

Abstract
Structural variants (SVs) are common in the human genome. Because approximately half of the human genome consists of repetitive, transposable DNA sequences, it is plausible that these elements play an important role in generating SVs in humans. Sequencing of the diploid genome of one individual human (HuRef) affords us the opportunity to assess, for the first time, the impact of mobile elements on SVs in an individual in a thorough and unbiased fashion. In this study, we systematically evaluated more than 8000 SVs to identify mobile element-associated SVs as small as 100 bp and specific to the HuRef genome. Combining computational and experimental analyses, we identified and validated 706 mobile element insertion events (including Alu, L1, SVA elements, and nonclassical insertions), which added more than 305 kb of new DNA sequence to the HuRef genome compared with the Human Genome Project (HGP) reference sequence (hg18). We also identified 140 mobile element-associated deletions, which removed ∼126 kb of sequence from the HuRef genome. Overall, ∼10% of the HuRef-specific indels larger than 100 bp are caused by mobile element-associated events. More than one-third of the insertion/deletion events occurred in genic regions, and new Alu insertions occurred in exons of three human genes. Based on the number of insertions and the estimated time to the most recent common ancestor of HuRef and the HGP reference genome, we estimated the Alu, L1, and SVA retrotransposition rates to be one in 21 births, 212 births, and 916 births, respectively. This study presents the first comprehensive analysis of mobile element-related structural variants in the complete DNA sequence of an individual and demonstrates that mobile elements play an important role in generating inter-individual structural variation.
Structural variants (SVs) in the human genome have been the subject of much recent research because of their ubiquity, their evolutionary significance, and their roles in diseases (Redon et al. 2006; Eichler et al. 2007; Lee et al. 2007b; McCarroll and Altshuler 2007). It is now recognized that SVs are common in human genomes, and most of them are, like single nucleotide polymorphisms (SNPs), selectively neutral residents of the genome (Jakobsson et al. 2008; McCarroll et al. 2008). Insertion/deletion polymorphisms, or indels, are the most common types of SVs, and the vast majority of them are relatively small in size (e.g., <10 kb) (Levy et al. 2007; Wheeler et al. 2008). Although indels have been characterized at the whole-genome level in multiple individual human genomes, most studies of indels to date have focused on relatively large events (usually >5 kb in size) using fosmid paired-end sequencing (FPES) (Tuzun et al. 2005; Kidd et al. 2008), paired-end mapping (PEM) (Korbel et al. 2007), array comparative genomic hybridization, or other microarray-based approaches (Sharp et al. 2005; Redon et al. 2006; Wong et al. 2007; Perry et al. 2008).
Mobile elements comprise approximately half of the human and primate genomes and have been a major factor in creating SVs and shaping the genome (for reviews, see Xing et al. 2007; Belancio et al. 2008; Goodier and Kazazian 2008). For example, mobile element insertions have contributed to a 15%–20% expansion of the human genome compared with strepsirrhine genomes (Liu et al. 2003). Several studies also suggest a correlation between mobile elements and the breakpoints of segmental duplications and SVs in the human genome (Bailey et al. 2003; Zhou and Mishra 2005; Kim et al. 2008; Lee et al. 2008). Although most mobile element-associated structural variants (MASVs) are thought to be selectively neutral, occasionally MASVs can cause human diseases. Since the first report of a Hemophilia A case caused by a de novo L1 insertion (Kazazian et al. 1988), more than 100 cases of documented MASVs have led to human diseases, including cases of Pelizaeus-Merzbacher disease, Lesch-Nyhan syndrome, Tay-Sachs disease, familial hypercholesterolemia, and Hunter syndrome (for reviews, see Deininger and Batzer 1999; Callinan and Batzer 2006; Chen et al. 2006).
Among all mobile element families, only retrotransposons, such as long interspersed element-1 (LINE-1, or L1), Alu element, SVA element (named after its main components, SINE-R, VNTR, and Alu), and endogenous retrovirus (ERV) are actively mobilizing in the human and primate genomes (Lander et al. 2001; Macfarlane and Simmonds 2004; Chimpanzee Sequencing and Analysis Consortium 2005; Wang et al. 2005; Mills et al. 2006; Han et al. 2007a). Non-LTR retrotransposons, including L1s, Alus, and SVAs, mobilize via RNA intermediates using a mechanism called target site-primed reverse transcription (TPRT) (Luan et al. 1993; Feng et al. 1996; Cost et al. 2002). In the TPRT process, an RNA copy is first generated from the original retrotransposon and subsequently reverse-transcribed back into the genome by a reverse transcriptase (for reviews, see Ostertag and Kazazian 2001a; Batzer and Deininger 2002; Wang et al. 2005). During the process, two short stretches of identical sequence, termed target site duplications (TSDs), are created on both ends of the new insertion. In some cases, genomic deletions are associated with the insertion events (Gilbert et al. 2002, 2005; Symer et al. 2002; Callinan et al. 2005; Han et al. 2005). In addition to canonical insertion events, retrotransposons can create genomic instability by several additional mechanisms, including nonallelic homologous recombination (NAHR) (Stankiewicz and Lupski 2002) mediated insertion/deletion between two retrotransposons from the same family, nonhomologous end-joining (NHEJ) mediated deletion, and nonclassical endonuclease-independent insertions of the retrotransposons (Deininger and Batzer 1999; Gilbert et al. 2002, 2005; Morrish et al. 2002; Symer et al. 2002; Kazazian 2004; Sen et al. 2006, 2007; Han et al. 2007b, 2008; Goodier and Kazazian 2008; Srikanta et al. 2008).
To date, a systematic evaluation of the impact of MASVs on the human genome has not yet been attempted at the individual level. With the sequence of the diploid genome of one individual human (HuRef; Levy et al. 2007), we are able to assess the impact of mobile element-associated structural variation in a thorough and unbiased fashion for the first time. In this study, we evaluated more than 8000 SVs that differ between the HuRef assembly and the haploid human genome reference sequence from the Human Genome Project (HGP). We demonstrate that an appreciable proportion of these SVs were mediated by mobile elements.
Results
Computational data mining and experimental validation
A total of 643,992 indels was initially identified by comparing the HuRef assembly and the HGP reference genome, including 559,473 homozygous indels, 6246 heterozygous indels, and 78,273 previously identified as “putative” indels (Levy et al. 2007). For homozygous and heterozygous loci, indels >100 bp were selected. Putative loci larger than 50 bp that contained complete sequence (i.e., no “N”s in the sequence) were also selected. These selection criteria resulted in a total of 8451 candidate indel loci. Then, we selected indels that contained mobile elements and manually inspected these loci along with their flanking sequence to determine the nature of these SVs.
The initial screening yielded more than 1000 “HuRef-specific” MASV candidates. These candidates represent mobile element insertions or mobile element-associated deletions in the HuRef genome that are not present in the HGP assembly. Similarly, a set of more than 1000 “HGP-specific” MASV candidates have also been identified in the HGP assembly that are not present in the HuRef genome. In this study, we focused on the “HuRef-specific” MASV candidates to assess the impact of mobile elements in an individual human.
Because many of the MASVs reside in the repeat-rich regions and because sequencing assembly errors can generate sequence artifacts similar to MASVs, we used two approaches to validate the candidate loci. First, we designed primers to amplify the candidate loci using PCR on a confirmation panel composed of the DNA samples from one common chimpanzee, one rhesus macaque, and five unrelated human individuals, including the HuRef donor, one African, one Asian, and two Europeans (Fig. 1). For the loci that were not amenable to primer design or that failed PCR amplification, we used several criteria to select loci that are most likely to be authentic MASV events based on their sequence and the orthologous loci in non-human primates (see Methods for details). In both validation approaches, we used the orthologous region in the chimpanzee genome, the orangutan genome (when available), and the rhesus macaque genome (when available) to determine the ancestral state of the candidate loci (i.e., no MASV present). Only MASVs that are present in the HuRef assembly but not present in either the HGP reference genome (hg18) or the chimpanzee genome are considered to be authentic “HuRef-specific” MASVs. It should be noted that although we use “HuRef-specific MASVs” in the following text for brevity, these MASVs are unlikely to be specific to the HuRef sequence (i.e., HuRef private SVs), but are simply absent from the HGP reference genome.
Figure 1.

PCR confirmation of the candidate MASVs. Four agarose gel chromatographs of the PCR products from a confirmation panel are shown. The DNA sample in each lane is labeled above the panel. (Arrows) Expected sizes (in bp) of the PCR amplicons. Diagrams representing the structure of each MASV allele are shown on the right of the panel. (Black line) Flanking DNA sequence, (filled arrows) mobile elements. (A) Locus 1104685335585, an Alu insertion that is heterozygous in the HuRef donor and absent in all other samples. The PCR products in the chimpanzee and the rhesus monkey are slightly smaller because of the smaller size of a (CA)n dinucleotide repeat in these genomes. (B) Locus 1104685664564, an Alu insertion that is present in all human samples tested but absent in the chimpanzee and rhesus macaque. (C) Locus 1104685512583, an L1 recombination-mediated indel. Because the HuRef sample is homozygous for the small size allele, as is the chimpanzee and rhesus macaque, this indel is likely to be caused by an insertion in the reference assembly. (Black box) The tandem duplication section inside the L1. (D) Locus 1104685523196, a false-positive Alu recombination-mediated deletion (ARMD) event where HuRef and all other samples are homozygous for the no-deletion allele.
For classical retrotransposon insertion candidates, we designed primers for all the L1 and SVA insertion loci that were amenable to PCR amplification and for 70 Alu insertions that are novel and not included in the database of human retrotransposon insertion polymorphisms (dbRIP) (Wang et al. 2006; http://dbrip.brocku.ca/). The PCR results show a 100% confirmation rate of all 124 loci that were successfully amplified (Supplemental Table 1). Two examples of the confirmation panel results are shown in Figure 1A,B. The high confirmation rate demonstrates both the validity of our computational approach for identifying classical mobile element insertion events and the high quality of the HuRef assembly.
For other types of MASVs, we designed primers for all loci that were amenable to PCR amplification. The PCR confirmation rates of other types of MASVs were lower than that of the canonical insertion events and varied from 100% to 44% for different types of MASVs (Supplemental Table 1). Some of the events were excluded because equal-sized fragments were amplified from the HuRef and the chimpanzee genome, suggesting that the insertion/deletion events occurred in the reference genome (Fig. 1C). Others failed to show the expected insertion/deletion in the HuRef genome (Fig. 1D). These events may have been caused by errors generated during the genome assembly process of either the HuRef or the HGP reference assembly. A total of 146 insertions and 100 deletion events were validated by PCR confirmation. Detailed information for each locus, including panel amplification results, primer sequences, annealing temperature, and PCR product sizes, are shown in Supplemental Table 2. An additional 560 insertion loci and 40 deletion loci passed our sequence structure analysis, yielding a total of 706 insertion events and 140 deletion events associated with mobile elements in the HuRef assembly (Table 1). A complete list of all MASVs can be found in the Supplemental Table 3.
Table 1.
MASVs in the HuRef genome

aNonclassical Alu insertion.
bNonclassical LINE insertion.
cAlu nonallelic homologous recombination-mediated deletion.
dAlu nonhomologous end-joining-mediated deletion.
eL1 nonallelic homologous recombination-mediated deletion.
fL1 nonhomologous end-joining-mediated deletion.
Human genetic diversity associated with MASVs
For loci that were successfully amplified on the five-person confirmation panel, we were able to assess heterozygosity in the panel and in the HuRef genome. Among the 146 validated insertion events for which we could assess HuRef genotypes, 59 (40%) are heterozygous and 82 (56%) are homozygous. Among the 100 validated deletion events, 32 (32%) are heterozygous in the HuRef genome and 68 (68%) are homozygous.
Next, we examined the diversity of these loci in the confirmation panel (Table 2). The majority of loci are polymorphic among the five human individuals for both insertions (71%) and deletions (75%), with a small proportion of events present only in the HuRef genome (Fig. 1A) or in all five human samples (Fig. 1B). Because only five human samples were tested, the events present only in the HuRef genome or present in all five human samples may still be polymorphic among human populations. To further assess the human genomic diversity associated with polymorphic insertions, we tested 50 confirmed Alu insertions on a population panel composed of 15 European individuals. Forty-three of the 50 loci had clear amplification in at least nine individuals. All 43 loci are polymorphic in our population panel. Among them, three insertions that are homozygous in the confirmation panel (five individuals) are polymorphic on the population panel (15 individuals). In addition, one L1 insertion (Locus ID 1104685647419) that is homozygous in all five individuals in our confirmation panel has been shown to be polymorphic on a larger human panel (Konkel et al. 2007). This result suggests that the majority of MASVs we identified are polymorphic among humans. The allele-frequency distribution of the 43 Alu insertion polymorphisms is skewed toward low insertion frequencies, in agreement with their absence in the HGP reference genome (Fig. 2).
Table 2.
HuRef heterozygosity and human diversity of MASVs

Figure 2.

Allele frequency distribution of 43 novel Alu insertions in 15 European individuals.
Mobile element-mediated insertions
Among the 706 insertion events, 650 are HuRef-specific retrotransposon insertions, including 584 Alu, 52 L1, and 14 SVA insertions (Table 1). We did not identify any new insertions of endogenous retroviruses or DNA transposons. Insertions are found in all chromosomes except the Y chromosome (Fig. 3A), and the number of insertions is highly correlated with the size of the chromosome (r = 0.85, P < 10−6, Spearman's rank correlation). Most insertions bear hallmarks of canonical retrotransposition: They end in a poly(A) tail and are flanked by TSDs, and some have 5′ truncations that are presumably created during the retrotransposition process. Six insertions (four Alu elements and two L1s) are associated with small deletions (13–117 bp) at the insertion sites. These insertions may represent the Alu/L1 insertion-mediated deletion (AIMD/L1IMD) events previously observed in the human and chimpanzee genomes (Gilbert et al. 2002, 2005; Symer et al. 2002; Callinan et al. 2005; Han et al. 2005).
Figure 3.

Genomic distribution of MASVs. Positions of MASVs are shown on a human ideogram. (Red dots, left side of each chromosome) Positions of insertions, (blue dots, right side of each chromosome) positions of deletions.
From our analysis of HuRef-specific insertions, we can estimate retrotransposition rates for these three mobile element families that are active in humans. Of the 70 Alu insertions validated through PCR confirmation, 35 were heterozygotes and 35 were homozygotes. Using this as an estimate for the proportion of homozygotes and heterozygotes among all 584 Alu insertions, we estimate that there are 438 Alu insertions in each haploid genome of HuRef with respect to the HGP reference sequence. Using the observed SNP diversity, we estimated the average time to the most recent common ancestor (TMRCA) between a haploid HuRef genomic locus and the HGP reference sequence to be 18,483 generations (see Methods for details). With 438 Alu insertions per haploid genome in 18,483 generations, we estimate the Alu retrotransposition rate at one in 21 births (95% confidence interval [CI] = 19.1–23.1). For the 52 L1 insertions, we validated 14 as heterozygotes and 29 as homozygotes, corresponding to an expected 43.5 L1 insertions per haploid genome in 18,483 generations, or one L1 insertion per 212 births (95% CI = 156–289). Due to the small number of SVA insertions that are successfully genotyped, we were unable to accurately estimate the proportion of homozygotes and heterozygotes for SVA insertions. Therefore, we opted for an indirect estimate by combining our Alu and L1 data, in which the heterozygosity estimate is 56%. Assuming the same level of heterozygosity in the 14 SVA loci we identified, we estimate that each haploid genome contains 10.1 SVA insertions, corresponding to a retrotransposition rate of one in 916 births (95% CI = 503–1927).
Next, we examined the subfamily composition and sequence structure of these insertions. All of the 584 HuRef-specific Alu insertions belong to the AluY subfamilies (Table 3), with the majority (∼70%) belonging to the AluYa subfamilies (AluYa5 and AluYa8) and AluYb subfamilies (AluYb8 and AluYb9). The AluYa5 subfamily is the most dominant subfamily, comprising >40% of all new insertions, while the AluYb8 subfamily comprises another 25% of the insertions. Other smaller AluY subfamilies, including AluY, AluYc1/2, AluYd3/8, AluYe5, AluYg6, AluYh9, and AluYi6, comprise the remaining 30% of the new insertions (Table 3). The dominance of the AluYa5 and AluYb8 subfamilies in HuRef-specific insertions is consistent with their high activity level in humans after the human–chimpanzee divergence (Hedges et al. 2004).
Table 3.
Subfamily composition of HuRef-specific retrotransposon insertions

For the 52 L1 insertions, in addition to the signatures of canonical retrotransposition, other typical structures associated with L1 insertions were identified: 11 insertions are inverted in the middle, presumably via the “twin-priming” mechanism (Ostertag and Kazazian 2001b); one element possesses an additional partial BC200 gene sequence at the 5′ end, possibly through 5′ transduction during retrotransposition or RNA–RNA hybridization (for review, see Kazazian 2004); and one insertion appears to be a 3′ transduction event, containing ∼70 bp of extra unique sequence at the 3′ end. The size distribution of new L1 insertions follow the typical “U”-shaped pattern observed in previous studies (Grimaldi et al. 1984; Pavlicek et al. 2002): Most insertions (77%) are heavily truncated and <2 kb in length, seven insertions (13%) are full-length or close to full-length, and only three insertions are 2–5 kb in length (Fig. 4A). Three full-length insertions contain intact ORF1 and ORF2 coding regions and could be autonomous elements that are capable of retrotransposition.
Figure 4.

(A) Size distribution of L1 insertions. The number of insertions in 500-bp bins is shown. (B) Size distribution of Alu- and L1-mediated deletions. The percentage of total events in 500-bp bins (except the last one) is shown.
Forty-nine out of 52 HuRef-specific L1 insertions belong to the L1HS (HS, human specific) lineage, and the other three elements belong to the older L1PA2 lineage. The L1HS lineage contains several subfamilies (e.g., L1 pre-Ta, Ta0, and Ta1 subfamilies) that have been active during different periods of human evolution (Boissinot and Furano 2005). To further explore the subfamily composition of the L1HS insertions, we aligned the 49 L1HS elements along with the consensus of the L1HS subfamilies. For the 33 elements that have enough sequence (>500 bp) for subfamily designation, we determined that six, seven, and 20 elements are derived from the L1HS preTa, Ta0, and Ta1 subfamilies, respectively (Table 3).
Fourteen polymorphic SVA insertions were identified, of which seven are full-length (i.e., contain all components of an SVA element) and average 1890 bp in length. The other seven insertions are truncated to various degrees, averaging 1487 bp in length. The 14 new SVA insertions belong to four SVA subfamilies, including one from SVA_D, four from SVA_E, seven from SVA_F, and two from the newly identified SVA_F1 subfamily (Table 3). Overall, more than 291 kb sequence was added to the HuRef assembly because of canonical retrotransposon insertions.
In addition to these canonical insertions, 56 events contain only internal fragments of Alu or LINE elements (i.e., missing both the 5′ and 3′ ends of the element). These insertions do not contain the hallmarks of TPRT: They have no poly(A) tails and are not flanked by identifiable TSDs. In addition, these events are sometimes associated with small deletions at the site of insertion. We collectively called these insertions nonclassical insertions (NCI), including 40 nonclassical Alu insertions (NCAIs) and 16 nonclassical LINE insertions (NCLIs). Based on their sequence structure, three types of events can be identified.
The most common type of NCI is located within a single Alu or LINE element, and the insertions represent a tandem duplication of a section of the element (see the diagram in Fig. 1C for an example). This type of event comprises 71% of all observed NCIs (24 of the NCAIs and all 16 NCLIs), with an average size of 217 bp. Several mechanisms, including strand-mispairing mediated replication slippage (Chen et al. 2005), fork stalling template switching (FoSTeS) (Lee et al. 2007a), or double-strand break (DSB)-induced homologous recombination (Liang et al. 1998) can all create this type of tandem duplication. In contrast, eight loci contain partial Alu insertions in the non-Alu regions. These events may have been created by the capture of retrotransposon RNAs at the DSB sites and the subsequent reverse transcription of the retrotransposon RNAs as a mechanism for DSB repairs (Morrish et al. 2002; Sen et al. 2007; Srikanta et al. 2008). The remaining seven loci appear to be the duplication products of the nonallelic homologous recombination process. Collectively, all NCI events added another 13,226 bp sequence to the HuRef genome.
Mobile element-mediated deletions
We identified 140 HuRef-specific mobile element-mediated deletions. These events were distributed across the whole genome with the exception of chromosomes 21 and Y. The correlation between the number of deletions and chromosome size is significant (r = 0.44, P < 0.05, Spearman's rank correlation) but much weaker than that of the insertion events. Further examination revealed that chromosomes 2 and 19 are the major outliers (two and 13 deletions on chromosomes 2 and 19, respectively). More than three-fourths of the deletion events are <1 kb in size, with an average of 787 bp and 1234 bp for Alu- and L1-mediated deletions, respectively (Fig. 4B). For the 100 loci that are confirmed on the confirmation panel, 75 loci (75%) are polymorphic among the five human samples and 19 loci are present as homozygous deletion in all five individuals. The deletion allele is present only in the HuRef sample for the remaining six loci (Table 2).
NAHRs between two Alu elements or two L1s produce the most common deletions associated with mobile elements. We identified and confirmed 98 Alu recombination-mediated deletions (ARMD) and nine L1 recombination-mediated deletions (L1RMD). The majority (82%) of NAHR-mediated deletions are <1 kb in size, with the largest two being one ARMD and one L1RMD event that deleted 7852 and 7953 bp from the HuRef genome, respectively. Among them, 16 ARMD events have each occurred within a single Alu element and appear to be a recombination between the left and right monomer of the same Alu element. We have confirmed these 16 events in the five-human confirmation panel, in which 14 out of the 16 events (88%) are polymorphic. For the other two events, one ARMD is present in all five individuals as homozygous deletions and one ARMD is only present in the HuRef genome as a heterozygous deletion. Overall, 98,533 bp have been removed from the HuRef genome due to the NAHR-mediated deletions (Table 1).
In addition to NAHR-mediated deletions, NHEJ accounted for a small number of deletions. The NHEJ-mediated deletions are characterized by “microhomology” between the breakpoints and are thought to be a product of the DSB repair mechanism (Moore and Haber 1996; Bentley et al. 2004; Yan et al. 2007). We identified 33 NHEJ-mediated deletion events that removed 27,344 bp of sequence. Twenty-two of the 26 L1-associated NHEJ events occurred within the L1 elements, suggesting that L1 elements may be subjected to a high frequency of DSBs.
Functional impact of MASVs
To determine if the MASVs have influenced gene structure or expression in the HuRef genome, we compared the locations of MASVs with the positions of all known genes in the RefSeq Gene database (http://www.ncbi.nlm.nih.gov/RefSeq/). Of the 706 insertion events, 238 (33.7%) are within genic regions. Of the 140 deletions, 60 (42.9%) are present in genic regions. By examining the genes containing SVs, we found that two Alu elements on chromosome 5 (Locus IDs 1104685725664 and 1104685203669) and one Alu element on chromosome 6 (Locus ID 1104685374124) have inserted into the exonic regions of the SPATA9 (spermatogenesis associated 9, HGNC ID 22988), C7 (complement component 7, HGNC ID 1346), and HCG26 (HLA complex group 26, HGNC ID 29671) genes, respectively.
We validated all three insertions on our confirmation panel using PCR and found that insertions in the SPATA9 and the HCG26 genes are polymorphic among the five human individuals, while the insertion in the C7 gene is present only as a heterozygote in the HuRef genome. Further examination revealed that the Alu insertions in the SPATA9 and C7 genes occurred in the 3′ untranslated region (3′ UTR) of each gene. The AluYa5 insertion in SPATA9 is located 117 bp downstream from the stop codon, and the partial AluYb8 insertion in the C7 gene is located 750 bp downstream from the stop codon. The positions of these insertions suggest that they do not change the coding sequence of these genes. The third insertion, an AluY element, inserted at the beginning of the noncoding HCG26 gene, and the TSD (AGTATTTTCCCTTTT) overlaps the transcription start site (TTTTCCCTTTT) of the gene. By searching the dbEST database, we found that at least one transcript (AW836456) from HCG26 contains the new AluY insertion.
Discussion
Our results demonstrate that mobile elements play an important role in creating new SVs in the human genome. These results were enhanced significantly by several unique attributes of the HuRef genome assembly. First, the HuRef genome was sequenced by the traditional Sanger sequencing method, while other currently available individual genomes are sequenced using second-generation sequencing techniques (Bentley et al. 2008; Wang et al. 2008; Wheeler et al. 2008). The Sanger method generates longer read lengths than the second-generation sequencing methods and is thus more suitable for studying SVs. Second, the HuRef assembly is a high-quality assembly that contains 68% fewer gaps as compared with the HGP assembly. In addition, we used sequence scaffolds instead of assembled chromosomes for the indel identification. The 188,394 HuRef scaffolds used in the comparison contain >3.03 billion bp of genomic sequence and cover >98% of the HGP autosomes on average (Levy et al. 2007). By using these high-quality sequence scaffolds, we decrease the possibility of missing indels, especially insertion events, in the HuRef assembly. Third, because the HuRef assembly provides diploid genotypes, we can easily assess homozygosity and heterozygosity of the HuRef variants. Furthermore, the availability of DNA from the HuRef donor permitted direct validation of the candidate MASV events.
To assess the MASVs in the HuRef assembly, we examined all indels that are >100 bp and are associated with mobile elements at their breakpoints. Several types of events were identified, including canonical retrotransposon insertions, NAHR-mediated insertions/deletions, NHEJ-mediated deletions, and nonclassical insertions (Fig. 5). These same types of MASVs have been observed when comparing human and chimpanzee genomes (Table 4). As expected, they account for a much larger number of events and sequence gain/loss in the human/chimpanzee comparison. Most of the HuRef-specific MASVs (91.5%) are <1 kb in size, and only 12 events are >5 kb. It is useful to compare results in the current study with those of previous studies of human SVs using the paired-end mapping (PEM) method (Korbel et al. 2007) or FPES methods (Kidd et al. 2008). Because of methodological differences in these studies, they have, to some extent, identified different groups of SV elements. For example, Alu insertions, which are ∼300 bp in length, were not identified by Korbel et al. (2007) or Kidd et al. (2008) because of the size of the libraries and fosmids used for PEM and FPES, respectively. On the other hand, our SV selection relied on a comparison of the two assemblies, and as a result it would miss complex SVs that have prevented an alignment of the two assemblies. Therefore, the size distribution of these events is complementary in these studies, and the combined results would provide a more complete picture of SVs in the human genome.
Figure 5.

Four types of common MASVs in the HuRef genome. (A) Classical retrotransposon insertion; (B) nonclassical insertions; (C) nonallelic homologous recombination-mediated insertion/deletion; (D) nonhomologous end-joining-mediated deletion. (TTAAAA) Standard L1 cleavage site for classical retrotransposition; (black lines) flanking regions, (gray lines) intervening regions, (dotted circles) homologous recombining regions, (red boxes) microhomology regions, (red arrow boxes) TSDs of each element.
Table 4.
HuRef-specific and human-specific MASVs

aNonclassical Alu insertion.
bThese are net sequence losses in human-specific nonclassical insertion events because many of these events are associated with deletions at the pre-insertion locus.
cNonclassical LINE insertion.
dAlu recombination-mediated deletion.
eNet sequence loss (deletion) is shown in parentheses.
fL1 recombination-mediated deletion.
gAlu insertion-mediated deletion.
hL1 insertion-mediated deletion.
Among all MASVs, retrotransposon insertions are the most abundant events. This is expected because retrotransposons have been actively transposed throughout primate evolution, and more than 7000 retrotransposons have inserted into the human genome since the divergence of human and chimpanzee (Chimpanzee Sequencing and Analysis Consortium 2005; Mills et al. 2006). As demonstrated here, retrotransposons are still actively transposing in individual human genomes, with estimated retrotransposition rates of one per 21 births (95% CI = 19.1–23.1) for Alu elements, one per 212 births for L1s (95% CI = 156–289), and one per 916 births for SVA elements (95% CI = 503–1927) over the last 450,000 yr. Our confidence intervals account for the stochasticity inherent in the process of retrotransposition, but could be subject to systematic bias from errors in the underlying parameter estimates. For example, the largest potential source of error is in our estimate of the average TMRCA between HuRef and the reference sequence, which is based on a single nucleotide mutation rate of 2.2 × 10−8 per generation (Nachman and Crowell 2000). In addition, our estimates could be biased due to insertions we were unable to observe, such as those in genomic regions of HuRef that could not be assembled. Finally, although we were able to determine the exact proportion of heterozygous and homozygous L1 insertions in HuRef by direct genotyping, we directly genotyped only a proportion of the Alu insertions, and the heterozygosity of SVA insertions was indirectly estimated. These heterozygosity estimates could bias the estimated retrotransposition rates.
Nevertheless, our estimate for the Alu retrotransposition rate is remarkably close to two recent estimates from Cordaux et al. (2006). In that study, the first estimated Alu retrotransposition rate was one per 22 births (95% CI = 17–27), based on the number of Alu elements specific to the human lineage since the human–chimpanzee divergence ∼6 million yr ago. The second estimate was one per 15 births (95% CI = 10–24), based on the number of disease-causing Alu insertions in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/). Along with our estimate, these estimates provide three snapshots of the Alu retrotransposition rate during human evolution (in the last 6 million yr, 450,000 yr, and recent/de novo events). The convergence of these estimates from different time periods suggests that the Alu retrotransposition rate has been relatively constant throughout recent human evolution. If so, this enhances the utility of these markers in studies of human population genetics.
By amplifying 146 retrotransposon insertions on a confirmation panel, we found that most of the new insertions we identified have occurred sometime during human evolution and are polymorphic among human populations. These insertions are informative for human population history and can be used in future population genetic studies. We also identified a small number of insertions that are present only in the HuRef genome among the five tested individuals. Most of these insertions should represent recent events that have low allele frequencies in human populations. Although a small number of these insertions may potentially be private events in the HuRef genome, a much larger number of human samples must be tested to accurately assess the prevalence and distribution of these low-frequency events.
In addition to classical insertion events, we identified 56 nonclassical insertion events and 140 deletion events. Based on their sequence structure, multiple mechanisms may have contributed to these structural rearrangements. For NCIs, the majority (71%) of insertions are tandem duplications of a section of an Alu/L1 element. Several mechanisms, including strand-mispairing mediated replication slippage (Chen et al. 2005), Fork Stalling Template Switching (Lee et al. 2007a), or DSB-induced homologous recombination (Liang et al. 1998), could have accounted for this type of NCIs. The remaining events appear to be generated either by NAHR-mediated insertions (14%) or endonuclease-independent reverse transcription during DSB repair (14%), as observed in previous studies (Sen et al. 2007; Srikanta et al. 2008). For mobile element-associated deletions, NAHR between similar elements is the major mechanism, due to the large number of mobile elements in the human genome (e.g., more than 1.1 million Alu elements and more than 500,000 L1s) (Sen et al. 2006; Han et al. 2008). We found that these deletions are usually small in size and sometimes even occur within the same element. NHEJ-mediated deletion represents yet another mechanism for MASVs. These events are thought to be the product of DSB repair and are initiated by 1–7 bp of homologous sequences at both ends of the DSB (termed “microhomology”) (Bentley et al. 2004; Guirouilh-Barbat et al. 2004; Yan et al. 2007). The presence of MASVs generated by a DSB repair process highlights the role of mobile elements in maintaining the integrity of the human genome. It should be noted that, although we invoke NAHR and NHEJ as the possible mechanisms responsible for these events, alternative mechanisms, including FoSTeS (Lee et al. 2007a) or replication slippage (Chen et al. 2005), could have generated some of the events. Further studies are needed to resolve the mechanisms underlying these MASVs.
Despite the small sizes of MASVs, the high frequency of these events makes them good candidates for altering gene content and expression. To date, at least 54 disease-causing mobile element insertions and 53 disease-causing mobile-element recombination events have been reported (for reviews, see Chen et al. 2006; Babushok and Kazazian 2007). If we divide this number by a total of 76,011 mutations in the HGMD (as of Dec. 2007; Stenson et al. 2008), we obtain an estimate that 0.14% of disease-causing mutations are associated with mobile elements. This estimate is remarkably similar to an earlier estimate of one in 670 (Kazazian and Moran 1998). In the HuRef genome, we found that more than one-third of the MASVs are within genic regions. Two Alu insertion events occurred in the 3′ UTR regions of the SPATA9 and C7 genes, and such insertions can in some cases suppress transcription. For example, an SVA insertion in the 3′ UTR region of the FKTN (formerly known as FCMD) gene has been shown to cause Fukuyama-type congenital muscular dystrophy (Kobayashi et al. 1998). In addition, mobile elements can alter the level of gene expression via other mechanisms. L1 and Alu elements can provide alternative splicing and polyadenylation sites if inserted inside a gene (for reviews, see Han et al. 2004; Wheelan et al. 2005; Belancio et al. 2008; Goodier and Kazazian 2008). If, as our data demonstrate, the average human genome contains nearly 1000 MASVs, mobile elements could represent a major factor in SV-related human diseases.
Overall, of the 8451 total HuRef SVs that are larger than 100 bp, 846 are HuRef-specific MASVs (706 insertions and 140 deletions). Similar numbers of “HuRef-specific” and “HGP-specific” MASV candidates were identified during our computational data-mining process. With a comparable validation rate of the candidate events, we infer that mobile elements are responsible for roughly 1700 (20%) of the indels >100 bp between HuRef and the HGP reference genome. It is noteworthy that although the HGP reference genome is a composite haploid sequence assembled from multiple individuals, the majority (∼75%) of the reference genome was based on one BAC library derived from a single individual (Lander et al. 2001). Therefore, our inferred number of MASVs may represent an estimate between two individual humans.
With recent advances in DNA sequencing, complete genomic sequences will be available for many more individuals in the near future (e.g., the ongoing 1000 Genomes Project, www.1000genomes.org). Genome-wide analysis of MASVs in multiple individuals will not only shed light on the impact of MASVs in human evolution but will also provide a large number of recent retrotransposon insertions that will be informative for fine-scale analysis of human population history.
Methods
Computational data mining and genomic distribution analysis
From the 643,992 indels that were initially identified by comparing the HuRef scaffolds and the HGP reference genome, indels were categorized into “homozygous indel,” “heterozygous indel,” and “putative indel.” We first selected homozygous and heterozygous indels that are >100 bp in size. Putative indels are often associated with gaps (i.e., stretches of “N”s) or mismatch sequence between the two assemblies. Therefore, we only selected putative indels with complete sequence (i.e., no “N”s in the sequence) and >50 bp difference between the two assemblies. These selection criteria resulted in a total of 8451 candidate loci. The repetitive element content of these loci along with their flanking regions was determined using RepeatMasker (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker). Loci containing mobile elements were then subjected to manual inspection.
For loci that were not amenable to PCR confirmation, we first used BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat) to determine their orthologous regions in the chimpanzee genome (panTro2), the orangutan genome (ponAbe2, when available), and the rhesus monkey genome (rheMac2, when available). Only loci that showed identical structure in the chimpanzee genome and the HGP reference genome were considered “HuRef-specific” loci. Next, we examined the sequence structure of these loci to determine the nature of the variants. For retrotransposon insertion events, we required the presence of a poly(A) tail and TSDs on both ends of the insertion. For recombination-mediated MASVs, we required the presence of homologous sequence (microhomology in the case of NHEJ-mediated deletions) at the breakpoints.
To determine the genomic distribution of MASVs and their positions relative to genic regions, sequence from human genome assembly 18 (hg18) was obtained from the UCSC Genome Bioinformatics Site (http://genome.ucsc.edu/). For the gene analysis, the gene definition from the Reference Sequence (RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq/) is used. The ideogram plotting and statistical analyses were performed using MATLAB (ver. R2008a).
PCR validation
Flanking oligonucleotide primers for PCR amplification of each locus were designed using Primer3 (Rozen and Skaletsky 2000; http://frodo.wi.mit.edu). The primers were subsequently screened using UCSC In-Silico PCR (http://genome.ucsc.edu/cgi-bin/hgPcr?command=start) to select primer pairs that produce a unique PCR product in the human genome. Oligonucleotide primer pairs were initially tested using HuRef DNA templates with temperatures of 55°C and 60°C to determine the appropriate annealing temperature for further analysis. All loci were screened on a confirmation panel that was composed of DNA samples from five human individuals (one African, one Asian, one Northern European, one Southern European, and the HuRef DNA sample), one common chimpanzee, one rhesus macaque, and one negative control. Because the quantity of genomic DNA sample for HuRef is limited, it was subjected to whole genome amplification using the REPLI-g whole genome amplification kit (Qiagen) following the manufacturer's instruction. The amplified samples were then purified and aliquoted for locus-specific PCR analysis. Fifty Alu insertion loci were genotyped on a population panel composed of 15 European individuals to assess the allele frequency of the insertions.
PCR amplification of each locus was performed as described previously (Xing et al. 2003). The resulting PCR products were run on 2% agarose gels with 0.25 μg of ethidium bromide and visualized using UV fluorescence. In cases where the expected size of the PCR product was >1.5 kb, iTaq (Bio-Rad), Ex Taq polymerase (TaKaRa), or KOD Hifi DNA polymerase (Novagen) were used, following the manufacturer's suggested protocols. Also, two separate PCRs were performed for some loci with large indels in an assay designed for L1 genotyping (Sheen et al. 2000) to determine their genotypes (Supplemental Table 2).
For the loci that were confirmed by sequencing, individual PCR products were directly sequenced on an ABI 3100 Genetic Analyzer as described previously (Xing et al. 2003). Sequences for each locus were aligned with the reference sequence and HuRef assembly using BioEdit (Hall 1999). Sequence alignments of these loci are available from our website (http://jorde-lab.genetics.utah.edu/) as Supplemental Alignments located under Published Data.
Retrotransposition rate estimates
Our retrotransposition rate estimates are derived by comparing the HuRef sequence with the HGP reference assembly. Because HuRef is a diploid sequence and the reference assembly is haploid, the most accurate measure would consider both haploid genomes in HuRef while accounting for shared genealogy in HuRef with respect to the reference sequence. However, this procedure is considerably more complicated than a pairwise haploid comparison and requires information about the HuRef genome that is currently unavailable, including the identification of maternal and paternal genomes and known phase for all markers across the genome. To simplify this problem, we instead compare the haploid reference sequence to the mean haploid genome in HuRef, represented by the average number of differences between each haploid HuRef genome and the reference sequence. Since we are averaging across both haploid genomes, the point estimates from this procedure are unbiased, but the size of the confidence regions may be underestimated if there are systematic differences in the relationships between the paternal and maternal HuRef genomes and the reference sequence.
The mean haploid genome contains all differences between HuRef and the reference sequence that are homozygous in HuRef, and half of the differences between HuRef and the reference sequence that are heterozygous in HuRef. Levy et al. (2007) identified 1,623,826 heterozygous SNPs and 1,450,860 homozygous SNPs between HuRef and the reference sequence out of a total of 2,782,357,138 nucleotides, for an average of 2,262,733 SNPs and nucleotide diversity of 8.13 × 10−4 per haploid genome comparison. Based on a single nucleotide mutation rate of 2.2 × 10−8 per generation (Nachman and Crowell 2000), the average TMRCA of the mean haploid HuRef genome and the reference sequence is 18,483 generations.
Confidence intervals for the retrotransposition rate estimates were derived from the relationship between the Poisson and χ2 distributions. For a Poisson process with n observed events, the (1 − α)% exact lower and upper bound confidence intervals (L and U, respectively) for the mean of the Poisson random variable is:
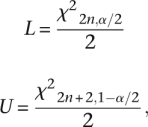 |
where χ2x,y is the χ2 deviate with x degrees of freedom and lower tail area y (Johnson and Kotz 1969). These intervals account for the randomness inherent in the process of retrotransposition but do not incorporate the variation in TMRCA across the genome or uncertainty in parameter estimates. While the original retrotransposition rate estimates from Cordaux et al. (2006) included interval ranges reflecting the uncertainty around their parameter estimates, no confidence intervals were included.
To allow a direct comparison with our data, we calculated confidence intervals around their original estimates. To ensure our confidence intervals accounted for the uncertainty in their original parameter estimates, we calculated the lower and upper confidence limits using the parameters from the respective lower and upper bounds of their retrotransposition rate estimates, resulting in a conservative 95% confidence interval.
Acknowledgments
We thank three anonymous reviewers for their useful comments. This work was supported by a grant to M.A.B. and L.B.J. from the National Institutes of Health (GM-59290).
Footnotes
[Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) under accession nos. FI569689–FI569698.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.091827.109.
References
- Babushok DV, Kazazian HH., Jr Progress in understanding the biology of the human mutagen LINE-1. Hum Mutat. 2007;28:527–539. doi: 10.1002/humu.20486. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Liu G, Eichler EE. An Alu transposition model for the origin and expansion of human segmental duplications. Am J Hum Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Belancio VP, Hedges DJ, Deininger P. Mammalian non-LTR retrotransposons: For better or worse, in sickness and in health. Genome Res. 2008;18:343–358. doi: 10.1101/gr.5558208. [DOI] [PubMed] [Google Scholar]
- Bentley J, Diggle CP, Harnden P, Knowles MA, Kiltie AE. DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 2004;32:5249–5259. doi: 10.1093/nar/gkh842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Furano AV. The recent evolution of human L1 retrotransposons. Cytogenet Genome Res. 2005;110:402–406. doi: 10.1159/000084972. [DOI] [PubMed] [Google Scholar]
- Callinan PA, Batzer MA. Transposable elements and human disease. In: Volff JN, editor. Genome dynamics. Karger; Basel, Switzerland: 2006. pp. 104–115. [DOI] [PubMed] [Google Scholar]
- Callinan PA, Wang J, Herke SW, Garber RK, Liang P, Batzer MA. Alu retrotransposition-mediated deletion. J Mol Biol. 2005;348:791–800. doi: 10.1016/j.jmb.2005.02.043. [DOI] [PubMed] [Google Scholar]
- Chen JM, Chuzhanova N, Stenson PD, Ferec C, Cooper DN. Meta-analysis of gross insertions causing human genetic disease: Novel mutational mechanisms and the role of replication slippage. Hum Mutat. 2005;25:207–221. doi: 10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- Chen JM, Ferec C, Cooper DN. LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease: Mutation detection bias and multiple mechanisms of target gene disruption. J Biomed Biotechnol. 2006;2006:56182. doi: 10.1155/JBB/2006/56182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Cordaux R, Hedges DJ, Herke SW, Batzer MA. Estimating the retrotransposition rate of human Alu elements. Gene. 2006;373:134–137. doi: 10.1016/j.gene.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Nickerson DA, Altshuler D, Bowcock AM, Brooks LD, Carter NP, Church DM, Felsenfeld A, Guyer M, Lee C, et al. Completing the map of human genetic variation. Nature. 2007;447:161–165. doi: 10.1038/447161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, Moran JV. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol. 2005;25:7780–7795. doi: 10.1128/MCB.25.17.7780-7795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Kazazian HH., Jr Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Grimaldi G, Skowronski J, Singer MF. Defining the beginning and end of Kpnl family segments. EMBO J. 1984;3:1753–1759. doi: 10.1002/j.1460-2075.1984.tb02042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, Lopez BS. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell. 2004;14:611–623. doi: 10.1016/j.molcel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- Han K, Sen SK, Wang J, Callinan PA, Lee J, Cordaux R, Liang P, Batzer MA. Genomic rearrangements by LINE-1 insertion-mediated deletion in the human and chimpanzee lineages. Nucleic Acids Res. 2005;33:4040–4052. doi: 10.1093/nar/gki718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Konkel MK, Xing J, Wang H, Lee J, Meyer TJ, Huang CT, Sandifer E, Hebert K, Barnes EW, et al. Mobile DNA in Old World monkeys: A glimpse through the rhesus macaque genome. Science. 2007a;316:238–240. doi: 10.1126/science.1139462. [DOI] [PubMed] [Google Scholar]
- Han K, Lee J, Meyer TJ, Wang J, Sen SK, Srikanta D, Liang P, Batzer MA. Alu recombination-mediated structural deletions in the chimpanzee genome. PLoS Genet. 2007b;3:e184. doi: 10.1371/journal.pgen.0030184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Lee J, Meyer TJ, Remedios P, Goodwin L, Batzer MA. L1 recombination-associated deletions generate human genomic variation. Proc Natl Acad Sci. 2008;105:19366–19371. doi: 10.1073/pnas.0807866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges DJ, Callinan PA, Cordaux R, Xing J, Barnes E, Batzer MA. Differential Alu mobilization and polymorphism among the human and chimpanzee lineages. Genome Res. 2004;14:1068–1075. doi: 10.1101/gr.2530404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC, Szpiech ZA, Degnan JH, Wang K, Guerreiro R, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- Johnson NL, Kotz S. Discrete distributions. Houghton Mifflin; Boston: 1969. [Google Scholar]
- Kazazian HH., Jr Mobile elements: Drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998;19:19–24. doi: 10.1038/ng0598-19. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–166. doi: 10.1038/332164a0. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PM, Lam HY, Urban AE, Korbel JO, Affourtit J, Grubert F, Chen X, Weissman S, Snyder M, Gerstein MB. Analysis of copy number variants and segmental duplications in the human genome: Evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008;18:1865–1874. doi: 10.1101/gr.081422.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- Konkel MK, Wang J, Liang P, Batzer MA. Identification and characterization of novel polymorphic LINE-1 insertions through comparison of two human genome sequence assemblies. Gene. 2007;390:28–38. doi: 10.1016/j.gene.2006.07.040. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007a;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007b;39:S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- Lee J, Han K, Meyer TJ, Kim HS, Batzer MA. Chromosomal inversions between human and chimpanzee lineages caused by retrotransposons. PLoS One. 2008;3:e4047. doi: 10.1371/journal.pone.0004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Han M, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhao S, Bailey JA, Sahinalp SC, Alkan C, Tuzun E, Green ED, Eichler EE. Analysis of primate genomic variation reveals a repeat-driven expansion of the human genome. Genome Res. 2003;13:358–368. doi: 10.1101/gr.923303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Macfarlane C, Simmonds P. Allelic variation of HERV-K(HML-2) endogenous retroviral elements in human populations. J Mol Evol. 2004;59:642–656. doi: 10.1007/s00239-004-2656-1. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39:S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- Mills RE, Bennett EA, Iskow RC, Luttig CT, Tsui C, Pittard WS, Devine SE. Recently mobilized transposons in the human and chimpanzee genomes. Am J Hum Genet. 2006;78:671–679. doi: 10.1086/501028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet. 2002;31:159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001a;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Twin priming: A proposed mechanism for the creation of inversions in l1 retrotransposition. Genome Res. 2001b;11:2059–2065. doi: 10.1101/gr.205701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek A, Paces J, Zika R, Hejnar J. Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: Implications for retrotransposition and pseudogene detection. Gene. 2002;300:189–194. doi: 10.1016/s0378-1119(02)01047-8. [DOI] [PubMed] [Google Scholar]
- Perry GH, Yang F, Marques-Bonet T, Murphy C, Fitzgerald T, Lee AS, Hyland C, Stone AC, Hurles ME, Tyler-Smith C, et al. Copy number variation and evolution in humans and chimpanzees. Genome Res. 2008;18:1698–1710. doi: 10.1101/gr.082016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics methods and protocols: Methods in molecular biology. Humana Press; Totowa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- Sen SK, Han K, Wang J, Lee J, Wang H, Callinan PA, Dyer M, Cordaux R, Liang P, Batzer MA. Human genomic deletions mediated by recombination between Alu elements. Am J Hum Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen SK, Huang CT, Han K, Batzer MA. Endonuclease-independent insertion provides an alternative pathway for L1 retrotransposition in the human genome. Nucleic Acids Res. 2007;35:3741–3751. doi: 10.1093/nar/gkm317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen FM, Sherry ST, Risch GM, Robichaux M, Nasidze I, Stoneking M, Batzer MA, Swergold GD. Reading between the LINEs: Human genomic variation induced by LINE-1 retrotransposition. Genome Res. 2000;10:1496–1508. doi: 10.1101/gr.149400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanta D, Sen SK, Huang CT, Conlin EM, Rhodes RM, Batzer MA. An alternative pathway for Alu retrotransposition suggests a role in DNA double-strand break repair. Genomics. 2008;93:205–212. doi: 10.1016/j.ygeno.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN. Human Gene Mutation Database: Towards a comprehensive central mutation database. J Med Genet. 2008;45:124–126. doi: 10.1136/jmg.2007.055210. [DOI] [PubMed] [Google Scholar]
- Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Wang H, Xing J, Grover D, Hedges DJ, Han K, Walker JA, Batzer MA. SVA elements: A hominid-specific retroposon family. J Mol Biol. 2005;354:994–1007. doi: 10.1016/j.jmb.2005.09.085. [DOI] [PubMed] [Google Scholar]
- Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: A highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 2006;27:323–329. doi: 10.1002/humu.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Guo Y, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelan SJ, Aizawa Y, Han JS, Boeke JD. Gene-breaking: A new paradigm for human retrotransposon-mediated gene evolution. Genome Res. 2005;15:1073–1078. doi: 10.1101/gr.3688905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, Eichler EE, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Salem AH, Hedges DJ, Kilroy GE, Watkins WS, Schienman JE, Stewart CB, Jurka J, Jorde LB, Batzer MA. Comprehensive analysis of two Alu Yd subfamilies. J Mol Evol. 2003;57:S76–S89. doi: 10.1007/s00239-003-0009-0. [DOI] [PubMed] [Google Scholar]
- Xing J, Witherspoon DJ, Ray DA, Batzer MA, Jorde LB. Mobile DNA elements in primate and human evolution. Am J Phys Anthropol. 2007;S45:2–19. doi: 10.1002/ajpa.20722. [DOI] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Mishra B. Quantifying the mechanisms for segmental duplications in mammalian genomes by statistical analysis and modeling. Proc Natl Acad Sci. 2005;102:4051–4056. doi: 10.1073/pnas.0407957102. [DOI] [PMC free article] [PubMed] [Google Scholar]