

Abstract
Population stratification has long been recognized as a confounding factor in genetic association studies. Estimated ancestries, derived from multi-locus genotype data, can be used to perform a statistical correction for population stratification. One popular technique for estimation of ancestry is the model-based approach embodied by the widely applied program structure. Another approach, implemented in the program EIGENSTRAT, relies on Principal Component Analysis rather than model-based estimation and does not directly deliver admixture fractions. EIGENSTRAT has gained in popularity in part owing to its remarkable speed in comparison to structure. We present a new algorithm and a program, ADMIXTURE, for model-based estimation of ancestry in unrelated individuals. ADMIXTURE adopts the likelihood model embedded in structure. However, ADMIXTURE runs considerably faster, solving problems in minutes that take structure hours. In many of our experiments, we have found that ADMIXTURE is almost as fast as EIGENSTRAT. The runtime improvements of ADMIXTURE rely on a fast block relaxation scheme using sequential quadratic programming for block updates, coupled with a novel quasi-Newton acceleration of convergence. Our algorithm also runs faster and with greater accuracy than the implementation of an Expectation-Maximization (EM) algorithm incorporated in the program FRAPPE. Our simulations show that ADMIXTURE's maximum likelihood estimates of the underlying admixture coefficients and ancestral allele frequencies are as accurate as structure's Bayesian estimates. On real-world data sets, ADMIXTURE's estimates are directly comparable to those from structure and EIGENSTRAT. Taken together, our results show that ADMIXTURE's computational speed opens up the possibility of using a much larger set of markers in model-based ancestry estimation and that its estimates are suitable for use in correcting for population stratification in association studies.
Population stratification has long been recognized as a confounding factor in genetic association studies (Li 1972; Knowler et al. 1988; Marchini et al. 2004). To correct for the effects of population stratification, association studies may take account of individuals' ancestries in their analyses, an approach known as “structured association” (Pritchard and Donnelly 2001). One simple technique is to incorporate ancestry as an additional covariate in an appropriate regression model (Price et al. 2006). Self-reported ancestries can be used for this purpose, but these are often vague or inaccurate. An alternative is to estimate ancestries from the genotypes actually collected in a study.
We offer the following taxonomy of ancestry estimation tools. At the highest level, we make a distinction between estimating “global ancestry” and “local ancestry.” In the local ancestry paradigm (Falush et al. 2003; Patterson et al. 2004; Tang et al. 2006; Sankararaman et al. 2008a,b), we imagine that each person's genome is divided into chromosome segments of definite ancestral origin. The goal then is to find the segment boundaries and assign each segment's origin. In the global ancestry paradigm (Pritchard et al. 2000; Tang et al. 2005), we are concerned only with estimating the proportion of ancestry from each contributing population, considered as an average over the individual's entire genome. Here we tackle estimation of global ancestry.
Under the broad heading of “global ancestry estimation,” there are two approaches: “model-based ancestry estimation” and “algorithmic ancestry estimation.” Model-based approaches, exemplified by structure (Pritchard et al. 2000), FRAPPE (Tang et al. 2005), and our program ADMIXTURE, estimate ancestry coefficients as the parameters of a statistical model. Algorithmic approaches use techniques from multivariate analysis, chiefly cluster analysis and principal component analysis (PCA), to discover structure within the data in a less parametric way. Cluster analysis directly seeks the ancestral clusters in the data, while principal component analysis constructs low-dimensional projections of the data that explain the gross variation in marker genotypes, which, in practice, is the variation between populations. EIGENSTRAT (Patterson et al. 2006; Price et al. 2006) is a popular implementation of PCA for ancestry inference.
Our approach is similar to structure's. Both programs model the probability of the observed genotypes using ancestry proportions and population allele frequencies. Like structure, ADMIXTURE simultaneously estimates population allele frequencies along with ancestry proportions.
structure takes a Bayesian approach and relies on a Markov chain Monte Carlo (MCMC) algorithm to sample the posterior distribution. We use the same likelihood model but focus on maximizing the likelihood rather than on sampling the posterior. Since high-dimensional optimization is much faster than high-dimensional MCMC, our maximum likelihood approach can accommodate many more markers. Of course, there is no single optimization algorithm suited to all occasions. The parameters of the ADMIXTURE model must satisfy linear constraints and bounds, and this requirement plus the large number of model parameters complicates matters. After considerable experimentation, we have settled on a block relaxation algorithm (de Leeuw 1994) that alternates between updating the ancestry coefficient matrix Q and the population allele frequency matrix F. Each update of Q itself involves sequential quadratic programming, a generalization of Newton's method suitable for constrained optimization. Finally, we accelerate convergence of block relaxation by a novel quasi-Newton method. Once point estimates are found, standard errors can be estimated, at the user's option, using the moving block bootstrap (Kunsch 1989).
Tang et al. (2005) take a similar approach in their program FRAPPE. They adopt the same model and estimate parameters by maximum likelihood using an EM algorithm. We show that FRAPPE's estimates are slightly inaccurate. These inaccuracies appear to be a result of FRAPPE's relaxed convergence criterion. Imposing a strict convergence criterion renders the EM algorithm computationally burdensome. In contrast, our algorithm is fast even with very strict convergence criteria.
In the Methods section, we present the underlying statistical model and describe the optimization techniques used to maximize the likelihood. We then sketch how we accelerate convergence. Finally, we review the block bootstrap and describe its use in estimating parameter standard errors. In the Results section, we compare ADMIXTURE's statistical performance to that of structure and FRAPPE on simulated and real data. We then briefly examine the numerical behavior of the EM and block relaxation algorithms and explore the effect the convergence criterion has on the accuracy of the estimates. We also examine the impact of a certain tuning parameter in quasi-Newton acceleration. We then compare the runtimes of structure and ADMIXTURE on the various data sets. The Results section ends with a simulated association study that shows that ADMIXTURE performs as well as EIGENSTRAT at statistically correcting for population structure. In the Discussion, we summarize our conclusions and suggest further directions for research.
Methods
A statistical model
The typical data set consists of genotypes at a large number J of single nucleotide polymorphisms (SNPs) from a large number I of unrelated individuals. These individuals are drawn from an admixed population with contributions from K postulated ancestral populations. Population k contributes a fraction qik of individual i's genome. Allele 1 at SNP j has frequency fkj in population k. As a matter of convention, one can choose allele 1 to be the minor allele and the alternative allele 2 to be the major allele. In our framework, both the qik and the fkj are unknown. We are primarily interested in estimating the qik to control for ancestry in an association study, but our approach also yields estimates of the fkj. Among other things, this allows us to estimate the degree of divergence between the estimated ancestral populations using the FST statistic.
In the likelihood model adopted by structure, individuals are formed by the random union of gametes. This produces the binomial proportions
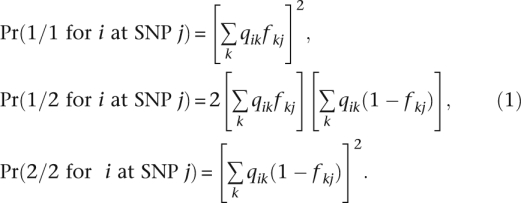 |
Our model makes the further assumption of linkage equilibrium among the markers. Dense marker sets should be pruned to mitigate background linkage disequilibrium (LD). This can be done informally, by thinning the marker set according to a minimum separation criterion or by pruning markers observed to be in linkage disequilibrium on the basis of common LD summary statistics such as D′ or r2. Neither pruning approach is a perfect remedy for linkage disequilibrium. Nonetheless, we consider the assumption of linkage equilibrium to be a useful approximation, one that is commonly used in model-based global ancestry estimation methods.
It is convenient to record the data as counts. Let gij represent the observed number of copies of allele 1 at marker j of person i. Thus, gij equals 2, 1, or 0 accordingly, as i has genotype 1/1, 1/2, or 2/2 at marker j. Since individuals are considered independent, the log-likelihood of the entire sample is
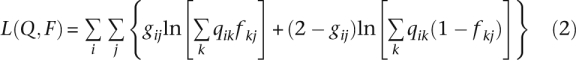 |
up to an additive constant that does not enter into the maximization problem. The parameter matrices Q = {qik} and F = {fkj} have dimensions I × K and K × J, for a total of K(I + J) parameters. For the realistic choices I = 1000, J = 10,000, K = 3, there are 33,000 parameters to estimate. The sheer number of parameters makes Newton's method infeasible. The storage space required for the Hessian matrix is prohibitively large, and the required matrix inversion is intractable.
Note that the log-likelihood (Equation 2) is invariant under permutations of the labels of the ancestral populations. Thus, the log-likelihood has at least K! equivalent global maxima. In practice, this is a minor nuisance and does not affect the convergence of well-behaved algorithms. The constraints 0 ≤ fkj ≤ 1, qik ≥ 0, and 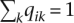 are more significant hindrances to contriving a good optimization algorithm.
are more significant hindrances to contriving a good optimization algorithm.
Point estimation algorithms
A wide variety of optimization methods exist. We have already ruled out Newton's method, so we now turn to algorithms that avoid manipulation and inversion of large matrices. Among the prime candidates is the EM algorithm (Dempster et al. 1977) as implemented in FRAPPE. We have already mentioned that the slow convergence of the EM algorithm makes it a poor candidate for a fast and highly accurate estimation procedure. A block relaxation algorithm turns out to be better suited to our purposes. It converges faster, and faster still under acceleration. We retain the EM algorithm to get us quickly to the vicinity of the maximum and then shift to accelerated block relaxation.
FRAPPE's EM algorithm
The EM algorithm of FRAPPE updates the parameters via
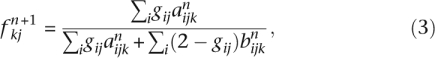 |
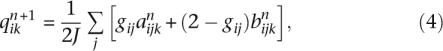 |
where for convenience we define
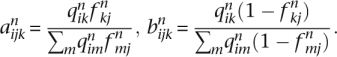 |
FRAPPE's EM algorithm converges slowly, as do many EM algorithms. FRAPPE compensates by using a fairly loose criterion for convergence. This approach permits fast termination of the algorithm, but at a cost of less precise parameter estimates. A simple convergence diagnostic strategy is to declare convergence once successive log-likelihoods satisfy
FRAPPE uses a convergence criterion that is effectively equivalent to Equation 5 with ∈ = 1. We found that FRAPPE's stopping criterion consistently results in slightly inaccurate estimates. Consequently, we choose ∈ = 10−4 as the default stopping criterion in ADMIXTURE. Such a strict convergence criterion entails thousands of additional EM iterates, in practice, often taking many more hours of computation on our test problems. This motivates consideration of non-EM-based algorithms.
Block relaxation algorithm
To achieve the goals of fast convergence and highly accurate parameter estimates, we turned to a block relaxation algorithm. Our block relaxation algorithm alternates updates of the Q and q parameters. It exploits the fact that the log-likelihood L(Q, F) (Equation 2) is concave in Q for F fixed and in F for Q fixed. Concavity makes block iteration amenable to convex optimization techniques. The block updates themselves are found iteratively by repeatedly maximizing the second-order Taylor's expansion of L(Q, F) around the current parameter vector. This technique is commonly referred to as “sequential quadratic programming” (Nocedal and Wright 2000); it coincides with Newton's method in the absence of constraints. For a general function f(x), each step of sequential quadratic programming finds the increment Δ = x − xn optimizing the quadratic approximation
subject to the constraints, and sets xn+1 = xn + Δ. Here df(x) and d2f(x) denote the first and second differentials (transposed gradient and Hessian) of f(x). A linear constraint 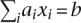 translates into the linear constraint
translates into the linear constraint 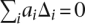 , and the bounds ci ≤ xi ≤ di translate into the bounds ci − xni ≤ Δi ≤ di − xni. There are many quadratic programming methods (Nocedal and Wright 2000). We use the simple pivoting strategy of Jennrich and Sampson (1978).
, and the bounds ci ≤ xi ≤ di translate into the bounds ci − xni ≤ Δi ≤ di − xni. There are many quadratic programming methods (Nocedal and Wright 2000). We use the simple pivoting strategy of Jennrich and Sampson (1978).
In the current application of block relaxation, the keys to success are the separation of parameters and the simple functional forms for the first and second differentials of L(Q, F). In the Q updates for F fixed, the admixture proportions for each individual i are optimized separately. In the F updates for Q fixed, the allele frequencies for each SNP are optimized separately. The entries of the first differentials are
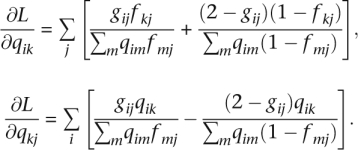 |
All entries of the second differentials vanish except for
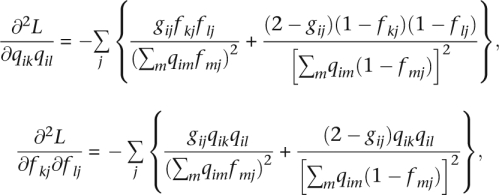 |
and for mixed partials involving a Q and an F parameter. Fortunately, the mixed partials do not enter into block relaxation.
The computational complexity for each iteration of this algorithm is O(IJK2), assuming the denominators in the formulas for the differentials are tabulated. The total runtime, however, depends on the number of iterations required for convergence, which cannot be formulated in terms of I, J, and K. In the data sets we explore in this paper, we usually found on the order of tens of iterations necessary, and never more than 200. The EM algorithm, in contrast, required thousands of iterations to converge according to our criterion.
Tang et al. (2005) also proposed a block relaxation algorithm similar to ours, which they found to perform poorly for large marker sets. We believe this is because (1) they did not take advantage of the block structure of the Hessian matrices within each block relaxation subproblem, and (2) they handle the parameter bounds differently. In contrast, our block relaxation scheme vastly outperforms the EM algorithm in all of our experiments.
Convergence acceleration
EM algorithms are known for their slow rates of convergence. Our block relaxation scheme is faster but still converges fairly slowly. Therefore, we turn to convergence acceleration. Considerable thought has been exercised on accelerating EM algorithms (Jamshidian and Jennrich 1993; Lange 1995; Varadhan and Roland 2008). Here we describe a more generic method.
Suppose an algorithm is defined by an iteration map xn+1 = M(xn). Since the optimal point is a fixed point of the iteration map, one can attempt to find the optimal point by applying Newton's method to the equation x − M(x) = 0. Because the differential dM(x) is usually unknown or cumbersome to compute, quasi-Newton methods seek to approximate it by secant conditions involving previous iterates. Our recent quasi-Newton method (H Zhou, DH Alexander, and K Lange, in prep.) is motivated by this strategy. It has the further advantages of avoiding the storage and inversion of large matrices and preserving parameter linear equality constraints. To keep computational complexity in check, we limit the number q of secant conditions carried along during acceleration. The ascent property of the EM algorithm and block relaxation are helpful in monitoring acceleration. Any accelerated step that leads downhill is rejected in favor of an ordinary step. Accelerated steps do not necessarily respect boundary constraints, so parameter updates falling outside their feasible regions need to be replaced by nearby feasible values. This is implemented by projecting an illegal update to the closest point in the feasible region, which for F and Q updates are the unit interval and the unit simplex, respectively. We also experimented with the squared extrapolation techniques of Varadhan and Roland (2008). These show good performance across a variety of high-dimensional problems. In ancestry estimation, the quasi-Newton acceleration performs about equally well as their best-performing SqS3 acceleration.
Standard errors
Standard errors for our parameter estimates are calculated using the moving block bootstrap (Kunsch 1989). As noted by Tang et al. (2005), bootstrap resampling assuming independence between observations, when they are, in fact, correlated, leads to overconfident (downward-biased) standard errors. The block bootstrap presents a natural way to account for the serial correlation between SNPs. Rather than resampling individual SNPs, one resamples blocks of SNPs. Under the block resampling scheme, blocks containing h consecutive SNP columns from the genotype matrix G are sampled with replacement. A total of ⌈J/h⌉ such blocks are assembled columnwise, and the first J columns of the assembly are taken as the resampled genotype matrix G*. The choice of h is tuned to capture the extent of correlation. Our default setting for h captures an average of 10 cM of genetic distance. This represents the typical span of admixture LD in a population with an admixture event 10 generations in the past (see Patterson et al. 2004). Our choice of h can be overridden by the user.
For each bootstrap resample G*, we re-estimate the parameters, using  and
and  as starting values. Convergence is usually rapid under acceleration. The sample standard errors of the resulting estimates
as starting values. Convergence is usually rapid under acceleration. The sample standard errors of the resulting estimates 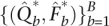 supply estimates of the parameter standard errors.
supply estimates of the parameter standard errors.
The computational time required for calculating these bootstrap standard errors is dominated by the parameter estimation for the bootstrap resamples. As a partial remedy, we have found that the estimation procedure for 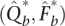 can be stopped after a few iterations with little loss of accuracy in computed standard errors. Early stopping promotes computational efficiency. There is a theoretical basis for a related “one-step bootstrap” procedure (Shao and Tu 1995) based on Newton's method. Here we offer empirical results to suggest that our comparable procedure is sound. Supplemental Figure S2 summarizes how the estimates of standard errors perform when re-estimation is terminated after one, two, or three steps, as compared to standard errors when re-estimation uses our strict convergence criterion. Termination after three steps yields reliable standard error estimates and is the default in ADMIXTURE.
can be stopped after a few iterations with little loss of accuracy in computed standard errors. Early stopping promotes computational efficiency. There is a theoretical basis for a related “one-step bootstrap” procedure (Shao and Tu 1995) based on Newton's method. Here we offer empirical results to suggest that our comparable procedure is sound. Supplemental Figure S2 summarizes how the estimates of standard errors perform when re-estimation is terminated after one, two, or three steps, as compared to standard errors when re-estimation uses our strict convergence criterion. Termination after three steps yields reliable standard error estimates and is the default in ADMIXTURE.
Results
Simulations
To ascertain how accurately ADMIXTURE recovers admixture coefficients, we performed a simulation study. As our ancestral populations, we chose the HapMap CHB, CEU, and YRI samples (The International HapMap Consortium 2005). We considered 13,262 arbitrary SNPs spaced at least 200 kb apart and having no more than 5% missing genotypes. The allele frequencies seen in the unrelated individuals of the three populations were used as the true values for the F matrix. The true values for the matrix Q of admixture coefficients were constructed by sampling from several different probability distributions on the unit simplex
In this manner, we generated admixture coefficients for 1000 simulated individuals in each experiment. The simulated genotype vector G was then constructed according to the binomial model (Equation 2). For each experimental realization of Q, we measured the accuracy of the estimates and by the estimated root mean squared error
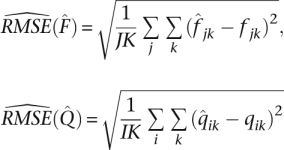 |
criteria. For the first set of experiments, we generated the qi independently from various symmetric Dirichlet distributions, Dir(α, α, α). Here the parameter α reflects the degree of admixture. When α < 1, most individuals show little admixture, while when α > 1, the opposite is true. structure uses these restricted Dirichlet distributions as priors on the admixture coefficients. Our analysis results, summarized in Table 1, indicate that both ADMIXTURE and structure provide fairly good estimates of Q and F, with the largest RMSEs being on the order of 0.025 for the case of α = 1. FRAPPE's estimates were slightly worse in all cases, most noticeably for the Q parameters. In the second set of experiments, we generated the qi independently from asymmetric Dirichlet distributions, Dir(α, β, γ). Again, the results indicate that both ADMIXTURE and structure provide good estimates of Q and F, while FRAPPE's estimates are slightly worse. The greater inaccuracies of FRAPPE'S estimates appear to stem from its convergence criterion. We will revisit this point further shortly.
Table 1.
Summary of simulation experiments using symmetric (Simulations 1–3) and asymmetric (4–6) Dirichlet distributions

Data gij were simulated from the binomial model (Equation 1) using known (Q, F). F was taken as the ancestral allele frequencies corresponding to the HapMap CHB, CEU, and YRI samples; the qi were sampled i.i.d. (independent and identically distributed) according to the Dirichlet distributions indicated. The figure indicates the points (qi1, qi2). Both ADMIXTURE and structure closely recovered the true (Q, F). FRAPPE'S estimates were less accurate.
Real data sets
HapMap Phase 3
Phase 3 of the HapMap Project (The International HapMap Consortium 2005) contains genotypes for individuals from additional populations. Of particular interest here, individuals with Mexican ancestry were sampled in Los Angeles (MEX), and individuals with African ancestry were sampled in the American Southwest (ASW). We chose a subset of the 1,440,616 available markers according to our two previous criteria: (1) to minimize background linkage disequilibrium, adjacent markers must be no closer than 200 kb apart, and (2) no more than 5% of the genotypes must be missing. Based on the genotypes for these markers for the unrelated individuals from the CEU, YRI, MEX, and ASW samples, we constructed a data set of 13,298 markers typed on 324 individuals. Henceforth we refer to this data set as HapMap3. To avoid complications stemming from missing data, we imputed all missing genotypes prior to performing the statistical analyses discussed below.
Figure 1 summarizes the results of analyzing HapMap3 with ADMIXTURE, structure, and EIGENSTRAT. ADMIXTURE, like structure and EIGENSTRAT, resolves the CEU and YRI samples and identifies the ASW sample as an admixture between the YRI and CEU samples, and the MEX sample as an admixture between the CEU sample and a third ancestral population. These results are in line with the current understanding of human population genetics (Jakobsson et al. 2008; Li et al. 2008). Historically, we would expect the third population for the MEX sample to represent one or more of the Native American groups from Mexico, from whom present-day Mexicans derive their non-European ancestry. It is interesting that the inferences about the MEX group made by admixture differ from those of structure. structure places the MEX sample centrally between the CEU and the third ancestral population, while ADMIXTURE places the MEX group further toward the third population. As noted by Tang et al. (2005), the admixture model tends to have difficulty identifying ancestral populations when the data set contains no individuals of unmixed ancestry. This problem is common to structure, FRAPPE, and ADMIXTURE. Inclusion of individuals from appropriate Native American groups to serve as proxies for the ancestral population could resolve questions involving the true degree of admixture in the MEX sample.
Figure 1.

Analyses of the HapMap3 data set. K = 3 for ADMIXTURE and structure. Plotted for each individual i are the point 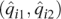 for ADMIXTURE (A) and structure (B), and the point (PC1i, PC2i) for EIGENSTRAT (C). Self-reported ancestries are indicated: (×) YRI; (○) ASW; (+) CEU; (●) MEX.
for ADMIXTURE (A) and structure (B), and the point (PC1i, PC2i) for EIGENSTRAT (C). Self-reported ancestries are indicated: (×) YRI; (○) ASW; (+) CEU; (●) MEX.
Although it may be of scientific interest to know the degree of admixture, the differences between the estimates found here are of little consequence in structured association testing. In fact, it appears that the estimates of qi2 from structure and ADMIXTURE are equivalent up to a change of scale. They are certainly highly correlated, with an R2 value of 0.9998.
Inflammatory Bowel Disease data set
The Inflammatory Bowel Disease (IBD) data set consists of 912 European-American controls genotyped as part of an IBD study conducted by the New York City Health Project (Mitchell et al. 2004). Subjects were genotyped on an Illumina HumanHap300. In addition to their genotypes, many of these individuals reported their ancestry. The availability of self-reported ancestry has made this data set appealing to researchers studying population stratification. For instance, Price et al. (2008) analyzed it with EIGENSTRAT in their study of European ancestry. They concluded that New Yorkers of European ancestry can be represented as an admixture of three ancestral populations: a northwestern European population, a southeastern European population, and an Ashkenazi Jewish population. Unfortunately, the IBD data set contains very few individuals of southeastern European ancestry. Inference of the existence of this third ancestral population group required Price and colleagues to perform a meta-analysis combining the IBD data set with other data sets that include substantial numbers of individuals from Greece and Italy.
We performed our own analysis of the IBD data set using ADMIXTURE, structure, and EIGENSTRAT on a subsample of 9378 of the available genotypes selected according to our previously stated criteria. Results are summarized in Figure 2. The self-reported ancestries of the sample individuals were classified according to criteria from Price et al. (2008) as IBD-AJreport (Ashkenazi Jewish), IBD-NWreport (northwestern European), or IBD-SEreport (southeastern European), and these classifications were used to color-code the figure. ADMIXTURE and structure, run with K = 3, easily differentiate the northwestern European and Ashkenazi Jewish individuals, but they do not clearly cluster the few individuals self-reporting southeastern European ancestry. Nor does EIGENSTRAT identify the southeastern European individuals as a distinct cluster. Given the small number (nine) of such sample individuals, this suggests that all three of these statistical approaches have difficulty resolving ancestry clusters represented by a very small number of individuals.
Figure 2.

Analyses of the IBD data set. K = 3 for ADMIXTURE and structure. In this data set, the evidence for a third population is weak. (A) ADMIXTURE; (B) structure; (C) EIGENSTRAT.
Since it might be argued that K = 3 ancestral populations incorrectly model the IBD data, we repeated our analysis assuming K = 2 ancestral populations. For EIGENSTRAT, note that choosing K = 2 corresponds to using only the first principal component. The results from this second round of analysis are shown in Figure 3. All three programs identify the Ashkenazi Jewish and northwestern European clusters for the self-reported individuals. The estimates from the programs were strongly correlated; R2 values were 0.988 (ADMIXTURE and structure), 0.999 (ADMIXTURE and EIGENSTRAT), and 0.987 (structure and EIGENSTRAT). However, R2 values do not tell the whole story. Pairwise scatterplots between the estimates (data not shown) reveal that structure's ancestry estimates are more skewed toward the boundaries 0 and 1 than ADMIXTURE's estimates. This behavior can be attributed to structure's prior distribution. Despite the small differences in ancestry estimates, the current analysis supports our claim that ADMIXTURE, structure, and EIGENSTRAT yield comparable results when applied to simple cases of admixture.
Figure 3.

Analysis of the IBD data set using K = 2 for structure and ADMIXTURE and the first principal component from EIGENSTRAT. The plots shown are histograms of the first estimated ancestry parameter (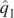 , or PC1) for the individuals, conditioned on self-reported ancestry. Only individuals self-reporting as Ashkenazi Jewish or northwestern European are shown. (A) ADMIXTURE; (B) structure; (C) EIGENSTRAT.
, or PC1) for the individuals, conditioned on self-reported ancestry. Only individuals self-reporting as Ashkenazi Jewish or northwestern European are shown. (A) ADMIXTURE; (B) structure; (C) EIGENSTRAT.
We note that for the IBD data set with K = 3, structure's Markov chain required roughly 10,000 burnin iterations to converge to its stationary distribution. In contrast we found 2000 burnin iterations to be roughly sufficient for our other analyses with structure. Supplemental Figure S1 depicts the trajectory of structure's Dirichlet parameter α for the IBD data set with K = 3 versus K = 2 ancestral populations.
Comparison of maximum likelihood point estimation algorithms
ADMIXTURE offers the user a choice of two point estimation algorithms: our block relaxation algorithm (the default) and a reimplementation of FRAPPE's EM algorithm. Our convergence acceleration technique is applicable to either algorithm. For both algorithms we use the convergence diagnostic (Equation 5) with the strict criterion ∈ = 10−4. As documented in Table 2A, the EM algorithm converges much more slowly than block relaxation. Even accelerated EM cannot match the speed of unaccelerated block relaxation. A strict convergence criterion clearly renders the EM algorithm computationally burdensome.
Table 2.
Comparison of various combinations of the optimization algorithms for the IBD data set with K = 2 ancestral populations

Experiments were performed on an Intel Xeon 2.8 GHz machine. Runtimes are reported as hours:minutes. The strict convergence criterion ∈ = 10−4 is used except where indicated otherwise. In A, acceleration is carried out by our quasi-Newton method with q = 3 secant conditions. In B, the case q = 0 represents unaccelerated block relaxation.
We also explored the effect of the convergence criterion used by FRAPPE. Although a loose convergence criterion will allow the estimation algorithm to terminate faster, it may jeopardize the accuracy of the resulting estimates and encourage bias. Indeed we note in Table 2A that the unaccelerated EM algorithm terminated much faster with the loose criterion than with the strict criterion. The estimates and found with the EM algorithm and loose convergence criterion had a slightly lower log-likelihood than the maximum likelihood estimates and found with the block relaxation algorithm and strict convergence criterion [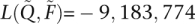 , versus
, versus 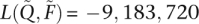 , an absolute difference of 54]. Estimated parameters also diverged substantially. Comparing the first components of the estimated ancestry vectors,
, an absolute difference of 54]. Estimated parameters also diverged substantially. Comparing the first components of the estimated ancestry vectors, 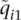 and
and 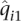 , we found that the median of
, we found that the median of 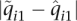 was 0.072. In other words, for half of the individuals in the IBD data set, the ancestry fraction attributed by FRAPPE's EM algorithm to the first population was off by at least 0.072. Similar results were found for other data sets.
was 0.072. In other words, for half of the individuals in the IBD data set, the ancestry fraction attributed by FRAPPE's EM algorithm to the first population was off by at least 0.072. Similar results were found for other data sets.
Note that our unaccelerated block relaxation algorithm under strict convergence runs faster than the EM algorithm under loose convergence. Our accelerated block relaxation algorithm converges faster still. In short, ADMIXTURE quickly delivers highly accurate parameter estimates. Our quasi-Newton acceleration depends on the number q of secant conditions used. Table 2B suggests that the degree of acceleration is fairly insensitive to the exact value of this tuning constant. Although ADMIXTURE's default value of q = 3 works well in practice, the user can override this choice.
We note that a forthcoming version of FRAPPE will tighten its default convergence criterion, allowing for more accurate results. However, as noted here, the EM algorithm proves impractically slow when used with a strict convergence criterion on genome-scale datasets.
Runtime comparison
It is difficult to make a direct comparison of runtimes between ADMIXTURE and structure. With structure, both the number of burnin iterations and the number of subsequent sampling iterations following burnin are configurable parameters set by the user. The proper values for these configuration parameters are essentially problem-dependent, but the structure documentation (Pritchard et al. 2007) advises that 10,000 to 100,000 burnin iterations are usually adequate for convergence to stationarity. The documentation also suggests that stationarity can be diagnosed by manually inspecting the periodic printouts of summary statistics, such as FST distances between inferred populations, for hints that the chain has stabilized.
Second, we have made a conscious decision to provide standard errors rather than interval estimates because most users will be satisfied with the point estimates, and accurate confidence intervals require significantly more bootstrap iterations than accurate standard errors. Conventional wisdom in the bootstrap literature (Efron and Tibshirani 1993) suggests that accurate interval estimation requires on the order of thousands of bootstrap samples, while accurate standard error estimation requires on the order of hundreds.
We are thus hesitant to make a definitive statement regarding the speed of ADMIXTURE versus structure. Let us simply state that in the experiments we have run on a 2.8 GHz Intel Xeon computer on data sets with around 1000 individuals and 10,000 markers, we have found that point estimation with ADMIXTURE typically took on the order of minutes, while point estimation with structure took on the order of hours. This is true even with a relatively small number of MCMC iterations, considerably fewer than advised by structure's documentation. Our runtime results are summarized in Table 3.
Table 3.
Runtimes for plain point estimation, as well as point estimation with interval or standard error estimates

structure was run using the ADMIXTURE model, while EIGENSTRAT and ADMIXTURE were run under their default settings. structure was allowed 2000 burnin iterations; thereafter, 200 iterations were used for point estimates, or 1000 iterations if credible intervals were required in addition. The one exception is IBD (K = 3), for which 10,000 burnin iterations were required as described earlier. ADMIXTURE'S standard error estimation used 200 bootstrap replicates. Experiments were performed on an Intel Xeon 2.8 GHz machine. Runtimes are given as hours:minutes.
The choice of K affects the runtimes of both structure and ADMIXTURE. The time it takes for either program to run is the product of the mean time per iteration and the number of iterations. The time per iteration scales as O(K2) for ADMIXTURE and as O(K) for structure (Falush et al. 2003) when I and J are held fixed. The number of iterations to convergence also tends to increase when K is increased. For example, Supplemental Figure S1 shows that convergence to stationarity for structure takes roughly 10,000 MCMC burnin iterations for K = 3 in the IBD data set, compared to 2000 for K = 2. Likewise, ADMIXTURE requires 153 iterations to converge for K = 3 compared to 23 for K = 2. Generally, we have observed that the number of iterations to convergence increases sharply when a value for K is chosen that is poorly supported by the data. This is precisely the situation with the IBD data set, where there seems to be strong support for only two ancestral populations.
The runtimes of ADMIXTURE and EIGENSTRAT are on the same order of magnitude. Indeed, on most of the data sets we have considered, ADMIXTURE's runtime was less than twice that of EIGENSTRAT, although we note that EIGENSTRAT can be made to run faster by disabling outlier detection.
Simulated association studies
Following Price et al. (2006), we simulated association studies to illustrate how the ancestry estimates from ADMIXTURE can be used to correct for population structure. Our simulation methods exactly parallel those described in their Table 1, so for the sake of brevity we omit simulation specifics and mention only highlights. An overview of our four experiments with two ancestral populations appear in the caption of Table 4. We performed “naive” association tests ignoring ancestry, as well as association tests incorporating either an estimate of ancestry (ADMIXTURE and EIGENSTRAT columns) or the true ancestry value (Ideal column). Ancestry estimates were based on 100,000 markers that were not tested for association. The significance level used was 0.0001.
Table 4.
Results from our simulated association study

The values tabulated are the average proportion of SNP markers of each class found to be significant at a level of 0.0001. Each class of SNPs within each experiment contained 1 million markers; each experiment was repeated 10 times, and averages were taken. Tests were conducted on a logistic regression model using a score test for the significance of each SNP marker. For Naive analyses, no additional predictors were included. For Ideal analyses, the true ancestries were included. For EIGENSTRAT and ADMIXTURE analyses, ancestry estimates were included. Experiment I, moderate case/control ancestry mismatching; Experiment II, more extreme mismatching; Experiment III, admixed population with ancestry risk r = 2; Experiment IV, r = 3.
Price et al. (2006) corrected for population structure by replacing both phenotypes and genotypes with the residuals formed by linear regression on the ancestry estimates. They then performed a modified Armitage trend χ2 test for association. We took the alternative approach of including ancestry as an additional predictor within a logistic regression model, where the first predictor for individual i is the minor allele count gi at the locus in question. For ADMIXTURE, K − 1 of the K entries of the individual ancestry estimate vector 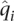 should be used as predictors. With EIGENSTRAT there is in principle no restriction on the number of principal components that can be used. Here we used a single entry from ADMIXTURE's estimate and the top principal component from EIGENSTRAT. Use of additional principal components did not improve the results noticeably.
should be used as predictors. With EIGENSTRAT there is in principle no restriction on the number of principal components that can be used. Here we used a single entry from ADMIXTURE's estimate and the top principal component from EIGENSTRAT. Use of additional principal components did not improve the results noticeably.
Each of our four experiments was conducted 10 times, with the average proportions of SNPs declared significant shown in Table 4. These results suggest that in simple cases of population structure, corrections using EIGENSTRAT and ADMIXTURE perform equally well, in terms of both observed Type I error and power. Corrections using EIGENSTRAT and ADMIXTURE both restored the observed Type I error rate to roughly the nominal level, while achieving essentially the same level of power attained by the Ideal analysis based on the true ancestry. The agreement between ADMIXTURE and EIGENSTRAT is a natural consequence of the high concordance we found between their ancestry estimates. The squared correlation coefficients between the estimates from the two programs were >0.9999 in all of the experiment runs we performed. A more detailed discussion of these results can be found in the Supplemental material.
Discussion
Our studies on simulated and real data sets show that ADMIXTURE performs as well as structure in ancestry estimation and runs considerably faster. ADMIXTURE's large speed advantage over structure opens up the possibility of using large sets of arbitrarily selected markers for ancestry determination, rather than focusing on a small number of ancestry-informative markers (AIMs) known a priori to have different allele frequencies in different populations. Since many populations, human and otherwise, have not been genotyped, AIMs are often unknown. Fortunately, the ancestral allele frequency estimates output by ADMIXTURE allow AIMs to be readily identified.
ADMIXTURE's speed in generating point estimates stems from the use of a relatively fast block relaxation strategy, coupled with quasi-Newton convergence acceleration. Interval and standard error estimation is costly for both ADMIXTURE and structure. We perform bootstrapping, while structure performs MCMC sampling. Both are computationally intensive. Our speed advantage with standard error estimation activated is due to a combination of (1) good starting values, (2) a judicious stopping rule in parameter estimation for each bootstrap resample, and (3) our decision to compute standard error estimates rather than confidence intervals.
Choice of an appropriate value for K is a notoriously difficult statistical problem. It seems to us that this choice should be guided by knowledge of a population's history. Because experimentation with different values of K is advisable, ADMIXTURE prints values of the familiar AIC (Akaike information criterion) and BIC (Bayesian information criterion) statistics, widely applied in model selection.
ADMIXTURE's model does not explicitly account for linkage disequilibrium (LD) between markers. The original version of structure also lacked support for markers in LD, as does FRAPPE, but structure 2.0 includes a linkage model capturing ADMIXTURE LD but not background LD. Falush et al. (2003) break LD down into three components: mixture LD, attributable to the variation in ancestry between individuals; admixture LD, attributable to recent admixture events; and background LD, attributable to population history. While thinning marker sets is beneficial in dealing with background LD, it is relatively helpless in ameliorating admixture LD, which can extend orders of magnitude further than background LD in recently admixed populations. In data sets where admixture LD is a significant factor, our likelihood can be considered a useful and tractable approximation. While the resulting estimates may be subject to some bias, we believe the biases stemming from unmodeled LD pose greater problems in local ancestry estimation than here. Another concern is the underestimation of standard errors using simple bootstrap tactics. Here the block bootstrap is a major corrective.
The speed of ADMIXTURE is on par with EIGENSTRAT's implementation of PCA, so geneticists can now choose a method for summarizing population structure based on considerations of statistical appropriateness alone. The model-based and PCA-based approaches are complementary; each offers its own advantages. PCA has the advantage of robustness. It does not specify an exact model and so may be more suitable in situations where the simple admixture model does not hold, for instance, when a population shows continuous spatial structure (Novembre and Stephens 2008; Novembre et al. 2008). Model-based estimates are more directly interpretable than PC coordinates and come with attached precisions. The model-based approach also directly provides allele frequency estimates for the ancestral populations.
Despite these differences, our analyses of real and simulated data show a high degree of concordance between the estimates from ADMIXTURE and EIGENSTRAT. In particular, we have observed a strikingly high degree of linear correlation between ancestry estimates from the two programs. Thus, while the two approaches are complementary, in simple settings, the resulting estimates are equally useful for statistical correction in association studies. More complicated population structure may reveal differences between the two programs' estimates.
In summary, we have presented a fast new algorithm and software suitable for inferring ancestry of individuals from stratified populations based on genotypes at a large number of arbitrary SNP markers. Our program ADMIXTURE is available as a stand-alone program and will soon also be available within the Mendel package. A free download is available at http://www.genetics.ucla.edu/software.
Acknowledgments
We thank Mike Boehnke, Nan Laird, Hua Tang, and Ravi Varadhan for helpful discussions, and two anonymous reviewers for their suggestions. We thank Nick Patterson for suggesting the block bootstrap. We thank the New York Health Project and the U.S. Inflammatory Bowel Disease Consortium for the IBD data set. This work was supported by grant number T32GM008185 (D.H.A.) from the National Institute of General Medical Sciences, and grant numbers GM53275 and MH59490 (K.L.) from the United States Public Health Service.
Footnotes
[Supplemental material is available online at http://www.genome.org. The ADMIXTURE program is freely available at http://www.genetics.ucla.edu/software.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.094052.109.
References
- de Leeuw J. Block relaxation algorithms in statistics. In: Bock H, et al., editors. Information systems and data analysis. Springer Verlag; New York: 1994. [Google Scholar]
- Dempster A, Laird N, Rubin D. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B Methodol. 1977;39:1–38. [Google Scholar]
- Efron B, Tibshirani R. An introduction to the bootstrap. CRC Press; Boca Raton, FL: 1993. [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multi-locus genotype data, linked loci, and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Scholz S, Scheet P, Gibbs J, VanLiere J, Fung H, Szpiech Z, Degnan J, Wang K, Guerreiro R, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- Jamshidian M, Jennrich R. Conjugate gradient acceleration of the EM algorithm. J Am Stat Assoc. 1993;88:221–228. [Google Scholar]
- Jennrich R, Sampson P. Some problems faced in making a variance component algorithm into a general mixed model program. In: Gallant A, Gerig T, editors. Proceedings of the Eleventh Annual Symposium on the Interface. Institute of Statistics; North Carolina State University: 1978. pp. 56–63. [Google Scholar]
- Knowler WC, Williams RC, Pettitt DJ, Steinberg AG. Gm3;5,13,14 and type 2 diabetes mellitus: An association in American Indians with genetic admixture. Am J Hum Genet. 1988;43:520–526. [PMC free article] [PubMed] [Google Scholar]
- Kunsch H. The jackknife and the bootstrap for general stationary observations. Ann Stat. 1989;17:1217–1241. [Google Scholar]
- Lange K. A Quasi-Newton acceleration of the EM algorithm. Statist Sinica. 1995;5:1–18. [Google Scholar]
- Li CC. Population subdivision with respect to multiple alleles. Ann Hum Genet. 1972;33:23–29. doi: 10.1111/j.1469-1809.1969.tb01625.x. [DOI] [PubMed] [Google Scholar]
- Li J, Absher D, Tang H, Southwick A, Casto A, Ramachandran S, Cann H, Barsh G, Feldman M, Cavalli-Sforza L, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat Genet. 2004;36:512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- Mitchell M, Gregersen P, Johnson S, Parsons R, Vlahov D. The New York Cancer Project: Rationale, organization, design, and baseline characteristics. J Urban Health. 2004;81:301–310. doi: 10.1093/jurban/jth116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocedal J, Wright SJ. Numerical optimization. 2nd ed. Springer; New York; 2000. [Google Scholar]
- Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 2008;40:646–649. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Hattangadi N, Lane B, Lohmueller KE, Hafler DA, Oksenberg JR, Hauser SL, Smith MW, O'Brien SJ, Altshuler D, et al. Methods for high-density admixture mapping of disease genes. Am J Hum Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price A, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A, Patterson N, Plenge R, Weinblatt M, Shadick N, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Price A, Butler J, Patterson N, Capelli C, Pascali VL, Scarnicci F, Ruiz-Linares A, Groop L, Saetta AA, Korkolopoulou P, et al. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Donnelly P. Case-control studies of association in structured or admixed populations. Theor Popul Biol. 2001;60:227–237. doi: 10.1006/tpbi.2001.1543. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Wen X, Falush D. Department of Human Genetics, University of Chicago; 2007. Documentation for structure software: Version 2.2. Tech. rep. [Google Scholar]
- Sankararaman S, Kimmel G, Halperin E, Jordan M. On the inference of ancestries in admixed populations. Genome Res. 2008a;18:668–675. doi: 10.1101/gr.072751.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Sridhar S, Kimmel G, Halperin E. Estimating local ancestry in admixed populations. Am J Hum Genet. 2008b;82:290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J, Tu D. The jackknife and bootstrap. Springer; New York: 1995. [Google Scholar]
- Tang H, Peng J, Wang P, Risch N. Estimation of individual admixture: Analytical and study design considerations. Genet Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- Tang H, Coram M, Wang P, Zhu X, Risch N. Reconstructing genetic ancestry blocks in admixed individuals. Am J Hum Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadhan R, Roland C. Simple and globally convergent methods for accelerating the convergence of any EM algorithm. Scand J Stat. 2008;35:335–353. [Google Scholar]