

Abstract
NAD+-dependent protein deacetylases (sirtuins) and other enzymes that produce nicotinamide are integral to many cellular processes. Yet current activity measurements involve expensive and time-consuming assays. Here, we present a spectroscopic assay that circumvents many issues of previous methods. This assay permits continuous product monitoring over time, allows determination of steady-state kinetic parameters, and is readily adaptable to high throughput screening. The methodology uses an enzyme-coupled system in which nicotinamide is converted to nicotinic acid and ammonia by nicotinamidase. The ammonia is transferred to α-ketoglutarate via glutamate dehydrogenase, yielding glutamate and the oxidation of NAD(P)H to NAD(P)+, which is measured spectrophotometrically at 340 nm. Utilizing this continuous assay with sirtuin-1 (Sirt1) and the ADP-ribosyl cyclase CD38, the resulting steady-state kinetic parameters are in excellent agreement with values obtained by other published methods. Importantly, this assay permitted determination of kcat and Km values with the native acetylated substrate acetyl-CoA synthetase-1, measurement of Sirt1, Sirt2, and Sirt3 activities from mammalian cell extracts, and determination of IC50 values of various Sirt1 inhibitors. This assay is applicable to any nicotinamide forming enzyme and will be an important tool to address many outstanding questions surrounding their regulation.
Keywords: sirtuin, Sirt1, nicotinamide, sir2, NAD+, deacetylase, acetyl, CD38, nicotinamidase, glutamate dehydrogenase
Introduction
Enzymes that produce nicotinamide from the catabolism of nicotinamide adenine dinucleotide (NAD+) are now appreciated to mediate many physiological processes that were nearly unimaginable just a decade ago. These enzymes include poly(ADP-ribosyl) polymerases (PARPs), mono(ADP-ribosyl)transferases, ADP-ribosyl cyclases, and sirtuin protein deacetylases. After DNA damage, PARPs consume NAD+ to form large ADP-ribose polymers and nicotinamide. Among other proposed functions, these polymers facilitate DNA double strand break repair [1]. Distinct from PARPs, mono(ADP-ribosyl)transferases transfer a single ADP ribose to protein residues such as arginine and glutamine, which can alter protein function [2]. ADP-ribosyl cyclases (e.g. CD38) utilize NAD+ to make cyclic ADP-ribose as a secondary messenger that has been implicated in insulin signaling, cell cycle control, and calcium signaling [3].
Sirtuin protein deacetylases utilize NAD+ as a co-substrate during catalysis to remove the acetyl group from acetyl-lysine residues of proteins, forming deacetylated protein, O-acetyl-ADP-ribose (OAADPr), and nicotinamide (for a review see [4]). Sirtuins stoichiometrically use NAD+ such that one molecule of nicotinamide is formed from NAD+ for every lysine residue deacetylated. Sirtuins are implicated in a multitude of cellular pathways, ranging from nuclear/epigenetic regulation to direct regulation of cellular metabolism. Humans possess seven sirtuin family members (Sirt1-7) that are localized to diverse cellular compartments, target multiple protein substrates, and affect many cellular processes such as cell differentiation, metabolism, apoptosis, and epigenetic signaling [4-6]. Although many different sirtuin protein substrates are known, the specificity and steady-state kinetic parameters of these substrates are uncharacterized. Determining these parameters would lead to a better understanding of sirtuin protein substrate recognition, sirtuin cellular regulation, and how acetylation affects the activity of these protein substrates.
Sirtuins are also attractive targets for drug discovery, and determination of specific sirtuin inhibitors and activators may provide treatments of many age-related neurodegenerative diseases and metabolic disorders (see for example [7-9]). Therefore, assays that accurately measure sirtuin activity (or more generally nicotinamide formation) in vitro and in vivo are important tools to unravel their cellular mechanisms and will aid in the discovery of new therapeutic agents.
Although several sirtuin enzymatic assays have been described, most require modified/artificial substrates and are non-continuous. Initially, sirtuin assays involved the use of radioactive substrates to monitor the transfer of a [3H]-labeled acetyl group from peptide or protein substrate to the [3H]OAADPr product (or [3H]acetate hydrolyzed from [3H]OAADPr). The products and reactants are separated by chromatography, ethyl acetate extraction, or charcoal binding and then detected by scintillation counting [10]. Alternatively, release of [14C]-nicotinamide from [14C]NAD+ can be measured by either HPLC separation, ethyl acetate extraction, or boronic acid resin filtration of the [14C]nicotinamide product followed by scintillation counting [10, 11]. Although useful and sensitive, these radioactive assays have the drawback of health hazards, cost, synthesis, and management of radioactive substrates and waste.
Other sirtuin assays utilize fluorescent substrates. The commercial Fluor de Lys assay employs 7-amino-4-methylcoumarin (AMC) that is quenched through conjugation to the C-terminal end of a short acetyl-lysine containing peptide. Upon deacetylation, the liberated ε-amino group of the lysine becomes a trypsin substrate. Trypsin cleavage releases the fluorophore resulting in an increase in fluorescence [12]. A similar principle underlies the Cbz-Lys(Ac)-AMC (Z-MAL) substrate [13], fluorescence-resonance-energy-transfer (FRET) substrates with a C-terminal fluorophore and an N-terminal quencher [14], and fluorescence polarization substrates with a C-terminal fluorophore and an N-terminal biotin [8]. Several Sirt1 inhibitors and activators have been identified through high-throughput screening using the Fluor de Lys assay [15, 16]. However, Sirt1 activation by some compounds in Fluor de Lys assays does not correlate with activity measured using non-fluorescent peptides or full-length proteins [17, 18]. For example, the putative Sirt1 activator, resveratrol, requires the AMC portion of the Fluor de Lys substrate to observe activation.
Additional sirtuin assays include those based on Caliper’s mobility shift assay technology [19], enzyme-linked immunosorbent assays (ELISA) [20], and capillary electrophoresis assays [21, 22]. Finally, a bioluminescence assay that measures consumption of NAD+ through a series of enzymatic reactions including luciferase has also been reported [19].
Although each of the above assays has advantages and disadvantages, all require unnatural fluorescent, radioactive, or otherwise difficult to synthesize substrates, additional enzymatic steps for detection of the deacetylation reaction, and specialized equipment. Importantly, all previously reported sirtuin assays are endpoint assays. Here, we report an assay that measures sirtuin (or other nicotinamide forming enzyme) activity with any peptide or bona fide protein substrate, does not use fluorescent or radioactive labels, requires no specialized instrumentation other than the common spectrophotometer, and continuously measures sirtuin activity. This assay requires no additional workup steps to detect enzymatic activity, and therefore is generally more rapid than existing methods. As we demonstrate with CD38, this assay can be used, in principle, to monitor any enzyme that produces nicotinamide.
Materials and methods
Chemicals and reagents
The acetyl-lysine peptide based on the sequence of the N-terminal tail of human histone H3 (H2N-KSTGGK(COCH3)APRKQ-OH; AcH3) was synthesized at the UW-Madison peptide synthesis center according to previously published procedures [23]. Elixir compound 1 was synthesized according to published procedures [16]. All other chemicals used were of the highest purity commercially available and were purchased from Sigma (St. Louis, MO), Aldrich (Milwaukee, WI), or Fisher Scientific (Pittsburgh, PA).
Enzymes
Human recombinant Sirt1 [17, 24], MBP-PncA (maltose binding protein fused to nicotinamidase from Salmonella enterica) [25], AceCS1 [24], and AcuA [24, 26] were expressed and purified from Escherichia coli as described previously. MBP-PncA was a generous gift from Jane Garrity and Jorge C. Escalante-Semerena (UW-Madison). Enzyme concentrations were determined using the method of Bradford and BSA as the standard [27]. Enzyme aliquots were stored at -20 °C until use. Glutamate dehydrogenase from bovine liver and glutamate dehydrogenase from Proteus were purchased from Sigma (St. Louis, MO). CD38 was purchased from R&D systems (Minneapolis, MN).
Cell lines and cell culture
Human embryonic kidney 293 (HEK 293) cell line was cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10 vol% Fetal Bovine Serum (FBS). Culturing was performed under standard conditions, 37 °C and 5% CO2. Cells were transfected with lipofectamine and 4 μg pcDNA3.1 containing Sirt1, Sirt2, or Sirt3 as described [24]. Cells were lysed by dounce homogenization in 10 mM HEPES pH 7.5, 175 mM NaCl, and HALT EDTA free Protease inhibitors (Pierce; Rockford, IL). Protein content was determined by the method of Bradford utilizing BSA as the standard [27].
General sirtuin enzyme coupled assay
Sirt1 activity was continuously measured using a Multiskan Ascent microplate reader (LabSystems; Franklin, MA). Typical assay mixtures contained 20-800 μM AcH3, 20-1000 μM NAD+, 0.2 mM NAD(P)H, 1 mM DTT, 3.3 mM α-ketoglutarate, 1-2 μM MBP-PncA (nicotinamidase), ∼2 units of glutamate dehydrogenase from Proteus or ∼3 units of glutamate dehydrogenase from bovine liver (one unit is defined by the manufacturer to reduce 1.0 μmol of α-ketoglutarate to glutamate per min), 0.2-1 μM Sirt1 in 20 mM potassium phosphate at pH 7.5. ADP-ribose inhibition reactions contained 150 μM NAD+ and 100 μM AcH3 with ADP-ribose varied from 1 μM to 3 mM. Elixir compound 1 [16] inhibition reactions contained 80 μM NAD+ and 100 μM AcH3 with inhibitor concentrations varied from 0.1 to 100 μM. Z’ factor reactions utilized NADH and glutamate dehydrogenase from bovine liver with standard assay conditions containing 100 μM AcH3, 150 μM NAD+, and 5% v/v DMSO with 0.5 μM Sirt1 for the positive control and no Sirt1 for the negative control. The Z’ factor was calculated as described previously [28]. Sirt1 reactions were carried out in a final volume of 300 μL per well in a flat-bottom clear 96-well plate. All assay components except Sirt1 or NAD+ were preincubated at 25 °C for 5 min or until absorbance at 340 nm stabilized and the reaction was initiated by addition of Sirt1 or NAD+. The rates were analyzed continuously for 10, 20, 30, or 60 min by measuring NAD(P)H consumption at 340 nm. Alternatively, NAD(P)H was quantified by its intrinsic fluorescence with excitation at ∼340 nm and emission at ∼460 nm in a solid black flat bottom 96-well plate. Rates were determined from the slopes of the initial linear portion of each curve using an extinction coefficient for NAD(P)H of 6.22 mM-1 cm-1 and a pathlength of 0.9 cm for 300 μL reactions. The background rates of reactions lacking either Sirt1 or NAD+ resulting from the spontaneous formation of nicotinamide or ammonia were subtracted from the initial velocities of the Sirt1-catalyzed reactions.
Sirt1 enzyme coupled assay using acetyl-CoA synthetase 1 as a substrate
Assays were carried out as described under general sirtuin coupled assay. Reaction mixtures contained 1-50 μM AceCS1, 10 μM AcuA, 250 μM acetyl-CoA, 750 μM NAD+, 0.2 mM NADPH, 1 mM DTT, 3.3 mM α-ketoglutarate, 2 μM MBP-PncA (nicotinamidase), ∼2 units of glutamate dehydrogenase from Proteus. All assay components except Sirt1 were preincubated at 25 °C for 10 min or until absorbance at 340 nm stabilized and the reaction was initiated by addition of Sirt1.
Measurement of sirtuin activity in HEK-293 cell extracts
Assays were carried out as described under general sirtuin coupled assay. Reaction mixtures contained 800 μM AcH3, 1 mM NAD+, 0.2 mM NADPH, 1 mM DTT, 3.3 mM α-ketoglutarate, 2 μM MBP-PncA (nicotinamidase), ∼2 units of glutamate dehydrogenase from Proteus, and 50 μL of a whole cell lysate from HEK-293 cells in 20 mM potassium phosphate at pH 7.5 in 300 μL total volume. All assay components except NAD+ were preincubated at 25 °C for at least 15 min until absorbance at 340 nm stabilized and the reaction was initiated by addition of NAD+. The rates were measured and analyzed as described above for the general sirtuin coupled assay.
Charcoal binding assay determination of Sirt1 activity
Reactions were performed in 80 μL containing NAD+, [3H]AcH3 prepared as previously described [10], 1 mM DTT, 0.2-0.5 μM Sirt1 in 50 mM Tris pH 7.5 at 25 °C. To determine Sirt1 steady-state kinetic parameters either NAD+ was varied from 20 to 640 μM at a saturating concentration of [3H]AcH3 (320 μM) or [3H]AcH3 was varied from 20 to 640 μM at saturating concentration of NAD+ (640 μM). ADP-ribose inhibition reactions contained 150 μM NAD+ and 100 μM [3H]AcH3 with ADP-ribose varied from 1 μM to 3 mM. Elixir compound 1 [16] inhibition reactions contained 80 μM NAD+ and 100 μM AcH3 with inhibitor concentrations varied from 0.1 to 100 μM. Reactions were initiated by addition of enzyme, incubated for 8-30 min, and quenched with activated charcoal. Inhibition was determined by measuring the initial forward rate of [3H]OAADPr formation using the charcoal-binding assay as previously described [10].
Saturation kinetics of CD38 with enzyme coupled assay
CD38 reaction mixtures contained 5-320 μM NAD+, 0.2 mM NADH, 1 mM DTT, 3.3 mM α-ketoglutarate, 1-2 μM MBP-PncA, ∼3 units of glutamate dehydrogenase from bovine liver, 2 nM CD38 in 20 mM potassium phosphate at pH 7.5 in 300 μL total volume. All assay components except CD38 were preincubated at 25 °C for 5 min or until absorbance at 340 nm stabilized and the reaction was initiated by addition of CD38. The rates were measured and analyzed as described above for the general sirtuin coupled assay.
Results and Discussion
General description of the enzyme coupled assay
Sirtuins catalyze protein deacetylation utilizing NAD+ as a co-substrate and generate the reaction products nicotinamide, deacetylated protein, and OAADPr (Figure 1) [4]. In this study, we designed a spectrophotometric assay that continuously measures nicotinamide formation. The rate of nicotinamide formation is measured using a coupled enzyme system with nicotinamidase and glutamate dehydrogenase. Nicotinamidase hydrolyses nicotinamide to nicotinic acid and ammonia. Glutamate dehydrogenase then converts ammonia, α-ketoglutarate, and NAD(P)H to glutamate and NAD(P)+. NAD(P)H oxidation/consumption is measured spectrophotometrically at 340 nm (Figure 1).
Figure 1.

General diagram of nicotinamidase/glutamate dehydrogenase coupled assay.
Enzyme linearity
The utility of the coupled assay was initially tested by measuring NADPH consumption at 340 nm due to Sirt1-dependent nicotinamide formation at a variety of Sirt1 concentrations. Figure 2A shows the kinetic traces of the nicotinamidase/glutamate dehydrogenase coupled Sirt1 reactions. When the reaction was initiated by Sirt1 addition, robust steady-state initial velocities were measured; only low background rates were observed in the absence of Sirt1. After initiation with Sirt1, a lag phase of approximately two minutes was observed that is likely due to the necessary build up of nicotinamide from the Sirt1 reaction before a linear response is achieved from the coupled enzyme system. Short lag phases in the kinetic traces are common in enzyme-coupled reactions [29]. The initial velocities were calculated from the linear portion of the curves.
Figure 2.

(A) Representative kinetic traces at increasing Sirt1 concentrations. (B) Linear dependence of coupled assay on Sirt1 concentration. Assays were performed as described under Materials and Methods. Reaction mixtures contained 640 μM AcH3, 640 μM NAD+, 0.2 mM NADPH, 1 mM DTT, 3.3 mM α-ketoglutarate, 2 μM MBP-PncA, ∼2 units of glutamate dehydrogenase from Proteus, and 0.2-1 μM Sirt1 in 20 mM potassium phosphate at pH 7.5. Background rates measured in the absence of Sirt1 were subtracted from the Sirt1 catalyzed reactions.
To verify the accuracy of the coupled assay and that the observed rates are not limited by the coupled reaction, initial velocities were measured with increasing Sirt1 concentrations. Figure 2B demonstrates a linear relationship between the NADPH consumption rate and Sirt1 concentration; that is, the observed rate depends only on Sirt1 activity - not nicotinamidase or glutamate dehydrogenase. To further demonstrate that the coupled assay does not limit the observed rate, concentrations of the coupled reaction components (i.e. α-ketoglutarate, nicotinamidase, and glutamate dehydrogenase) were decreased two to four-fold and the reactions repeated. Decreasing α-ketoglutarate concentrations from 3.33 mM to 1 mM, glutamate dehydrogenase levels two-fold, or nicotinamidase concentrations from 2 μM to 0.5 μM did not significantly affect the observed rates (see Figure S1A-C in supplementary material), indicating that the original conditions were sufficient and that the coupled reaction does not limit the observed initial rates.
Determination of sirtuin saturation kinetics
Having demonstrated Sirt1 activity could be accurately measured, we used this spectrophotometric assay to obtain the relevant steady-state rate constants. First, we measured the ability of an 11-mer acetyl-lysine peptide based on the N-terminal tail of histone H3 acetylated at lys-14 (AcH3) to saturate Sirt1 catalyzed reactions. The concentration of AcH3 was varied at saturating NAD+ concentrations, and the initial velocities were determined by measuring the continuous consumption of NADPH at 340 nm. Background rates in the absence of AcH3 were subtracted from the rates determined in the presence of AcH3. The data were fitted to the Michaelis-Menten equation to yield kcat and Km values of 0.062 ± 0.002 s-1 and 109 ± 9 μM (Figure 3A; Table 1) and compared to values of 0.065 ± 0.004 s-1 and 127 ± 20 μM that we obtained with the radioactive charcoal-binding assay and the same Sirt1 preparation (Table 1). We also measured the ability of NAD+ to saturate Sirt1 activity. NAD+ concentrations were varied and the resulting initial velocities were fitted to the Michaelis-Menten equation (Figure 3B), yielding kcat and Km values of 0.060 ± 0.003 s-1 and 171 ± 23 μM were compared to values of 0.064 ± 0.004 s-1 and 154 ± 28 μM measured with the radioactive charcoal-binding assay. These measured steady-state kinetic parameters reveal excellent agreement between the coupled and radioactive assays.
Figure 3.

Steady-state kinetics of Sirt1 varying (A) AcH3 or (B) NAD+ concentrations. Reaction mixtures either contained 20-640 μM AcH3 with 640 μM NAD+ or 20-640 μM NAD+ with 640 μM AcH3 in 0.2 mM NAD(P)H, 1 mM DTT, 3.3 mM α-ketoglutarate, 2 μM MBP-PncA, ∼2 units of glutamate dehydrogenase from Proteus, 0.5 μM Sirt1 in 20 mM potassium phosphate at pH 7.5. Background rates measured in absence of the varied substrate were subtracted from the Sirt1 catalyzed reactions. To determine kcat and Km values, the plot of steady-state nicotinamide formation rates versus substrate concentration was fit to the Michaelis-Menten equation [v = (kcat · [S])/(Km + [S])] using KaleidaGraph (Synergy Software, Reading, PA).
Table 1.
Comparison of kinetic parameters with coupled and charcoal binding sirtuin assays
| Assay | kcat (s-1) | Km (μM) | ADP-ribose IC50 (μM) | Elixir compound 1 IC50 (μM) | ||
|---|---|---|---|---|---|---|
| Acetyl-lysine peptide | NAD+ | Acetyl-lysine peptide | NAD+ | |||
| Coupled | 0.062 ± 0.002 | 0.060 ± 0.003 | 109 ± 9 | 171 ± 23 | 77 ± 4 | 0.74 ± 0.25 |
| Charcoal binding | 0.065 ± 0.004 | 0.064 ± 0.004 | 127 ± 20 | 154 ± 28 | 80 ± 15 | 1.18 ± 0.24 |
Measurement of sirtuin activity with native protein substrates
Having demonstrated Sirt1 activity and kinetic parameters can be accurately measured using the coupled assay, we used this spectrophotometric assay to obtain the relevant steady-state rate constants for a native protein substrate. Direct measurement of sirtuin activity with native protein substrates has previously been problematic because of the difficulty in specifically labeling proteins with radioactive isotopes or fluorophores. Here, we utilized the protein lysine acetyltransferase, AcuA, to specifically acetylate the Sirt1 protein substrate acetyl-CoA synthetase 1 (AceCS1) at lysine-661 and to recycle the deacetylated AceCS1 [24]. We then measured Sirt1-dependent deacetylation at variable AceCS1 and saturating NAD+ concentrations. Spontaneous background rates (without Sirt1) were subtracted from rates determined in the presence of Sirt1. Importantly, AceCS1 deacetylation reactions exhibited linear NADPH consumption rates that far exceeded the initial AceCS1 concentration in each reaction (data not shown), confirming that AceCS1 is continuously acetylated and recycled in the assay. Recycling of acetylated AceCS1 permitted accurate rates to be determined with very low (less than 10 μM) AceCS1 concentrations. The data were fitted to the Michaelis-Menten equation to yield kcat and Km values of 0.081 ± 0.010 s-1 and 12.0 ± 4.2 μM (Figure 4). The kcat values were in excellent agreement between the AceCS1 and AcH3 assays. Interestingly, the Km value for AceCS1 was 10-fold less than that of AcH3, which is not surprising given that AceCS1 is a bona fide in vivo substrate of Sirt1 [24]. To our knowledge this is the first direct measurement of the steady-state parameters for a native-protein sirtuin substrate.
Figure 4.
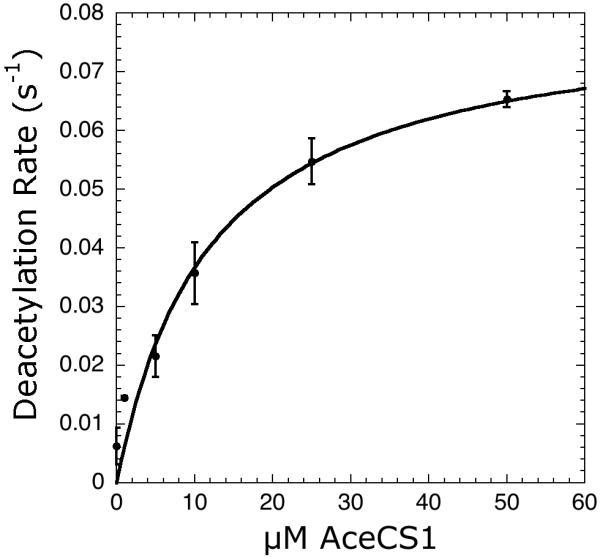
Steady-state kinetics of Sirt1 varying concentrations of native AceCS1. Reaction mixtures either contained 0-50 μM AceCS1 with 10 μM AcuA, 250 μM acetyl-CoA, in 750 μM NAD+, 0.2 mM NADPH, 1 mM DTT, 3.3 mM α-ketoglutarate, 2 μM MBP-PncA, ∼2 units of glutamate dehydrogenase from Proteus, 0.5 μM Sirt1 in 20 mM potassium phosphate at pH 7.5. Background rates measured in absence of the varied substrate were subtracted from the Sirt1 catalyzed reactions. To determine kcat and Km values, the plot of steady-state nicotinamide formation rates versus substrate concentration was fit to the Michaelis-Menten equation [v = ([E]0 kcat · [S])/(Km + [S])] using KaleidaGraph (Synergy Software, Reading, PA).
Direct measurement of sirtuin activity in human cell extracts
Different strategies to measure protein lysine deacetylase activity in cells or tissue have focused on antibody-based techniques such as immunohistochemistry [30], western blotting [31], and enzyme-linked immunosorbent assays [32], or assays utilizing small molecule substrates with either magnetic resonance [33] or fluorescence detection [34]. However, these assays are either unselective or otherwise not viable for directly measuring sirtuin activity over other protein lysine deacetylases from biological samples. In addition, none of these assays are capable of measuring sirtuin activity in a continuous manner. Therefore, we investigated the feasibility of directly measuring sirtuin activity from whole cell extracts. Because the coupled assay detects nicotinamide formation and utilizes a general sirtuin acetyl-lysine peptide substrate, it is selective for sirtuins (Class III) over other classes (I, II, IV) of deacetylases that do not use NAD+.
The human sirtuins Sirt1, Sirt2, and Sirt3 were transiently transfected into HEK 293 cells, and soluble cellular lysates were used to determine the deacetylase activity in a variety of extract samples. Utilizing saturating NAD+ and AcH3 concentrations, the relative activity of each sirtuin extract was determined in μM sec-1 μg-1 of total protein (Figure 5A). Activity was determined from three biological replicates to determine the reliability and repeatability of measuring activity from cellular extracts. To determine the expression levels of the transiently-transfected sirtuins, the extracts were further resolved by SDS-PAGE, western blotted, and detected with anti-FLAG antibody to confirm overexpression of the transfected sirtuin (Figure 5B). The measured deacetylase activity in extracts from HEK 293 cells overexpressing Sirt1, Sirt2, and Sirt3 demonstrated a substantial increased (≥3-fold) relative to the mock-transfected cells. A low but significant level of activity was observed in the mock-transfected cells due to endogenous sirtuin activity or another activity that forms nicotinamide or ammonia from NAD+. Sirt1, Sirt2, and Sirt3 are expressed endogenously in HEK 293 cells ([35] and unpublished observations). Importantly, only a slow rate of nicotinamide formation, which was not significantly above background, was observed when NAD+ was not added to the reaction mixture. Thus, the coupled assay can be used to monitor both endogenous and over-expressed sirtuin activity in whole-cell protein extracts.
Figure 5.


Direct measurement of sirtuin activity in cell extracts. Assays were carried out as described in Materials and Methods. Reaction mixtures contained 800 μM AcH3, 1 mM NAD+, 0.2 mM NADPH, 1 mM DTT, 3.3 mM α-ketoglutarate, 2 μM MBP-PncA, ∼2 units of glutamate dehydrogenase from Proteus, and 50 μL of a whole cell lysate from HEK 293 cells in 20 mM potassium phosphate at pH 7.5. All assay components except NAD+ were preincubated at 25 °C for at least 15 min until absorbance at 340 nm stabilized and the reaction was initiated by addition of NAD+. The measured rates were normalized to overall protein concentration using the method of Bradford with BSA as the standard [27] and are expressed in μM nicotinamide per second per μg total protein in the cell extract.
(B) Soluble protein extracts from HEK 293 cells overexpressing flag tagged Sirt1, Sirt2, and Sirt3 were separated by SDS-PAGE, western blotted and detected with Anti-flag antibody, and anti-actin antibody. Protein was normalized by using the method of Bradford with BSA as the standard [27].
Evaluation of sirtuin inhibitors
The coupled assay was evaluated further for use in the analysis of sirtuin inhibitors. Using AcH3 and NAD+ concentrations below their respective Km values, the potency of ADP-ribose as a Sirt1 inhibitor was determined. With the coupled assay utilizing absorbance detection, ADP-ribose inhibited Sirt1 with an IC50 value of 77 ± 4 μM (Figure 6; Table 1). This IC50 value is consistent with the value of 80 ± 15 μM determined by the charcoal-binding assay (Table 1). Because some small molecule inhibitors might absorb in the same spectral region as NAD(P)H, we also measured ADP-ribose inhibition utilizing the intrinsic fluorescence of NAD(P)H (λex = 340 nm; λem = 460 nm) as a readout. Indeed, monitoring NADPH fluorescence a similar ADP-ribose IC50 value of 61 ± 21 μM (compared to 77 ± 4 μM for absorbance) was obtained.
Figure 6.
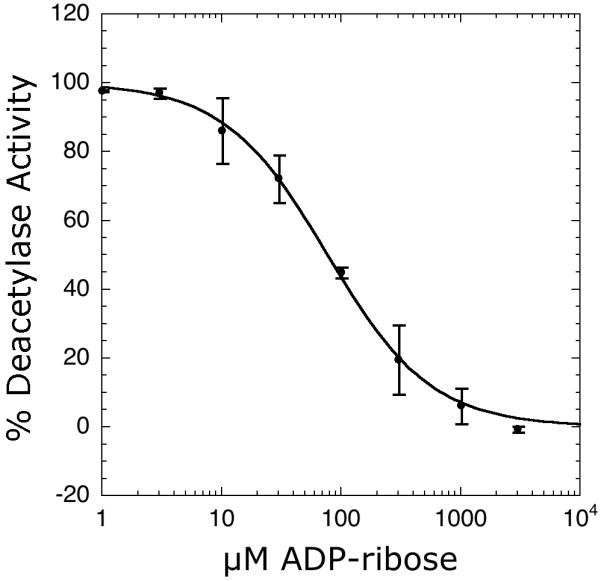
Inhibition of Sirt1 by ADP-ribose determined by the coupled assay. Reaction mixtures contained 1-3000 μM ADP-ribose, 100 μM AcH3, 150 μM NAD+, 0.2 mM NADPH, 1 mM DTT, 3.3 mM α-ketoglutarate, 2 μM MBP-PncA, ∼2 units of glutamate dehydrogenase from Proteus, 1 μM Sirt1 in 20 mM potassium phosphate at pH 7.5. To determine IC50 values, the plot of steady-state nicotinamide formation rates versus ADP-ribose concentration were fit to the equation: v = Vmax(1 - ([I]/(IC50 + [I]))) using KaleidaGraph (Synergy Software, Reading, PA).
It is important to utilize proper controls to ensure that the coupling enzymes are not inhibited by the addition of the small molecule effectors being evaluated. A simple control would be to add a standard concentration of nicotinamide to the assay mixture and assess whether the compound yields a lower rate compared to the mock treated sample (e.g. DMSO). Sirtuin inhibitors that bind in the nicotinamide binding pocket could pose problems; such inhibitors might inhibit or be a substrate of nicotinamidase. This can easily be examined as described above. In cases where the coupled assay is compromised, another assay (e.g. charcoal binding assay) should be utilized. To provide an example for an inhibitor that might present such issues, we examined Elixir compound 1 [16] (see Figure S2A in supplementary material) which is a potent Sirt1 inhibitor and is similar in structure to nicotinamide and likely binds to the sirtuin nicotinamide binding pocket [16]. In addition, Elixir compound 1 contains a primary amide and could serve as a nicotinamidase substrate. Despite these potential issues, we were able to measure an IC50 value of 0.74 ± 0.25 μM (see Figure S2B in supplementary material; Table 1) with the coupled assay, in good agreement with a value of 1.18 ± 0.24 μM determined from the charcoal binding assay as well as with previously reported values [16, 19]. We observed higher background rates (absence of Sirt1) in the coupled assay at higher inhibitor concentrations, which made it necessary to measure and subtract background rates at all inhibitor concentrations. These variable background rates resulted in larger errors compared to those of the charcoal-binding assay (see Figure S2B in supplementary material), though equivalent IC50 values were obtained with both assays.
Applicability of the coupled assay to high throughput screening
Although several other sirtuin high throughput screens have been reported [8, 12, 14, 19, 36], the coupled assay could be adapted to a high throughput format. Importantly, the coupled assay is a practical secondary screen to identify potential false positives from other high throughput screens. The coupled assay would be particularly useful in cases where no other viable assay exists. As a case in point, there is significant controversy surrounding the ability of resveratrol to activate Sirt1 [17, 18, 22]. In vitro Sirt1 activation by resveratrol has never been observed when applied outside of the Fluor de Lys or other fluorophore based assays. The use of a secondary screen could have eliminated this ‘activator’ from obfuscating current research.
Using a continuous glutamate dehydrogenase coupled assay, Pfizer has developed an entire high-throughput screening program targeting fatty acid amide hydrolase (FAAH) [37-39]. FAAH activity was measured continuously by following the production of ammonia generated from FAAH catalyzed hydrolysis of oleamide. As employed in our assay, glutamate dehydrogenase catalyzes the condensation of ammonia and α-ketoglutarate to glutamate with a concomitant conversion of NADH to NAD+, followed as an absorbance change at 340 nm. To date, Pfizer has screened ∼500,000 compounds utilizing this assay [40]. A continuous assay avoids many of the artifacts associated with fluorescent and end-point assays. This is primarily because product development (signal) is followed over time, minimizing issues from background small molecule absorbance or fluorescence.
To assess the potential of adapting our coupled assay to a high throughput format, we determined a Z’-factor under typical assay conditions for testing inhibitors (see Materials and methods). Under these conditions, a Z’-factor of 0.86 was determined, which is equal to or better than all other Z’-factors reported for protein lysine deacetylase high-throughput screens that range from 0.69-0.88 [12, 19, 34] and is indicative of an ‘excellent’ assay [28]. However, one drawback to a continuous assay is the additional time required to measure each well compared to endpoint assays, which may limit the throughput of our coupled assay. In addition, because small molecule compounds are typically dissolved in DMSO, the tolerance of the coupled assay to DMSO concentration was determined. With 2 μM Sirt1, 80 μM NAD+, and 100 μM AcH3, no significant decrease in enzyme activity was observed up to 20% v/v DMSO (see Figure S1D in supplementary material).
Measuring other nicotinamide forming enzymes via the enzyme coupled assay
As the nicotinamidase/glutamate dehydrogenase enzyme coupled assay detects nicotinamide, this assay should prove useful in assaying any nicotinamide forming enzyme. To test this hypothesis, we assayed CD38 activity. CD38 is a dual-activity ectoenzyme that possesses both ADP-ribosyl cyclase and NAD+ hydrolase activity, both of which form nicotinamide. We used the nicotinamidase/glutamate dehydrogenase coupled assay to measure the kcat and Km values of CD38 using NAD+ as substrate and compared these values to those previously obtained via an HPLC assay [41]. Using the coupled assay, we determined a kcat value of 98 ± 9 s-1 and a Km value of 17 ± 6 μM (Figure 7). These values agreed very favorably with the previously reported values of 96 ± 5 s-1 and 16 ± 1 μM using an HPLC assay [41], demonstrating the utility of the coupled assay with other nicotinamide forming enzymes.
Figure 7.
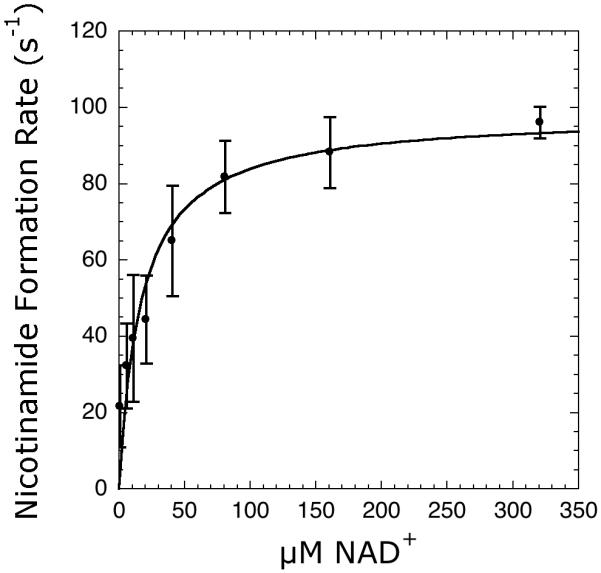
Steady-state kinetics of CD38 utilizing the coupled assay. Reaction mixtures contained 5-320 μM NAD+, 0.2 mM NADH, 1 mM DTT, 3.3 mM α-ketoglutarate, 1-2 μM MBP-PncA, ∼3 units of glutamate dehydrogenase from bovine liver, 2 nM CD38 in 20 mM potassium phosphate at pH 7.5. The rates were measured and analyzed as described under Materials and Methods.
Considerations in selecting a glutamate dehydrogenase homolog, NADH or NADPH, and absorbance or fluorescence detection
The flexibility of our assay, through utilizing either NADH or NADPH and either glutamate dehydrogenase from bovine liver or from Proteus, provides researchers the ability to tailor the assay to their specific application without changing the general protocol. To optimize the coupled assay for a specific application several considerations are necessary in choosing the appropriate glutamate dehydrogenase homolog, as well as in the decision to use NADH or NADPH. Glutamate dehydrogenase from bovine liver accepts both NADH and NADPH as substrates, whereas glutamate dehydrogenase from Proteus is selective for NADPH. To aid in such decisions, we provide a decision flow chart (Figure 8). When preparing to use the coupled assay with an uncharacterized nicotinamide-forming enzyme, one must first determine if NADH or NADPH significantly inhibits the enzyme of interest. Given the excellent agreement of kinetic parameters obtained between the coupled and radioactive assays, this is clearly not an issue with Sirt1. We have previously measured an NADH IC50 value of 28 mM compared to the >2000-fold lower Km value of 12 μM for NAD+ [42] with a yeast Sirt1 homolog, Hst2. These results support the inability of NADH or NADPH to significantly inhibit sirtuins.
Figure 8.

Decision flow chart to aid in choosing a glutamate dehydrogenase, NADH or NADPH, and absorbance or fluorescence detection in the coupled assay.
The next step in selecting a glutamate dehydrogenase is to determine if the enzyme of interest is selective for NAD+ over NADP+. Because the coupled assay will form NAD(P)+ from NAD(P)H oxidation, it is important that NADP+ is not a substrate that competes for NAD+ binding in the enzyme being assayed. For Sirt1, the Km value for NADP+ is > 2.5 mM (data not shown) in line with the previously reported Km value for NADP+ of 1.6 mM with Hst2 [42]. Therefore, the Km value for NAD+ is at least 15-fold lower than that of NADP+ for Sirt1, and NADPH can be utilized as a substrate for glutamate dehydrogenase when assaying Sirt1 (or Hst2) activity. However, CD38 is capable of utilizing NAD+ and NADP+ as substrate with similar efficiencies [43, 44], so NADH and glutamate dehydrogenase from bovine liver must be utilized if rates from NAD+ turnover are desired.
A subsequent point to consider is the NAD+ concentrations that will be used in the assay. If saturating NAD+ concentrations will be used, then either glutamate dehydrogenase from bovine liver or glutamate dehydrogenase from Proteus may be used to obtain identical results. If not, then utilization of glutamate dehydrogenase from Proteus and NADPH is ideal to allow tight control over NAD+ levels since premixing everything except enzyme will form a small amount of NADP+ from any residual ammonia or nicotinamide present in the assay mixture. One exception is if NAD+ levels of less than 10 μM will be used. If this is the case, then glutamate dehydrogenase from bovine liver and NADH should be used to afford continuous regeneration of NAD+ from NADH oxidation, such that linear rates are measured for concentrations of NADH consumption exceeding the initial NAD+ concentration in the reaction mixture. With NAD+ concentrations less than 10 μM, caution must be used to minimize formation of NAD+ from NADH oxidation prior to enzyme addition due to the presence of ammonia or nicotinamide or the spontaneous hydrolysis of NAD+ in the assay mixture, otherwise elevated initial rates will be measured from the additional NAD+ formed. In practice however, this is not a major issue as evident by the excellent agreement in steady-state kinetic parameters obtained for CD38 between the coupled assay and an HPLC assay.
Finally, the utilization of absorbance or fluorescence detection should be considered. In general, absorbance detection of NAD(P)H at 340 nm is the simplest and most robust option. If a small molecule added to the assay mixture also absorbs at 340 nm and precludes continuous product detection at this wavelength, then detection of the intrinsic fluorescence of NAD(P)H is a viable alternative. If the small molecule also interferes with the intrinsic fluorescence of NAD(P)H, then an alternative assay must be utilized. However, as the absorption and emission wavelength of AMC are similar to NAD(P)H, these small molecules may be problematic in assays based on AMC fluorescence. It should be noted that NAD(P)H fluorescence is not as bright as that of AMC and other common fluorophores [45], and the sensitivity of NAD(P)H absorbance detection is less than other sirtuin assays that utilize fluorescence or radioactive detection.
Conclusions
In this study, we developed a continuous, nonradioactive, spectrophotometric assay for sirtuins and other nicotinamide-forming enzymes. This assay system utilizes nicotinamidase and glutamate dehydrogenase as coupling enzymes. Our investigation indicates that the assay is rapid, inexpensive, accurate, and a safer means of measuring steady-state kinetic parameters compared to commonly used HPLC, radioactive, or fluorescent sirtuin assays. Importantly, the kinetic constants obtained with the coupled assay were comparable to those obtained with a radioactive, charcoal-binding assay, which has been a gold standard for mechanistic studies. This new coupled assay is amenable to high-throughput analysis of acetylated substrates or inhibitors as was evident from utilization of a microplate format in the current study. Although the coupled assay is susceptible to false positives from inhibition of nicotinamidase or glutamate dehydrogenase, the assay possesses the significant advantage of continuously measuring product formation over time which minimizes interference from compounds that absorb in the same spectral region as NAD(P)H. Therefore, this coupled assay is well suited for high-throughput screening for small molecule sirtuin inhibitors or activators. In addition, this assay could serve as a secondary assay to validate potential sirtuin inhibitors or activators discovered through high-throughput screening.
In addition, although native AceCS1 protein deacetylation has been measured by activation of AceCS1 activity [24], this coupled assay is the first assay to directly measure deacetylase activity on native protein substrates, permitting detailed kinetic analysis of specific sirtuin substrates and facilitating the development of substrate specific inhibitors and activators. We also demonstrated that the coupled assay can accurately measure deacetylase activity from mammalian cell lysates. The ability to assay sirtuins in cell extracts will enable researchers to monitor the regulation of sirtuin activity under various cellular pharmacological treatments, with endogenous regulators such as AROS and DBC1 [46-48], as well as when comparing normal and disease states. This continuous spectrophotometric assay will be widely applicable for studying the diverse families of nicotinamide forming enzymes.
Supplementary Material
Acknowledgments
We thank Dr. Gary Case for help with peptide syntheses. We thank Jörg Schemies for suggestions and assistance with assays. We thank Jane Garrity and Dr. Jorge C. Escalante-Semerena for providing the plasmid for MBP-PncA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- [2].Di Girolamo M, Dani N, Stilla A, Corda D. Physiological relevance of the endogenous mono(ADP-ribosyl)ation of cellular proteins. Febs J. 2005;272:4565–4575. doi: 10.1111/j.1742-4658.2005.04876.x. [DOI] [PubMed] [Google Scholar]
- [3].Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- [4].Smith BC, Hallows WC, Denu JM. Mechanisms and molecular probes of sirtuins. Chem Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vaquero A, Sternglanz R, Reinberg D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene. 2007;26:5505–5520. doi: 10.1038/sj.onc.1210617. [DOI] [PubMed] [Google Scholar]
- [6].Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- [7].Milne JC, Denu JM. The Sirtuin family: therapeutic targets to treat diseases of aging. Curr Opin Chem Biol. 2008 doi: 10.1016/j.cbpa.2008.01.019. [DOI] [PubMed] [Google Scholar]
- [8].Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- [10].Borra MT, Denu JM. Quantitative assays for characterization of the Sir2 family of NAD(+)-dependent deacetylases. Methods Enzymol. 2004;376:171–187. doi: 10.1016/S0076-6879(03)76011-X. [DOI] [PubMed] [Google Scholar]
- [11].McDonagh T, Hixon J, DiStefano PS, Curtis R, Napper AD. Microplate filtration assay for nicotinamide release from NAD using a boronic acid resin. Methods. 2005;36:346–350. doi: 10.1016/j.ymeth.2005.03.005. [DOI] [PubMed] [Google Scholar]
- [12].Wegener D, Hildmann C, Riester D, Schwienhorst A. Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal Biochem. 2003;321:202–208. doi: 10.1016/s0003-2697(03)00426-3. [DOI] [PubMed] [Google Scholar]
- [13].Heltweg B, Dequiedt F, Verdin E, Jung M. Nonisotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Anal Biochem. 2003;319:42–48. doi: 10.1016/s0003-2697(03)00276-8. [DOI] [PubMed] [Google Scholar]
- [14].Marcotte PA, Richardson PL, Guo J, Barrett LW, Xu N, Gunasekera A, Glaser KB. Fluorescence assay of SIRT protein deacetylases using an acetylated peptide substrate and a secondary trypsin reaction. Anal Biochem. 2004;332:90–99. doi: 10.1016/j.ab.2004.05.039. [DOI] [PubMed] [Google Scholar]
- [15].Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- [16].Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, Thomas RJ, Kates M, Jones S, Navia MA, Saunders JO, DiStefano PS, Curtis R. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- [17].Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- [18].Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, Bedalov A, Kennedy BK. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- [19].Liu Y, Gerber R, Wu J, Tsuruda T, McCarter JD. High-throughput assays for sirtuin enzymes: a microfluidic mobility shift assay and a bioluminescence assay. Anal Biochem. 2008;378:53–59. doi: 10.1016/j.ab.2008.02.018. [DOI] [PubMed] [Google Scholar]
- [20].Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins sirt3 and sirt5. J Mol Biol. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- [21].Fan Y, Ludewig R, Imhof D, Scriba GK. Development of a capillary electrophoresis-based assay of sirtuin enzymes. Electrophoresis. 2008;29:3717–3723. doi: 10.1002/elps.200800361. [DOI] [PubMed] [Google Scholar]
- [22].Fan Y, Ludewig R, Scriba GK. 9-Fluorenylmethoxycarbonyl-labeled peptides as substrates in a capillary electrophoresis-based assay for sirtuin enzymes. Anal Biochem. 2009;387:243–248. doi: 10.1016/j.ab.2009.01.038. [DOI] [PubMed] [Google Scholar]
- [23].Smith BC, Denu JM. Sir2 deacetylases exhibit nucleophilic participation of acetyl-lysine in NAD+ cleavage. J Am Chem Soc. 2007;129:5802–5803. doi: 10.1021/ja070162w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem. 2007;282:30239–30245. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- [26].Gardner JG, Escalante-Semerena JC. Biochemical and mutational analyses of AcuA, the acetyltransferase enzyme that controls the activity of the acetyl coenzyme a synthetase (AcsA) in Bacillus subtilis. J Bacteriol. 2008;190:5132–5136. doi: 10.1128/JB.00340-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- [28].Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- [29].Rudolph FB, Baugher BW, Beissner RS. Techniques in coupled enzyme assays. Methods in Enzymology. 1979;63:22–42. doi: 10.1016/0076-6879(79)63004-5. [DOI] [PubMed] [Google Scholar]
- [30].Mitic T, McKay JS. Immunohistochemical analysis of acetylation, proliferation, mitosis, and apoptosis in tumor xenografts following administration of a histone deacetylase inhibitor--a pilot study. Toxicol Pathol. 2005;33:792–799. doi: 10.1080/01926230500459435. [DOI] [PubMed] [Google Scholar]
- [31].Stockwell BR, Haggarty SJ, Schreiber SL. High-throughput screening of small molecules in miniaturized mammalian cell-based assays involving post-translational modifications. Chem Biol. 1999;6:71–83. doi: 10.1016/S1074-5521(99)80004-0. [DOI] [PubMed] [Google Scholar]
- [32].Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sankaranarayanapillai M, Tong WP, Maxwell DS, Pal A, Pang J, Bornmann WG, Gelovani JG, Ronen SM. Detection of histone deacetylase inhibition by noninvasive magnetic resonance spectroscopy. Mol Cancer Ther. 2006;5:1325–1334. doi: 10.1158/1535-7163.MCT-05-0494. [DOI] [PubMed] [Google Scholar]
- [34].Ciossek T, Julius H, Wieland H, Maier T, Beckers T. A homogeneous cellular histone deacetylase assay suitable for compound profiling and robotic screening. Anal Biochem. 2008;372:72–81. doi: 10.1016/j.ab.2007.07.024. [DOI] [PubMed] [Google Scholar]
- [35].Grubisha O, Rafty LA, Takanishi CL, Xu X, Tong L, Perraud AL, Scharenberg AM, Denu JM. Metabolite of SIR2 reaction modulates TRPM2 ion channel. J Biol Chem. 2006;281:14057–14065. doi: 10.1074/jbc.M513741200. [DOI] [PubMed] [Google Scholar]
- [36].Heltweg B, Trapp J, Jung M. In vitro assays for the determination of histone deacetylase activity. Methods. 2005;36:332–337. doi: 10.1016/j.ymeth.2005.03.003. [DOI] [PubMed] [Google Scholar]
- [37].Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- [38].Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci U S A. 2008;105:12820–12824. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ahn K. personal communication.
- [41].Sauve AA, Munshi C, Lee HC, Schramm VL. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry. 1998;37:13239–13249. doi: 10.1021/bi981248s. [DOI] [PubMed] [Google Scholar]
- [42].Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme Specificity of Sir2 Protein Deacetylases: Implications for physiological regulation. Journal of Biological Chemistry. 2004;279:40122–40129. doi: 10.1074/jbc.M407484200. [DOI] [PubMed] [Google Scholar]
- [43].Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem. 1995;270:30327–30333. doi: 10.1074/jbc.270.51.30327. [DOI] [PubMed] [Google Scholar]
- [44].Preugschat F, Tomberlin GH, Porter DJ. The base exchange reaction of NAD+ glycohydrolase: identification of novel heterocyclic alternative substrates. Arch Biochem Biophys. 2008;479:114–120. doi: 10.1016/j.abb.2008.09.005. [DOI] [PubMed] [Google Scholar]
- [45].Lavis LD, Raines RT. Bright ideas for chemical biology. ACS Chem Biol. 2008;3:142–155. doi: 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- [47].Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.