hindbrain boundary and exhibit a shortened body axis, consistent with roles of FGF in brain, trunk, and tail development (Griffin and Kimelman, 2003). Additionally, hearts of embryos continuously exposed to SU5402 resemble ace mutant hearts morphologically and in terms of cell number (Fig. 1B,C and Fig. 3E,L,M; Supplemental Table 3). The hearts of embryos treated during gastrulation appear slightly larger than the hearts of continuously treated embryos (Fig. 3E,G), though they are still small and dysmorphic relative to wild-type (Fig. 3C). Consistent with their morphology, the hearts of embryos treated during gastrulation exhibit significant deficiencies in both ventricular and atrial cell numbers (Fig. 3L,M; Supplemental Table 3). It is noteworthy that both continuous treatment and treatment during gastrulation reduce ventricular cell number more significantly then atrial cell number, as seen in ace. The same trend is seen in embryos exposed to 9 μM SU5402 during gastrulation (Supplemental Table 4). Comparing the effectiveness of the different SU5402 treatments, we observe similar effects on atrial cell number in ace mutants, embryos exposed to SU5402 continuously, and embryos exposed to SU5402 during gastrulation (Fig. 1C and Fig. 3L,M). In contrast, ventricular cell number appears more affected in ace mutants and in embryos treated continuously with SU5402 than it is in embryos treated only during gastrulation (Fig. 1C and Fig. 3L,M). Taken together, our results indicate that FGF signaling during gastrulation stages plays an important role in promoting the formation of both ventricular and atrial cardiomyocytes.
Figure 3.
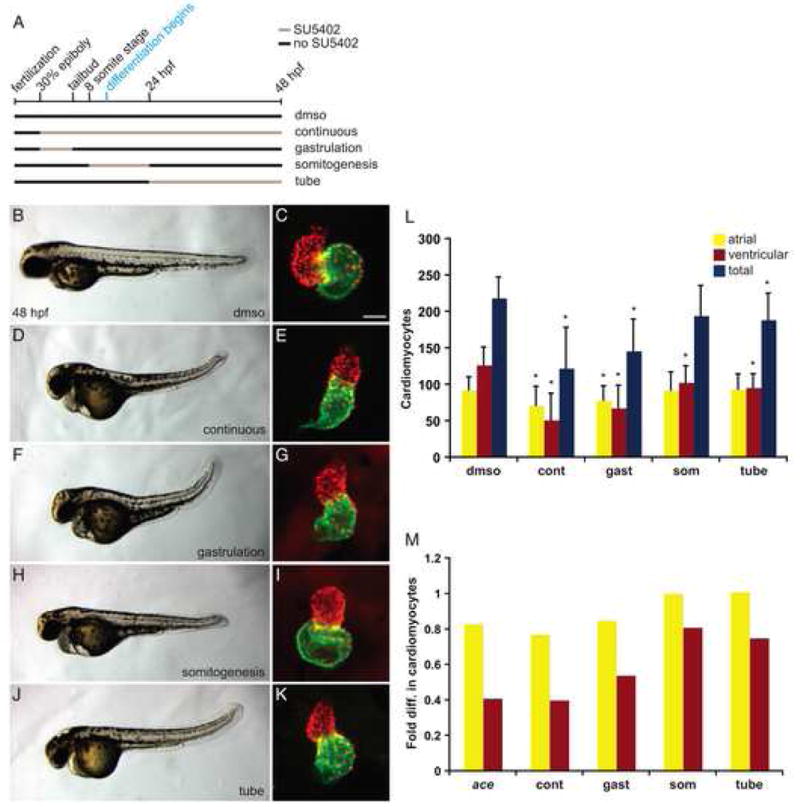
Transient reduction of FGF signaling leads to differential reduction of cardiomyocyte numbers. (A) Schematic depicts the transient periods of FGF signaling inhibition caused by addition and removal of SU5402. Black lines represent time intervals with normal FGF signaling, and tan lines represent intervals of SU5402 treatment. Control embryos were treated only with DMSO, “continuous” SU5402 treatment extended from 30% epiboly (3 hpf) until 48 hpf, “gastrulation” treatment began at 30% epiboly and ended at the tailbud stage (10 hpf), “somitogenesis” treatment began at the 8-somite stage (13 hpf) and ended at 24 hpf, and “tube” treatment extended from 24 hpf to 48 hpf. (B–K) Representative 48 hpf embryos, lateral views, exhibiting morphology resulting from each type of SU5402 treatment, coupled with respective frontal views of hearts in which immunofluorescence detects DsRed (red) in all cardiomyocyte nuclei and Amhc (green) in atrial cells, as in Figure 1. Scale bar represents 50 μm; all images of hearts are shown at the same magnification. Embryos treated continuously or during gastrulation occasionally exhibited severe tail truncations characteristic of higher SU5402 concentrations (Griffin and Kimelman, 2003); severely truncated embryos were excluded from quantification of cardiomyocytes. The concentration of SU5402 used did not induce nonspecific apoptosis, as indicated by TUNEL (SRM and DY, unpublished results). (L) Quantification of cardiomyocytes at 48 hpf after each type of SU5402 treatment. Graph indicates mean and standard deviation for each data set; asterisks indicate statistically significant differences relative to DMSO-treated controls (p<0.005, Student’s t-test). Note that the total number of cardiomyocytes following somitogenesis treatment is also significantly different from the control number, although with a larger p value (p<0.01). (M) Graph indicates fold difference in mean values relative to wild-type for ace mutant embryos and embryos treated with SU5402. n=33 for DMSO, n=8 for continuous treatment, n=28 for gastrulation treatment, n=28 for somitogenesis treatment, and n=22 for tube treatment; see also Supplemental Table 3.
A continuing influence of FGF signaling on ventricular cardiomyocyte number
While FGF signaling during gastrulation clearly has a potent effect on cardiomyocyte formation, our data also imply that the impact of FGF signaling on the ventricular lineage extends beyond these stages, since we find that continuous treatment with SU5402 has a stronger effect than treatment only during gastrulation on the number of ventricular cardiomyocytes (Fig. 3L,M). The locations of myocardial progenitors after gastrulation continue to correlate with the locations of fgf8 expression, with particular proximity of ventricular progenitors to sources of fgf8 (Reifers et al., 2000). Following gastrulation, myocardial progenitors integrate into the LPM, with ventricular progenitors located more medially than atrial progenitors (Schoenebeck et al., 2007). Robust expression of precardiac markers like nkx2.5 begins around the 6–8-somite stage, at which time fgf8 expression is medially adjacent to and overlapping with the precardiac portion of the LPM (Reifers et al., 2000). Terminal differentiation of cardiomyocytes begins around the 13-somite stage, when these cells initiate expression of myocardial markers like cmlc2 (Yelon et al., 1999). Heart tube assembly then facilitates the merger of bilateral cardiomyocyte populations and creates discrete domains of ventricular and atrial cardiomyocytes within the linear tube by 24 hpf (Auman et al., 2007). Even after myocardial differentiation is underway, fgf8 expression persists in ventricular cells and is apparent in the ventricular portion of the heart tube (Reifers et al., 2000). Thus, the fgf8 expression pattern suggests multiple opportunities for FGF signaling to influence ventricular development.
To test whether FGF signaling is required for cardiomyocyte formation after gastrulation stages, we examined the effects of inhibiting FGF signaling during somitogenesis stages (from the 8-somite stage to 24 hpf; hereafter referred to as somitogenesis treatment) and after the heart tube has formed (from 24 hpf to 48 hpf; hereafter referred to as tube treatment) (Fig. 3A). Embryos exposed to SU5402 during somitogenesis and tube stages show disorganized development of tail somites (Fig. 3H,J), consistent with reported roles for FGF in posterior myogenesis (Hamade et al., 2006). Additionally, somitogenesis treatment results in tubular ventricles with expanded atria (Fig. 3I) and causes a significant loss of ventricular cardiomyocytes (Fig. 3L,M; Supplemental Table 3); however, atrial cell number is unchanged. A similar, though not statistically significant, trend is observed in embryos exposed to 9 μM SU5402 during somitogenesis stages (Supplemental Table 4). Tube treatment results in mild defects in cardiac looping and chamber morphology (Fig. 3K) and also causes a significant reduction in ventricular cell number (Fig. 3L,M, Supplemental Table 3); like somitogenesis treatment, tube treatment does not affect atrial cell number. Together, these results reveal that FGF signaling has a continuing influence on ventricular cell number that extends well beyond its role during gastrulation.
Ectopic FGF signaling prior to terminal differentiation creates a cardiomyocyte surplus
Our loss-of-function data point to a general correlation between levels of FGF signaling and the number of cardiomyocytes generated. Prior studies have shown that ectopic FGF signaling is sufficient to induce cardiac gene expression; for example, FGF8-soaked beads induce nkx2.5 in chick (Alsan and Schultheiss, 2002) and zebrafish (Reifers et al., 2000). However, it is not known whether induction of gene expression by ectopic FGF signaling results in a surplus of cardiomyocytes. To test this hypothesis, we employed transgenic embryos carrying Tg(hsp70:ca-fgfr1), which allows heat-inducible expression of a constitutively active form of Fgfr1 (see Materials and Methods). Heat shock of transgenic embryos rapidly induces high levels of FGF signaling throughout the embryo: ectopic and robust expression of the FGF pathway target gene pea3 begins as early as 2 hours after heat shock and is maintained for at least 8 hours (Fig. 4A,B; SRM, YL, KDP, and DY, unpublished data).
Figure 4.
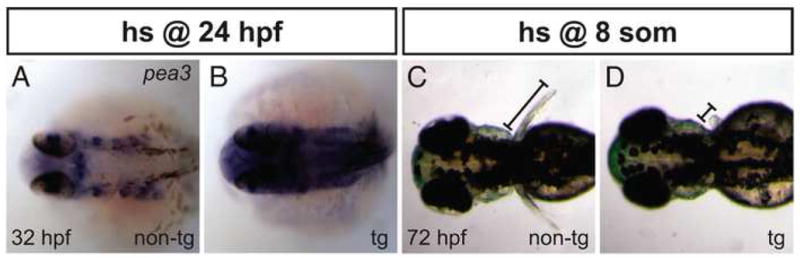
Heat shock of Tg(hsp70:ca-fgfr1) embryos causes ectopic FGF signaling and perturbs pectoral fin development. (A,B) In situ hybridization depicts pea3 expression at 32 hpf; dorsal views, anterior to the left. (A) After heat shock at 24 hpf, non-transgenic (non-tg) embryos show a wild-type restriction of pea3 expression to specific regions, including the midbrain-hindbrain boundary and the pharyngeal pouches. (B) In contrast, heat shock of transgenic (tg) embryos causes strong and ectopic expression of pea3. (C,D) Dorsal views at 72 hpf of embryos after heat shock at the 8-somite stage. (C) Non-transgenic embryos exhibit pectoral fins of normal length (bar). (D) Transgenic embryos lack at least one pectoral fin, as shown here (n=14/50), or have two small fins (n=31/50).
We increased FGF signaling through heat shock of transgenic embryos at the beginning of each time interval analyzed with SU5402 treatment. First, we found that heat shock of transgenic embryos at 30% epiboly results in severely dorsalized embryos consistent with what has been previously reported for fgf8 RNA injection (Fürthauer et al., 1997), preventing us from analyzing ventricular and atrial cell numbers at 48 hpf (SRM, YL, KDP, and DY, unpublished data). In contrast, heat shock of transgenic embryos at the 8-somite stage does not grossly disrupt embryonic axis formation (Fig. 5A,C), although it does cause body elongation (Fig. 5C), yolk extension abnormalities (Fig. 5C), and pectoral fin (forelimb) defects ranging from loss of fins to reduction of fin size (Fig. 4C,D). Heat shock at the 8-somite stage also results in severe pericardial edema (Fig. 5C) and elongated cardiac chambers (Fig. 5B,D). By counting ventricular and atrial cardiomyocytes at 48 hpf, we found that activation of FGF signaling at the 8-somite stage can lead to an approximately 25% increase in the numbers of both ventricular and atrial cells (Fig. 5G; Supplemental Table 5).
Figure 5.
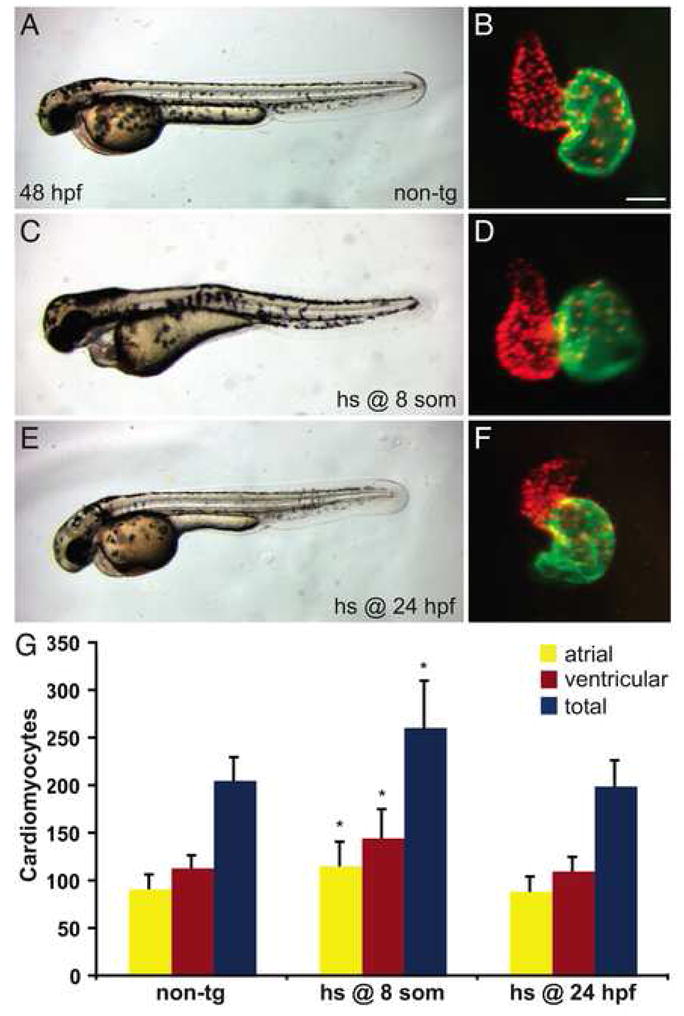
Increased FGF signaling prior to terminal differentiation induces a surplus of cardiomyocytes. (A–F) Representative non-transgenic (A) or Tg(hsp70:ca-fgfr1) (C,E) sibling embryos at 48 hpf, lateral views, exhibiting morphology resulting from heat shock at the 8-somite stage (C) or at 24 hpf (E), coupled with respective frontal views of hearts in which immunofluorescence detects DsRed (red) in all cardiomyocyte nuclei and Amhc (green) in atrial cells, as in Figure 1. Scale bar represents 50 μm; all images of hearts are shown at the same magnification. (C,D) Although heat shock at the 8-somite stage causes pericardial edema (C) and dysmorphic cardiac chambers (D), heat shock at 24 hpf does neither (E,F). (G) Quantification of cardiomyocytes at 48 hpf reveals that increased FGF signaling causes a significant cardiomyocyte surplus only in embryos heat shocked at the 8-somite stage. Graph indicates mean and standard deviation for each data set; asterisks indicate statistically significant differences relative to non-tg controls (p<0.005, Student’s t-test). n=24 for non-tg, n=18 for heat shock at the 8-somite stage, and n=14 for heat shock at 24 hpf; see also Supplemental Table 5.
The cardiomyocyte surplus caused by heat shock at the 8-somite stage is evident even a few hours following overexpression of the constitutively active receptor: heat shocked embryos exhibit posterior expansions of cmlc2, vmhc, and amhc expression within the LPM prior to heart tube assembly (Fig. 6A–F). Based on the observed expansion, it is interesting to consider that increased FGF signaling may recruit cells into the myocardial lineage from positions posterior to the heart field. Coincident with the appearance of enlarged heart fields in embryos heat shocked at the 8-somite stage, we also observe reduction of the pectoral fin fields, bilateral zones of tbx5 expression located posterior to the heart fields within the LPM (Fig. 6G,H) (Ahn et al., 2002; Begemann and Ingham, 2000). This loss of pectoral fin precursors is consistent with the observed reduction or absence of pectoral fins at later stages (Fig. 4C,D) and suggests the possibility that ectopic FGF signaling can transform pectoral fin progenitors into myocardial progenitors.
Figure 6.
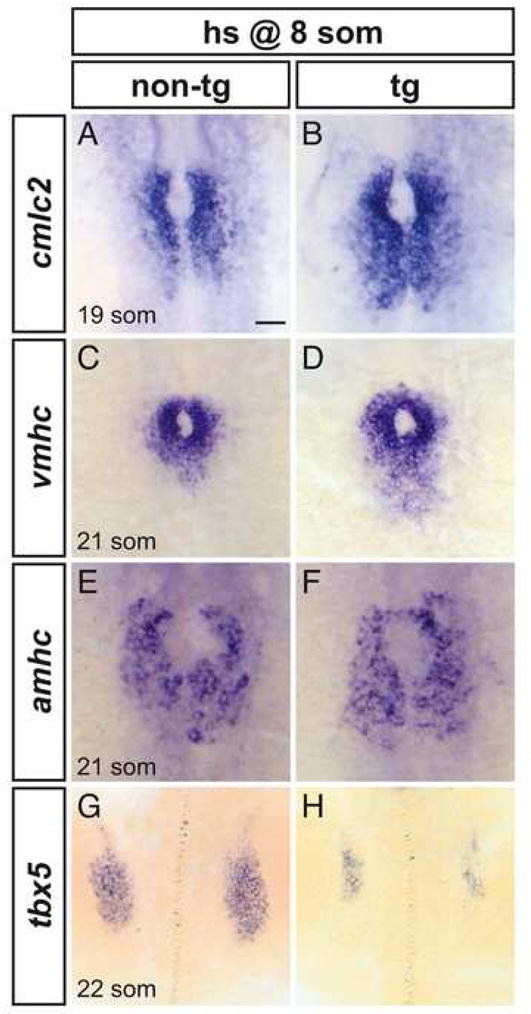
Increased FGF signaling enhances formation of cardiomyocyte populations and inhibits formation of the pectoral fin field. (A–H) In situ hybridization depicts expression of cmlc2, vmhc, amhc and tbx5 at the 19-somite or 21-somite stage in non-transgenic (A,C,E,G) and Tg(hsp70:ca-fgfr1) (B,D,F,H) sibling embryos after heat shock at the 8-somite stage; dorsal views, anterior to the top. Scale bar represents 50 μm; all images are shown at the same magnification. (A,B) Ectopic FGF signaling leads to a posterior expansion of cmlc2-expressing cardiomyocytes (n=30/37). (C,D) Ectopic FGF signaling leads to a posterior expansion of vmhc-expressing ventricular cardiomyocytes (n=16/19). (E,F) Ectopic FGF signaling leads to a mild posterior expansion of amhc-expressing atrial cardiomyocytes (n=8/10). (G,H) Ectopic FGF signaling leads to reduction of the pectoral fin field, as demarcated by tbx5 expression (n=18/18).
Loss of FGF signaling does not enlarge the pectoral fin field
The notion that ectopic FGF signaling may recruit cells from the forelimb field into the heart field suggests that endogenous FGF signaling might play a role in distinguishing the developmental potential of the heart and forelimb fields. In this scenario, loss of FGF signaling would cause expansion of the forelimb field at the expense of the heart field. However, ace mutants develop normal pectoral fins (Reifers et al., 1998), and fgf24 mutants display normal tbx5 expression (Fischer et al., 2003) at 24 hpf. It has also been reported that exposure to SU5402 from the 1-somite stage until the 23-somite stage does not perturb tbx5 expression (Mercader et al., 2006). However, prior studies have not addressed whether loss of FGF signaling during gastrulation stages could result in pectoral fin field expansion.
We examined tbx5 expression in the forelimb fields and in the pectoral fins of embryos treated with SU5402 continuously, during gastrulation, or during somitogenesis (Fig. 7). None of these treatments cause expansion of the area of tbx5 expression in the pectoral fin field at 20 hpf (Fig. 7A,C,E,G). Pectoral fin size at 48 hpf also appears normal in embryos treated with SU5402 during gastrulation (Fig. 7F) or somitogenesis (Fig. 7H), although fin morphology appears variably aberrant following somitogenesis treatment. Embryos continuously treated with SU5402 lack pectoral fins at 48 hpf (Fig. 7D), consistent with a role for Fgf24 in pectoral fin development after 24 hpf (Fischer et al., 2003; Mercader et al., 2006). Thus, while loss of FGF signaling results in a reduction of the heart field, we cannot detect a concomitant expansion of the forelimb field. Taken together, these results do not support a role for FGF signaling in regulating a decision between heart and forelimb fates.
Figure 7.
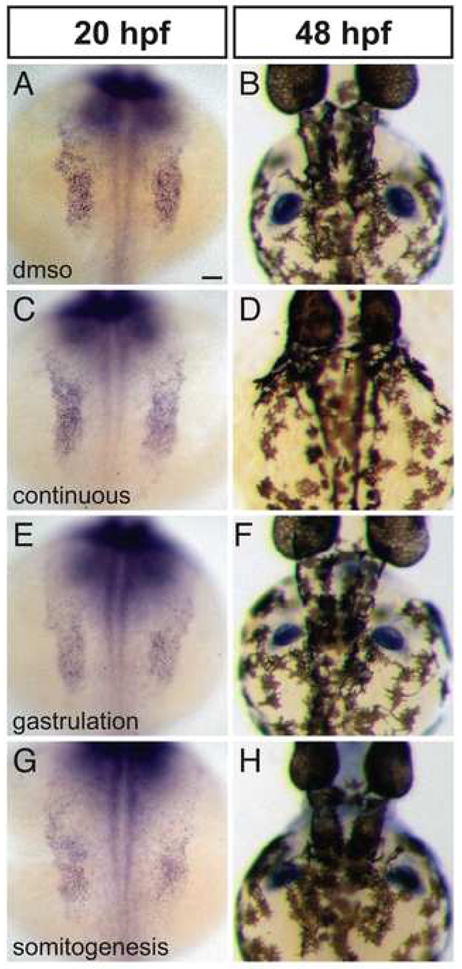
Reduction of FGF signaling does not expand the forelimb field. (A–H) In situ hybridization depicts expression of tbx5 at 20 hpf (A,C,E,G) and 48 hpf (B,D,F,H); dorsal views, anterior to the top. Scale bar represents 50 μm; all images are shown at the same magnification. (A–D) Compared to control embryos treated only with DMSO (A,B), embryos exposed to continuous SU5402 treatment exhibit normal areas of tbx5 expression in the pectoral fin fields at 20 hpf (C; n=8/8) but lack pectoral fins at 48 hpf (D; n=15/15). (E,F) Embryos treated with SU5402 during gastrulation exhibit normal areas of tbx5 expression in the pectoral fin fields at 20 hpf (n=16/16) and normal pectoral fins at 48 hpf (n=18/18). (G,H) Embryos treated with SU5402 during somitogenesis do not display increased areas of tbx5 expression in the pectoral fin fields at 20 hpf (n=9/9) and do not have detectable defects in pectoral fin size at 48 hpf (n=15/16), although fin morphology varies between treated embryos. It is noteworthy that the level of tbx5 expression generally appears higher in wild-type embryos than in SU5402-treated embryos.
Induction of ectopic FGF signaling after heart tube assembly does not affect cardiomyocyte number
Generation of a cardiomyocyte surplus by ectopic FGF signaling suggests that some developmental plasticity remains within the organ fields of the LPM during somitogenesis stages. We wondered whether this plasticity is maintained after myocardial differentiation begins. To address this, we chose to induce ectopic FGF signaling via heat shock of Tg(hsp70:ca-fgfr1) embryos at 24 hpf. Like heat shock at the 8-somite stage, heat shock of transgenic embryos at 24 hpf does not disturb general embryonic morphology (Fig. 5E). However, in contrast to the effects of heat shock at the 8-somite stage, pectoral fin formation appears to progress normally after heat shock at 24 hpf (SRM and DY, unpublished data). In an additional contrast to heat shock at the 8-somite stage, heat shock at 24 hpf only subtly affects heart morphology and looping (Fig. 5B,F) and does not affect ventricular or atrial cell number (Fig. 5G; Supplemental Table 5). Thus, our data suggest that, although ectopic FGF signaling at early stages can generate enlarged cardiomyocyte populations, this responsiveness to heightened FGF signaling is lost after the onset of myocardial differentiation.
Discussion
Our data demonstrate that FGF signaling plays an essential part in establishing the proper numbers and types of cardiomyocytes through a series of roles, beginning when myocardial progenitor specification is underway and continuing after the heart tube has formed. First, during gastrulation, FGF signaling promotes the formation of both ventricular and atrial lineages (Fig. 8A,B). Although atrial cell number does not depend on FGF signaling once gastrulation is complete, FGF signaling continues to promote development of the ventricular lineage while myocardial progenitor cells reside within the LPM (Fig. 8C,D). Even after the initiation of myocardial differentiation and the assembly of the heart tube, FGF signaling is important for the formation of the proper number of ventricular cardiomyocytes (Fig. 8E,F). Thus, reiterative roles of the FGF signaling pathway contribute to the regulation of both overall heart size and chamber proportionality.
Figure 8.
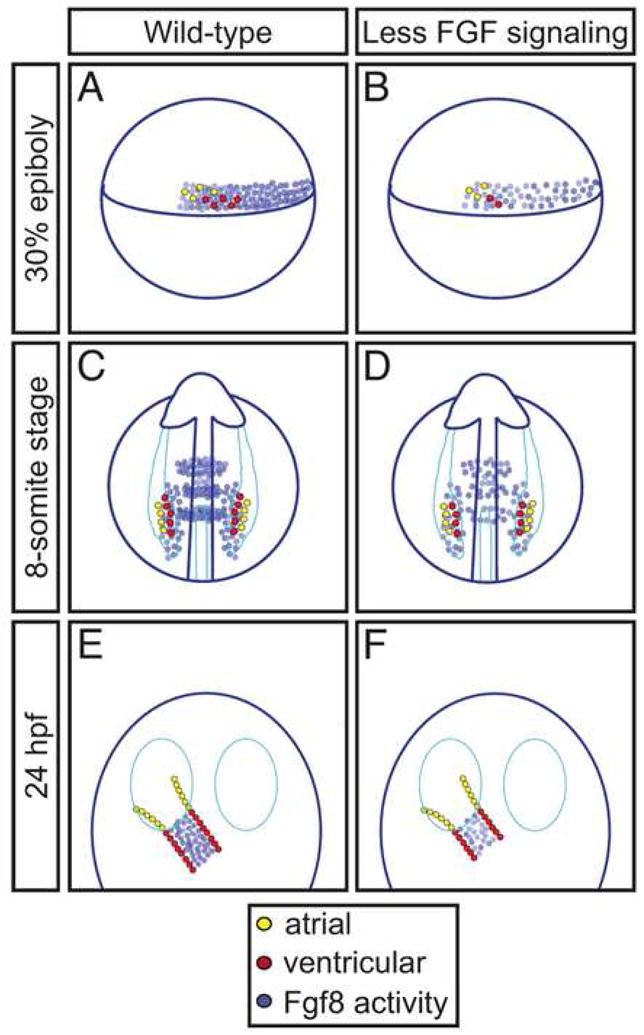
FGF signaling plays reiterative roles in the establishment of heart size and chamber proportionality in zebrafish. (A–F) Schematics depict a model for the roles of FGF signaling indicated by our data. Each image represents the quantities of ventricular (red) or atrial (yellow) progenitor cells or cardiomyocytes and the locations of Fgf8 activity at the 30% epiboly stage (A,B, lateral views), the 8-somite stage (C,D, dorsal views), and 24 hpf (E,F, dorsal views), either in wild-type embryos (A,C,E) or in embryos with reduced FGF signaling (B,D,F). Locations of ventricular and atrial cells and Fgf8 activity in wild-type embryos are based on established fate map data and gene expression patterns (Auman et al., 2007; Fürthauer et al., 1997; Keegan et al., 2004; Reifers et al., 1998; Reifers et al., 2000; Schoenebeck et al., 2007). Differences depicted in (B,D,F) are inferred from the results presented here. (A,B) During gastrulation, FGF signaling along the margin influences both heart size and chamber proportionality by promoting specification or proliferation of myocardial progenitors, particularly ventricular progenitors. (C,D) During somitogenesis, FGF signaling near the medial LPM continues to influence ventricle proportion by promoting maintenance, proliferation, or differentiation of ventricular progenitors. (E,F) In the ventricular portion of the heart tube, FGF signaling influences ventricle proportion by regulating maintenance or augmentation of the ventricular cardiomyocyte population.
Each of the temporally distinct roles of FGF signaling during establishment of cardiomyocyte populations correlates with the dynamic expression pattern of fgf8, in the sense that high levels of fgf8 expression are located near the lineages affected by a loss of FGF signaling (Fig. 8). Moreover, given the similarities between the cardiac phenotypes of ace mutant embryos and embryos continuously treated with SU5402, it seems likely that Fgf8 is the major FGF family member with an influence on heart size and chamber proportionality in zebrafish. We cannot rule out the contribution of other FGFs, such as fgf3 and fgf24, which are coexpressed with fgf8 during gastrulation (Fürthauer et al., 2004); however, neither the fgf3 nor fgf24 mutant phenotypes are known to include cardiac defects (Fischer et al., 2003; Herzog et al., 2004). Hereafter, for simplicity of discussion, we refer to Fgf8 as the primary ligand of interest.
The temporally distinct effects of Fgf8 signaling on cardiomyocyte numbers provide suggestions for the cellular mechanisms responsible for establishing heart size and chamber proportionality. The early impact of Fgf8 signaling on heart size likely reflects a role of Fgf8 during the initial specification of ventricular and atrial myocardial progenitors or during the proliferation of these progenitors. During gastrulation stages, the multipotential mesendodermal progenitor cells that will ultimately give rise to the ventricular or atrial lineages are thought to be in the process of integrating the inductive signals that regulate their fate assignment (Brand, 2003; Zaffran and Frasch, 2002). Although our data do not address the cell autonomy of the requirement for receiving Fgf8 signaling, the known proximity of fgf8 expression to the origins of myocardial progenitors indicates an opportunity for inductive signal reception by progenitor cells. Therefore, we propose that Fgf8 signaling helps to establish heart size in zebrafish by promoting the formation of a properly sized myocardial progenitor pool, consistent with the roles of FGF signaling during cardiac specification in Drosophila and Ciona (Davidson et al., 2006; Michelson et al., 1998).
Similar to its influence on heart size, the impact of Fgf8 signaling on chamber proportionality begins during gastrulation. The ventricular lineage is more sensitive than the atrial lineage to reduction of Fgf8 signaling during gastrulation, and the sensitivity of ventricular cells continues during somitogenesis and tube assembly, well beyond the sensitivity of the atrial population. The reiterative importance of Fgf8 signaling for establishment of ventricular cardiomyocyte numbers is consistent with the previously reported suggestion of a continuous requirement for Fgf8 signaling during zebrafish heart development (Reifers et al., 2000). Our data confirm and extend this prior work, providing a quantitative assessment of both ventricular and atrial cell number phenotypes in loss-of-function and gain-of-function scenarios at multiple stages.
Do the reiterative roles of FGF signaling in the establishment of chamber proportionality, like the role of FGF signaling in establishing heart size, reflect regulation of progenitor specification? During gastrulation and somitogenesis stages, the proximity of ventricular progenitors to the highest levels of fgf8 expression (Fig. 8A,C) suggests an appealing model in which Fgf8 controls a ventricular/atrial fate decision: higher levels of Fgf8 signaling could induce ventricular identity, whereas lower levels of signaling could induce atrial identity. However, neither gastrulation treatment nor somitogenesis treatment with SU5402 leads to enhanced numbers of atrial cells, as would be expected if reduced Fgf8 signaling favored atrial specification over ventricular specification. Additionally, ectopic FGF signaling at the 8-somite stage leads to increases in both ventricular and atrial populations, instead of increasing the ventricular population at the expense of the atrial population. Therefore, although Fgf8 signaling may promote ventricular specification, particularly at gastrulation stages, its impact on chamber proportionality is more consistent with a particular influence on the ventricular lineage than with a role in regulating a binary decision between ventricular and atrial identities.
The comparison of FGF pathway loss-of-function and gain-of-function phenotypes during somitogenesis provides additional ideas for cellular mechanisms regulating ventricle proportion. Rather than reflecting a role in ventricular progenitor specification, the functions of Fgf8 signaling at these stages may indicate its importance for maintaining a population of ventricular progenitors, or for regulating the differentiation or proliferation of this population. These explanations fit well with the location of fgf8 expression medially adjacent to the ventricular progenitor population (Fig. 8C). However, the ability of ectopic FGF signaling at the 8-somite stage to increase both the atrial and ventricular cardiomyocyte populations indicates an effect that extends beyond nurturing a population of ventricular progenitors. This expansion of both atrial and ventricular cells suggests that a degree of plasticity is retained in the LPM even after the initiation of nkx2.5 expression, such that cell identity can still be swayed by high levels of FGF signaling.
Like reduction of Fgf8 signaling during somitogenesis, reduction of Fgf8 signaling after heart tube assembly results in a significant decrease in ventricular cell number but has no effect on atrial cell number. This is not likely to reflect a role of FGF signaling in promoting ventricular progenitor specification, since ectopic FGF signaling at 24 hpf is unable to increase the number of cardiomyocytes. Additionally, although cardiomyocyte proliferation would be an attractive mechanism for ventricular growth, the failure of ectopic FGF signaling to enhance cardiomyocyte cell number argues against its regulation of ventricular proliferation. Furthermore, we have detected very few proliferating cells with BrdU incorporation assays or with phospho-histone-3 immunohistochemistry in either wild-type or SU5402-treated cardiomyocytes between 24 and 48 hpf (SRM and DY, unpublished data). This is consistent with prior studies showing that, even though the number of cardiomyocytes increases by as much as 50% between 24 and 36 hpf (Rohr et al., 2006; Sato et al., 2006; Shu et al., 2003), the cardiomyocyte mitotic index is no greater than 10% during this time period (Rohr et al., 2006).
Instead of suggesting a late role of Fgf8 in ventricular specification or proliferation, our results point toward a role of Fgf8 in maintaining the proper number of differentiated ventricular cardiomyocytes, perhaps by promoting ventricular cardiomyocyte survival. However, TUNEL analysis of embryos treated with SU5402 during tube stages did not reveal an increase in apoptotic cardiomyocytes, and the ventricular cell number deficiency in ace mutant embryos is not ameliorated by treatment with caspase antagonists at tube stages (SRM and DY, unpublished data). Therefore, rather than invoking a role for Fgf8 in ventricular cardiomyocyte survival, it is interesting to consider another possibility, in which Fgf8 plays a role in the recruitment of additional cardiomyocytes into the heart tube after 24 hpf. Consistent with this model, tissue-specific Fgf8 knockout mice have implicated Fgf8 in the regulation of proliferation and survival of progenitor cells within the anterior heart field, which contributes cardiomyocytes to the outflow pole after the formation of the primitive heart tube (Ilagan et al., 2006; Park et al., 2006). Perhaps Fgf8 signaling regulates the addition of cells from a secondary source to the outflow pole of the zebrafish heart, potentially by promoting their survival, differentiation, or migration. This secondary source could be the zebrafish equivalent to the amniote anterior heart field or, alternatively, might relate to the neural crest populations that have been suggested to contribute to the zebrafish myocardium (Li et al., 2003; Sato et al., 2006; Sato and Yost, 2003).
Taken together, our data suggest a model in which a single signaling pathway, and perhaps even a single growth factor, can act reiteratively to coordinate heart size and chamber proportionality. Fgf8 signaling acts early to create a cardiac progenitor pool of appropriate total size and ventricular/atrial proportionality. However, ventricle proportion is not regulated solely through generation of progenitor cells; it is also enforced by the continuing chamber-specific effects of Fgf8 that impact ventricular progenitor maintenance, ventricular differentiation, or recruitment of additional ventricular cardiomyocytes. The effects of FGF signaling extend even further into the more mature ventricle, in which FGFs influence ventricular growth, homeostasis, and regeneration (Lavine et al., 2005; Lepilina et al., 2006; Wills et al., 2008). In future studies, it will be important to determine whether the same downstream effectors of FGF signaling are utilized in each temporal context and to elucidate how the FGF pathway networks with other pathways that contribute to myocardial specification, differentiation, and maintenance during the establishment of heart size and chamber proportionality.
Supplementary Material
Acknowledgments
We thank J. Schoenebeck for helpful observations and input, R. Friesel for sharing the Xenopus K562E construct, K. Rohr and J. Bakkers for reagents, J. Castro Lopes for support, and members of the Yelon laboratory for feedback. This work was supported by grants from the National Institutes of Health to DY and KDP. SRM received support from the GABBA Program and the Portuguese Foundation for Science and Technology (POCI 2010-FSE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Issa R, et al. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–25. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Ahn DG, et al. T-box gene tbx5 is essential for formation of the pectoral limb bud. Nature. 2002;417:754–8. doi: 10.1038/nature00814. [DOI] [PubMed] [Google Scholar]
- Alsan BH, Schultheiss TM. Regulation of avian cardiogenesis by Fgf8 signaling. Development. 2002;129:1935–43. doi: 10.1242/dev.129.8.1935. [DOI] [PubMed] [Google Scholar]
- Auman HJ, et al. Functional Modulation of Cardiac Form through Regionally Confined Cell Shape Changes. PLoS Biol. 2007;5:e53. doi: 10.1371/journal.pbio.0050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auman HJ, Yelon D. Vertebrate organogenesis: getting the heart into shape. Curr Biol. 2004;14:R152–3. [PubMed] [Google Scholar]
- Begemann G, Ingham PW. Developmental regulation of Tbx5 in zebrafish embryogenesis. Mech Dev. 2000;90:299–304. doi: 10.1016/s0925-4773(99)00246-4. [DOI] [PubMed] [Google Scholar]
- Beiman M, et al. Heartless, a Drosophila FGF receptor homolog, is essential for cell migration and establishment of several mesodermal lineages. Genes Dev. 1996;10:2993–3002. doi: 10.1101/gad.10.23.2993. [DOI] [PubMed] [Google Scholar]
- Berdougo E, et al. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development. 2003;130:6121–9. doi: 10.1242/dev.00838. [DOI] [PubMed] [Google Scholar]
- Böttcher RT, Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocr Rev. 2005;26:63–77. doi: 10.1210/er.2003-0040. [DOI] [PubMed] [Google Scholar]
- Brand T. Heart development: molecular insights into cardiac specification and early morphogenesis. Dev Biol. 2003;258:1–19. doi: 10.1016/s0012-1606(03)00112-x. [DOI] [PubMed] [Google Scholar]
- Brown LA, et al. Molecular characterization of the zebrafish PEA3 ETS-domain transcription factor. Oncogene. 1998;17:93–104. doi: 10.1038/sj.onc.1201911. [DOI] [PubMed] [Google Scholar]
- Cook M, Tyers M. Size control goes global. Curr Opin Biotechnol. 2007;18:341–50. doi: 10.1016/j.copbio.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Crump JG, et al. An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development. 2004;131:5703–16. doi: 10.1242/dev.01444. [DOI] [PubMed] [Google Scholar]
- Davidson B, et al. FGF signaling delineates the cardiac progenitor field in the simple chordate, Ciona intestinalis. Genes Dev. 2006;20:2728–38. doi: 10.1101/gad.1467706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Era P, et al. Fibroblast growth factor receptor-1 is essential for in vitro cardiomyocyte development. Circ Res. 2003;93:414–20. doi: 10.1161/01.RES.0000089460.12061.E1. [DOI] [PubMed] [Google Scholar]
- Deng CX, et al. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–57. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- Fischer S, et al. The zebrafish fgf24 mutant identifies an additional level of Fgf signaling involved in vertebrate forelimb initiation. Development. 2003;130:3515–24. doi: 10.1242/dev.00537. [DOI] [PubMed] [Google Scholar]
- Fürthauer M, et al. A role for FGF-8 in the dorsoventral patterning of the zebrafish gastrula. Development. 1997;124:4253–64. doi: 10.1242/dev.124.21.4253. [DOI] [PubMed] [Google Scholar]
- Fürthauer M, et al. Fgf signalling controls the dorsoventral patterning of the zebrafish embryo. Development. 2004;131:2853–64. doi: 10.1242/dev.01156. [DOI] [PubMed] [Google Scholar]
- Griffin KJ, Kimelman D. Interplay between FGF, one-eyed pinhead, and T-box transcription factors during zebrafish posterior development. Dev Biol. 2003;264:456–66. doi: 10.1016/j.ydbio.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Hamade A, et al. Retinoic acid activates myogenesis in vivo through Fgf8 signalling. Dev Biol. 2006;289:127–40. doi: 10.1016/j.ydbio.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Herzog W, et al. Fgf3 signaling from the ventral diencephalon is required for early specification and subsequent survival of the zebrafish adenohypophysis. Development. 2004;131:3681–92. doi: 10.1242/dev.01235. [DOI] [PubMed] [Google Scholar]
- Hidalgo A, ffrench-Constant C. The control of cell number during central nervous system development in flies and mice. Mech Dev. 2003;120:1311–25. doi: 10.1016/j.mod.2003.06.004. [DOI] [PubMed] [Google Scholar]
- Higashijima S, et al. High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev Biol. 1997;192:289–99. doi: 10.1006/dbio.1997.8779. [DOI] [PubMed] [Google Scholar]
- Ilagan R, et al. Fgf8 is required for anterior heart field development. Development. 2006;133:2435–45. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- Keegan BR, et al. Organization of cardiac chamber progenitors in the zebrafish blastula. Development. 2004;131:3081–91. doi: 10.1242/dev.01185. [DOI] [PubMed] [Google Scholar]
- Kurita R, et al. Suppression of lens growth by alphaA-crystallin promoter-driven expression of diphtheria toxin results in disruption of retinal cell organization in zebrafish. Dev Biol. 2003;255:113–27. doi: 10.1016/s0012-1606(02)00079-9. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, et al. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lepilina A, et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127:607–19. doi: 10.1016/j.cell.2006.08.052. [DOI] [PubMed] [Google Scholar]
- Li YX, et al. Cardiac neural crest in zebrafish embryos contributes to myocardial cell lineage and early heart function. Dev Dyn. 2003;226:540–50. doi: 10.1002/dvdy.10264. [DOI] [PubMed] [Google Scholar]
- Linzbach A. Heart failure from the point of view of quantitative anatomy. Am J Cardiol. 1960:370–82. doi: 10.1016/0002-9149(60)90084-9. [DOI] [PubMed] [Google Scholar]
- Lopez-Sanchez C, et al. Induction of cardiogenesis by Hensen’s node and fibroblast growth factors. Cell Tissue Res. 2002;309:237–49. doi: 10.1007/s00441-002-0567-2. [DOI] [PubMed] [Google Scholar]
- Mably JD, et al. heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol. 2003;13:2138–47. doi: 10.1016/j.cub.2003.11.055. [DOI] [PubMed] [Google Scholar]
- Maroon H, et al. Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Development. 2002;129:2099–108. doi: 10.1242/dev.129.9.2099. [DOI] [PubMed] [Google Scholar]
- Mercader N, et al. Prdm1 acts downstream of a sequential RA, Wnt and Fgf signaling cascade during zebrafish forelimb induction. Development. 2006;133:2805–15. doi: 10.1242/dev.02455. [DOI] [PubMed] [Google Scholar]
- Michelson AM, et al. Dual functions of the heartless fibroblast growth factor receptor in development of the Drosophila embryonic mesoderm. Dev Genet. 1998;22:212–29. doi: 10.1002/(SICI)1520-6408(1998)22:3<212::AID-DVG4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, et al. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–60. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- Nechiporuk A, et al. Endoderm-derived Fgf3 is necessary and sufficient for inducing neurogenesis in the epibranchial placodes in zebrafish. Development. 2005;132:3717–30. doi: 10.1242/dev.01876. [DOI] [PubMed] [Google Scholar]
- Neilson KM, Friesel R. Ligand-independent activation of fibroblast growth factor receptors by point mutations in the extracellular, transmembrane, and kinase domains. J Biol Chem. 1996;271:25049–57. doi: 10.1074/jbc.271.40.25049. [DOI] [PubMed] [Google Scholar]
- Nijhout HF. The control of body size in insects. Dev Biol. 2003;261:1–9. doi: 10.1016/s0012-1606(03)00276-8. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Dahm R. Zebrafish, A Practical Approach. Oxford University Press; 2002. [Google Scholar]
- Park EJ, et al. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–33. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Xu T. Mechanisms of size control. Curr Opin Genet Dev. 2001;11:279–86. doi: 10.1016/s0959-437x(00)00191-x. [DOI] [PubMed] [Google Scholar]
- Raff MC. Size control: the regulation of cell numbers in animal development. Cell. 1996;86:173–5. doi: 10.1016/s0092-8674(00)80087-2. [DOI] [PubMed] [Google Scholar]
- Reifers F, et al. Fgf8 is mutated in zebrafish acerebellar (ace) mutants and is required for maintenance of midbrain-hindbrain boundary development and somitogenesis. Development. 1998;125:2381–95. doi: 10.1242/dev.125.13.2381. [DOI] [PubMed] [Google Scholar]
- Reifers F, et al. Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar) Development. 2000;127:225–35. doi: 10.1242/dev.127.2.225. [DOI] [PubMed] [Google Scholar]
- Roehl H, Nüsslein-Volhard C. Zebrafish pea3 and erm are general targets of FGF8 signaling. Curr Biol. 2001;11:503–7. doi: 10.1016/s0960-9822(01)00143-9. [DOI] [PubMed] [Google Scholar]
- Rohr S, et al. Heart and soul/PRKCi and nagie oko/Mpp5 regulate myocardial coherence and remodeling during cardiac morphogenesis. Development. 2006;133:107–15. doi: 10.1242/dev.02182. [DOI] [PubMed] [Google Scholar]
- Sato M, et al. Semaphorin3D regulates invasion of cardiac neural crest cells into the primary heart field. Dev Biol. 2006;298:12–21. doi: 10.1016/j.ydbio.2006.05.033. [DOI] [PubMed] [Google Scholar]
- Sato M, Yost HJ. Cardiac neural crest contributes to cardiomyogenesis in zebrafish. Dev Biol. 2003;257:127–39. doi: 10.1016/s0012-1606(03)00037-x. [DOI] [PubMed] [Google Scholar]
- Schoenebeck JJ, et al. Vessel and blood specification override cardiac potential in anterior mesoderm. Dev Cell. 2007;13:254–67. doi: 10.1016/j.devcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X, et al. Na,K-ATPase is essential for embryonic heart development in the zebrafish. Development. 2003;130:6165–73. doi: 10.1242/dev.00844. [DOI] [PubMed] [Google Scholar]
- Stainier DY, et al. Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Development. 1993;119:31–40. doi: 10.1242/dev.119.1.31. [DOI] [PubMed] [Google Scholar]
- Sun X, et al. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–46. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, et al. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 2001;414:768–73. doi: 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- Wills AA, et al. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development. 2008;135:183–92. doi: 10.1242/dev.010363. [DOI] [PubMed] [Google Scholar]
- Yelon D, et al. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol. 1999;214:23–37. doi: 10.1006/dbio.1999.9406. [DOI] [PubMed] [Google Scholar]
- Yelon D, et al. The bHLH transcription factor hand2 plays parallel roles in zebrafish heart and pectoral fin development. Development. 2000;127:2573–82. doi: 10.1242/dev.127.12.2573. [DOI] [PubMed] [Google Scholar]
- Zaffran S, Frasch M. Early signals in cardiac development. Circ Res. 2002;91:457–69. doi: 10.1161/01.res.0000034152.74523.a8. [DOI] [PubMed] [Google Scholar]
- Zhu X, et al. Evidence that FGF receptor signaling is necessary for endoderm-regulated development of precardiac mesoderm. Mech Ageing Dev. 1999;108:77–85. doi: 10.1016/s0047-6374(99)00003-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.