

Abstract
We describe the genome sequencing of an anonymous individual of African origin using a novel ligation-based sequencing assay that enables a unique form of error correction that improves the raw accuracy of the aligned reads to >99.9%, allowing us to accurately call SNPs with as few as two reads per allele. We collected several billion mate-paired reads yielding ∼18× haploid coverage of aligned sequence and close to 300× clone coverage. Over 98% of the reference genome is covered with at least one uniquely placed read, and 99.65% is spanned by at least one uniquely placed mate-paired clone. We identify over 3.8 million SNPs, 19% of which are novel. Mate-paired data are used to physically resolve haplotype phases of nearly two-thirds of the genotypes obtained and produce phased segments of up to 215 kb. We detect 226,529 intra-read indels, 5590 indels between mate-paired reads, 91 inversions, and four gene fusions. We use a novel approach for detecting indels between mate-paired reads that are smaller than the standard deviation of the insert size of the library and discover deletions in common with those detected with our intra-read approach. Dozens of mutations previously described in OMIM and hundreds of nonsynonymous single-nucleotide and structural variants in genes previously implicated in disease are identified in this individual. There is more genetic variation in the human genome still to be uncovered, and we provide guidance for future surveys in populations and cancer biopsies.
The sequencing of the human genome provided the springboard to understand the role of genetic variation on disease and human evolution (Lander et al. 2001; Venter et al. 2001). More recently, the HapMap project surveyed the frequencies of ∼4 million highly ascertained single nucleotide variants on a sample of four major human populations (The International HapMap Consortium 2005; Frazer et al. 2007), providing useful background information to design and analyze genetic association studies for common variants. However, a hypothesis-free view of the full complement of genetic variants on individual genomes has been explored in only a few instances (Levy et al. 2007; Bentley et al. 2008; Ley et al. 2008; Wang et al. 2008; Wheeler et al. 2008), mostly due to cost and time constraints. Yet, these five studies, together with other recent population studies (Redon et al. 2006; Korbel et al. 2007; Kidd et al. 2008), have revealed a much more dynamic picture of genomic variation in the human species, suggesting that the prevalence of structural variation has been largely underestimated. Therefore, the picture of human genomic variation is far from complete, and the introduction of second-generation, high-throughput, and inexpensive sequencing opens the possibility of sequencing hundreds to thousands of human genomes to gain a better understanding of the full extent of human variation (Kaiser 2008). Here, we introduce the use of massively parallel short-read sequencing by ligation of mate-paired and fragment libraries to uncover single nucleotide, insertion and deletion (indel), and inversion variants of the genome from an anonymous donor from Yoruba in Ibadan, Nigeria, who was part of the International HapMap Project (Frazer et al. 2007). We also explore the benefits of a novel error correction system that aids in single nucleotide polymorphism (SNP) discovery.
Several techniques for massively parallel DNA sequencing have recently been described (Ronaghi et al. 1999; Brenner et al. 2000; Braslavsky et al. 2003; Margulies et al. 2005; Shendure et al. 2005; Ju et al. 2006; Gibbs et al. 2007; Bentley et al. 2008; Eid et al. 2009). They broadly fall into two assay categories (polymerase- or ligase-based) and two detection categories (“asynchronous single molecule” and “synchronous multi-molecule,” often referred to as “ensemble” readouts). SOLiD (sequencing by oligo ligation detection) sequencing, the method used here, is a DNA ligase-based synchronous ensemble detection method utilized to read 500 million to over 1 billion reads per instrument run (Cloonan et al. 2008; Valouev et al. 2008).
All of these techniques are theoretically compatible with mate-paired sequencing, but they differ in how they generate the mate-paired reads. Campbell et al. (2008) utilized an approach that generates short pairs from cluster PCR colonies often referred to as “paired-ends.” These paired-end reads have limited insert sizes due to the efficiency and representation of PCR amplification of long amplicons via cluster PCR. Consequently, very few paired-end reads are generated that are longer than a Sanger capillary electrophoresis read (<10× clone coverage in pairs >1.0 kb). Korbel et al. (2007) and Bentley et al. (2008) utilized DNA circularization and random shearing, circumventing the need to PCR amplify the entire pairing distance at the cost of more input DNA. These pairs each differ substantially in their tag length due to the random shearing step. The asymmetrical tag lengths reduce the pairing efficiency and often contaminate the library prep with a high number of 200-bp inserts; thus, no more than 100× clone coverage is obtained, and many tags are sequenced that are not paired or are paired in the wrong distance or orientation. Furthermore, these techniques may result in many inverted molecules that complicate the detection of inversions.
The preferred pairing method would provide both high sequence coverage and high clone or “physical” coverage with flexible insert sizes such that SNPs, small indels, larger structural variations, and copy number variants (CNVs) could be surveyed in one method. Here, we utilize two pairing methods that retain less variable tag lengths while enabling both high sequence coverage (14.9×) and high clone coverage (297×) of the human genome to enable the broadest survey of variation possible.
Use of ligases for massively parallel short-read DNA sequencing of human genomes offers several unique attributes next to polymerases. Most notable is the use of an error-correcting probe-labeling scheme (two-base encoding, or 2BE), which provides error correction concurrent with the color-called alignment of the data (i.e., without having to resequence the reads). This correction property has specific utility in bisulfite sequencing, de novo assembly, indel detection, and SNP detection, but in this manuscript we will focus only on the SNP detection attributes.
Here, we demonstrate that SOLiD sequencing is capable of efficiently surveying single nucleotide polymorphisms and many forms of structural variation concurrently at relatively modest coverage levels. The unprecedented clone coverage allows us to uncover a significantly larger number of structural variants in a size range not efficiently explored in previous studies, helping to complete the picture of functional variants in this genome. The observed pattern of putatively functional genetic variation in the Yoruba genome recapitulates population signatures of natural selection and suggests a higher than expected individual load of potentially deleterious variants in the human genome.
Results
Sequencing performance with short reads
We generated 76.53 Gb of mate-paired reads that align to the reference human genome (hg18), of which 49.07 Gb have both tags mapping to the genome as a uniquely placed pair. 46.55 Gb of these pairs align at the expected distance, order, and orientation, and, of these, 42.56 Gb are comprised of mate pairs in which at least one tag has a unique start point and are therefore derived from unique and unamplified molecules (Supplemental Table S1). In addition, we generated “fragment” reads (45–50 bp), of which ∼10.5 Gb and 8.6 Gb align and align uniquely to hg18, respectively.
Overall, we cover on average 17.9-fold the haploid genome length. While the genomic regions covered by uniquely placed mate-paired and fragment reads overlap substantially (80.35% of the covered regions), the mate-paired reads are mapped to an additional 19.59% of the genome not accessible by the fragments, whereas only 0.06% of the genome is covered exclusively with fragments.
Over 98.57% and 96.58% of the autosomal genome is covered by at least one and three reads, respectively, while >99.6% and 95% is spanned by a single clone and 124 clones, respectively, when we require that a mate pair be placed uniquely in the genome (Fig. 1). When we remove this criteria of uniqueness, >99.8% and 99.0% of the autosomal genome is covered by at least one and three reads, respectively, while 100% and 95% of the genome is spanned by a single clone and 212 clones, respectively. Ns in the reference assembly are excluded. Further investigation of the increased coverage gained from mate-paired libraries demonstrates that a more comprehensive sampling of the human genome is achieved with uniquely placed mate pairs than with the unique placement of each of the independent tags (Fig. 2). The coverage per chromosome is lower on the sex chromosomes, as expected. While theoretically mapping all possible 25-mers allowing up to two mismatches uniquely back to the autosomal reference leaves 75.38% of the reference covered, or “mappable,” we cover 77.1% of the genome with 25-bp reads allowing up to two mismatches. This performance is likely due to the presence of sequence that is missing from the reference. Color space alignments yield greater specificity than base space in which the genome is 70.86% uniquely mappable with 25-mers allowing up to two mismatches. The mate pair coverage nearly matches what is expected based on the estimated mappability of mate-paired tags (using a constant insert size and sampling every 25 bases).
Figure 1.

Cumulative plot of sequence and clone coverage from uniquely placed fragments and uniquely placed mate pairs. The sequence coverage is derived from the fragment, 2 × 25 mate-paired, and 2 × 50 mate-paired libraries while the clone coverage is from only the mate-paired libraries (2 × 25 and 2 × 50).
Figure 2.

Uniquely placed mate pairs provide a more comprehensive sampling of the human genome than the unique placement of each of the tags independently. The coverage is separated by mate-paired data treated as single tags before pairing (mate pairs, unpaired; blue) and mate-paired data treated as mate pairs (mate pairs, paired; pink).
The uncovered genomic regions tend to be of low GC content and enriched in repetitive sequences, in particular long repeats and segmental duplications (Supplemental Table S2). This is expected since it is difficult to uniquely map short reads to segmental duplications and other repeats; when the requirement of uniqueness is removed, these features are much less prominent or the correlation reverses. These uncovered regions also tend to be enriched in dbSNP entries, which suggests many of these are likely to be pseudo-SNPs generated by assembly or mapping artifacts as previously reported (Tuzun et al. 2005). Longer repeats are particularly enriched in areas not covered by uniquely placed mate-paired reads, and this is certainly due to an inability to cross over the repeat at the insert sizes used to generate the libraries in this work (the longest being 3.5 kb) and suggests that using longer inserts would help to overcome this limitation.
To assess the sampling of sequence tags across the genome, we plotted the average sequence coverage across the genome according to GC content (see Methods) along with the tenth and 90th centiles and compared these with what is expected according to the Poisson distribution. While the 2 × 25 mate pairs and fragments lack the most in low GC content and the 2 × 50 mate pairs lack the most in high GC content, the combination of all three library types compensates for one another and has good coverage in all but the most extreme GC content (Supplemental Fig. S4).
Identification of SNPs
The NA18507 genome has not been Sanger sequenced to more than 0.5× sequence coverage (Kidd et al. 2008). However, it has been extensively genotyped as part of the HapMap project (Frazer et al. 2007), and some regions were shotgun sequenced to higher depth as part of the ENCODE project (The ENCODE Project Consortium 2007). As a result, false-negatives can only be approximated by estimating how much of the hg18 genome is left uncovered (Fig. 1), but false-positives can be assessed with several techniques. Once reads are uniquely mapped to the genome, sequence variants can be discovered by comparing read sequence with the reference genome, and taking into account the redundancy obtained to distinguish natural sequence variation from sporadic sequencing errors. Using two-base encoding (2BE), only adjacent sequencing or color differences to the reference can be candidates for single SNPs. Thus the majority (>92%) of our sequencing errors can be eliminated as putative SNPs. Adjacent color differences that can encode a base change are termed “valid adjacent” and are explained in more detail in the Supplemental material on color space.
Across the entire genome, we call 3,866,085 SNPs, of which 81% are in dbSNP (release 129). Due to the error-correcting qualities of the color space reads, we only need to see each allele twice to call a variant, in contrast to other methods that require three and four alleles (Bentley et al. 2008; Wang et al. 2008). This allows us to call a comparable number of SNPs in the human genome at lower coverage levels without sacrificing accuracy, as suggested by the high dbSNP concordance. To assess the false-discovery rate in an unknown genome, we performed an analysis similar to that of Wheeler et al. (2008) in which we counted the number of times our sample contains an allele that is not annotated at a bi-allelic locus in dbSNP 129. Using this as a proxy for false discovery, we observe that 99.88% of dbSNP loci in our sample are called as one of the two annotated alleles, and a nonannotated third allele is called only 0.12% of the time.
Comparing new sequencing technologies with array-based genotyping assays that have selected SNPs with high assay conversion rates can be misleading. This does not address our goal of understanding our ability to detect all polymorphisms in a human genome. As a result, we choose to focus our laboratory validation on SNPs that are novel as indicated by an absence in dbSNP. To confirm our findings, we randomly selected 333 of our 734,662 novel SNP calls (those not found in dbSNP 129) for validation with SNPlex genotyping assays (Tobler et al. 2005). There were 34 assay failures, which leaves us with 299 successful assays. 86% of our novel SNP calls are heterozygous, and 94% of the successful assays (280 of the 299) are heterozygous. The SNPlex genotypes are in >95% agreement with the SOLiD-detected genotypes. There are 14 cases in which SNPlex disagrees with the SOLiD-detected genotype. Two calls are SOLiD-detected homozygous SNPs that SNPlex calls as heterozygous, indicating that SOLiD undercalled the heterozygous state in favor of the variant allele (in both cases SOLiD detects the reference allele but the variant allele is present in greater numbers than the reference allele, and the SNP was called a homozygous variant). These calls are expected due to the lower likelihood of sampling both alleles proportionately at fairly low coverage. The remaining disagreements are SOLiD-detected homozygous (one) and heterozygous (11) SNPs in which SNPlex detects the genotypes as homozygous for the reference allele. 12/299 (4.01%) of our novel SNP calls detect one allele that is unsubstantiated by SNPlex and could be considered to be true false-positives (FPs). Since our novel SNP calls are made up of ∼19% of all of our SNP calls, it can be inferred that 0.76% of all of our SNP calls are FPs. Eight of the 11 heterozygous FP calls are low coverage (5× to 8×) in which the variant allele is on only one strand. If we discount these from the analysis, then 1.34% of our novel SNP calls are indicated as FP, and it can be inferred that 0.25% of all of our SNPs are FPs. Previous validation of SNP detection with the fragment, 2 × 25 mate-paired, and a different set of 2 × 50 mate-paired data with manually reviewed Sanger CE sequencing confirmed 111 out of 112 assayed novel heterozygous SNPs that were not in dbSNP.
Our goal is not only to detect SNPs (alternative alleles to the reference), but also to infer whether the sample is heterozygous or homozygous at a given position, the most challenging being the detection of a heterozygous state, given the sampling introduced by the shotgun process and the bias induced by mapping to a reference sequence. However, this task is facilitated by the error detection and correction scheme of the SOLiD 2BE sequencing chemistry, which reduces the average sequencing error rate to <0.1% (Supplemental Table S1).
To further assess our ability to call genotypes correctly by sequencing, we compared our data with those from the 3.9M SNP calls for this individual in the HapMap r26 data release. Out of the 3,026,465 HapMap homozygous genotypes called (there are 3,054,399 in HapMap genotyped for NA18507, but some are not called by us due to low coverage or noise; we make calls for >99%), we had an overall 99.16% agreement with HapMap. Removing SNPs with a minor allele frequency (MAF) of <5%, where genotypes are less confident in the HapMap due to genotyping assay artifacts (Welch et al. 2008), the agreement climbs to 99.68%.
Higher coverage is required to adequately sample two alleles rather than one and thus call a heterozygote rather than a homozygote SNP. Reads that contain variant alleles utilize two color mismatches to cover the variant; therefore, these reads are allowed fewer sequencing errors than reads with no variants. As a result, more reads match the reference alleles than the variant alleles. Approximately 57% of all reads that map to either allele at HapMap r26 heterozygous loci map to the reference allele. We are assessing mapping valid adjacent mismatches as a single mismatch to compensate for this.
Figure 3 shows the dependency of homozygous and heterozygous calling with coverage and suggests that both types of calling are highly concordant with HapMap even at modest coverage levels and despite a tendency for reads to overmap to the reference. For the cases where we do not detect a heterozygous SNP at known HapMap r26 loci despite having adequate coverage, we have demonstrated that diBayes, an alternative algorithm, can identify them in most cases.
Figure 3.

Dependence of genotype calling on depth of sequence coverage. The NA18507 genotypes called by SOLiD at all HapMap loci are compared with the HapMap genotypes by SOLiD coverage per genome position (average 18× coverage). Coverage includes alleles representing the reference or a valid base change; i.e., alleles with single or invalid adjacent mismatches are not included. No prior information about SNP presence or SNP alleles was used in making SOLiD gentoype calls. The number of HapMap loci with a given level of SOLiD coverage (“Count”) are shown and the percentage of these loci for which SOLiD gives the same genotype as HapMap for homozygotes and heterozygotes is represented by the colored lines (graphed using the left-hand y-axis and referred to as “% Concordance”) using two genotyping algorithms: Consensus Caller and diBayes. diBayes is more sensitive at heterozygous SNP detection and yields a lower false-negative rate than Consensus Caller, but we did not attempt to estimate the false-positive rate of diBayes with validation data. SOLiD genotypes that differ from HapMap gentoypes are nearly always heterozygous undercalls (i.e., the position is called homozogyous for one of the two alleles) or called as N (insufficient evidence to make a confident genotype call).
We call 60.3% of the SNPs we identify as heterozygous; among these, 67.3% are transitions and 32.7% are transversions—a 2.05 ratio, which is very close to the expectation for the human genome (Lander et al. 2001; Venter et al. 2001). 27.2% of the heterozygotes we call are novel, and amongst these 65.3% are transitions and 34.7% are transversions. SNPs are more densely represented on autosomes than on the X and Y chromosomes.
Single base changes have the potential to be disruptive to gene structure when they are within exons. Amongst the SNPs that we identify in NA18507, 68,624 of them are in a known exon and 16.1% of these SNPs within exons are novel. 12.5% of exons contain at least one SNP, and one exon contains 49 SNPs. The distribution of the number of SNPs identified per exon is shown in Supplemental Figure S6. From these, we identify 9902 SNPs that produce nonsynonymous changes in coding sequences (ns-SNPs). While exons comprise 2.6% of the total genome, only 1.78% of all SNPs we identified are in exons, indicating that there is an underrepresentation of SNPs in exons due to their deleterious potential (Lander et al. 2001; Venter et al. 2001).
Resolving haplotype phases with mate-paired data
Mate-paired data with accurate reads can be used to resolve haplotype phases when two reads in a pair cover alleles at different loci (Kidd et al. 2008). We combined our 3,759,673 genome-wide autosomal reference-variant SNP calls with autosomal HapMap r26 genotypes (which includes NA18507 genotypes for homozygous reference-allele loci) to obtain 6,184,461 distinct potential genotypes. We evaluated the number of mate-paired reads that cover two or more of these loci to establish the upper limit for potentially resolving phases with the 14.89× mate-paired data. We observed 4,027,548 potential genotypes (65.12%) covered by 6,767,943 pairs (1.1% of all mate pairs) with 24.03% of the mate pairs calling SOLiD-detected heterozygous/heterozygous, 35.90% heterozygous/homozygous, and 40.07% homozygous/homozygous pairing events. 3,734,035 pairs (55.17%) have both a Forward (F3) and a Reverse (R3) tag covering 3,580,440 distinct genotype loci, and the remaining 44.83% have only a F3 or R3 tag covering 1,205,779 distinct genotype loci. Nearly two-thirds of the genotypes for this individual are covered by at least one mate-paired read that is in phase with another genotyped location, and 43% that we detect as heterozygous are in phase with another that we also detect as heterozygous.
We evaluated all mate pairs that cover HapMap r22 phased heterozygous genotypes and determined that our phases agree with 98.95% of the HapMap phases and we cover 21.74% of the HapMap-phased heterozygous genotypes with at least one mate pair. We also looked for mate pairs in which both a novel heterozygous locus and a HapMap-phased heterozygous locus are covered to determine if the phases of the novel loci are in agreement with the HapMap loci. 76,300 of our novel heterozygotes fall into this category, and for the 15,946 pairs where both alleles of the novel heterozygotes are covered, the alleles are in opposite HapMap phases compared with each other, as expected, 99.52% of the time. Mate pairs can be collapsed into longer haplotype “blocks” of sizes up to 215 kb (Supplemental Fig. S5). In principle, these physically phased blocks can be used together with HapMap genotype data and statistical phasing algorithms to produce more accurate and complete haplotype phases for this individual.
Intra-read or “split-read” insertions and deletions
The most prevalent class of small insertions and deletions are those <5 bp in length (Mills et al. 2006; Levy et al. 2007), with 57% of the events being single-base indels that were undetectable with the approach taken by Wheeler et al. (2008). Since SOLiD is a terminating chemistry, it can accurately call single and multiple base indels under the sequence read by mapping the indel read to the reference genome. We find 226,529 indels, including 89,679 insertions of up to three bases, 124,024 deletions of up to 11 bases, and 12,826 larger indels (Fig. 4).
Figure 4.

Length distributions of small and medium insertions and deletions under sequencing reads with respective concordances. Deletions are detected up to 500 bp and insertions up to 20 bp. A high prevalence of small indels, even-sized indels, and Alu-sized deletions (300–350 bp) are found in this genome. Larger indels (deletions 12 bp and higher and insertions 4 bp and higher) are called with more restrictive settings (see Methods) than smaller ones.
Approximately 67% of the small indels found (insertions up to 3 bp and deletions up to 11 bp) are present in dbSNP, and 49% are seen in data from the nine individuals in Kidd et al. (2008) that had 2.78× total sequence coverage. The concordance with Kidd et al. (2008) drops to 37% and 22% when considering only Yoruba samples (1.46× sequence coverage) and only NA18507 (0.50× sequence coverage), respectively. This low concordance is expected as dbSNP underrepresents multiple nucleotide variants as seen in Kidd et al. (2008), in which 75% of the discovered indels <100 bp are novel, and the low sequence coverage of Kidd et al. (2008) prevents much of the genome from being accessible to small indel detection. Additionally, we find 10,525 insertions of length four to 14, with dbSNP and Kidd concordances of 74% and 64%, respectively, and 233 insertions of length 15–19, with concordances of 52% and 51%, respectively. We also detect 2068 deletions from size 12–498, with dbSNP and Kidd concordances of 41% and 38%, respectively.
With regard to the impact of small indels (insertions up to 3 bp and deletions up to 11 bp) on gene structure, we find that 2788 exons contain at least one indel, while 392 exons have more than one indel and 2241 genes contain an indel within an exon (Supplemental Fig. S6). We observe an overabundance of indels in first and last exons as observed with SNPs. In total, 76% of 2241 genes containing indels have them in the first and last exon (362 in the first, 1492 in the last, and 159 in their only exon). There is a noticeable preference for even-sized indels across the genome due to the existence of dinucleotide and tetranucleotide repeats as reported by Mills et al. (2006) and Levy et al. (2007) (Fig. 4). However, this preference is skewed in favor of 3- and 6-base (3n) indels in coding exons (Supplemental Fig. S7) in which 34.3% of the indels within and 12.7% of the indels outside of coding exons are 3 bp or 6 bp in size. The observation that indels in coding exons show a size distribution that favors 3- and 6-base codon skipping mutations when the rest of the genome exhibits even-sized indels is expected due to purifying selection against frameshifts in coding regions.
Large inter-read insertions and deletions
We have identified 1515 insertions and 4075 deletions by clustering of mate-paired reads with discordant distances when mapped to the human reference assembly hg18 (see Methods). Deletions range in size from 86 to 96,957 bp and insertions from 30 to 1287 bp. Figure 5 illustrates the size distributions of the insertions and deletions and highlights the abundance of variations in the size range of Alu and LINE elements. The insertions and deletions have clones with discordant distances spanning them that are deviated by at least six standard deviations from expectation at the given level of clone coverage. Supplemental Figure S8 illustrates the size limit of detection of inter-read insertions and deletions at various levels of clone coverage at six standard deviations of significance given our library insert size of 1400 bp with a standard deviation of 199 bp.
Figure 5.

Length distributions of large insertions and deletions identified between mate-paired tags. There is an abundance of insertions and deletions in the size range of Alus as well as a spike in the number of deletions in the size range of LINEs (6000 bp).
Comparing the large deletions to the Venter (Levy et al. 2007), Watson (Wheeler et al. 2008), and YH (Wang et al. 2008) genomes, we find that 40% of the deletions ≥200 bp and 30% of the deletions ≥1000 bp have been previously identified in these genomes. Common to all four of the genomes are 353 deletions ≥200 bp and 49 deletions ≥1000 bp. These deletions in common in all four genomes from three ethnically distinct populations indicate the possibility of the presence of a minor allele in the hg18 reference sequence. A pairwise comparison of the number of deletions identified in each of the four genomes is presented in Supplemental Table S3. While we detect more deletions in NA18507 than have been detected in the other genomes, in each pairwise comparison >20% of the deletions ≥200 bp detected in NA18507 have been detected in the other genome. We also detect >38% of the deletions ≥200 bp that have been detected in each of the other genomes, significantly more than when considering most of the other pairs of genomes with respect to each other.
Deletions in NA18507 identified by both intra- and inter-read approaches
The long tag mate-paired data allow us to identify some of the same deletions by both the intra- and inter-read approaches. Amongst the 2068 deletions ranging in size from 12 to 498 bases detected within reads, 193 of them are in common with the deletions identified by discordant read-pair clusters (Fig. 6). Amongst these deletions, 60 of them have also been identified in the Venter, Watson, and YH genomes. Figure 6 also illustrates a 328-bp deletion that has been identified by both the intra- and inter-read approaches in NA18507 by SOLiD and in each of the other three genomes. The ability to identify deletions with both inter- and intra-read approaches bridges the gap between these two methods, which not only allows the corroboration of the identified events, but also permits the detection of the full range of sizes of deletions in the human genome.
Figure 6.

The distribution of the 193 deletions identified in NA18507 with SOLiD by both the intra-read and inter-read approaches. (Inset) A 328-bp deletion detected using both the inter- and intra-read approaches. Four nonredundant molecules identify the deletion with the intra-read approach while 81 clones identify the deletion with the inter-read approach. This deletion has also been found in the Venter, Watson, and YH genomes.
Inversions
While inversion detection is sensitive to both sequence coverage and clone coverage, the clone coverage is especially important. If a read is in the center of an inversion, it is more likely that its mate is flipped with respect to itself the farther away its pair is located and therefore able to detect an inversion breakpoint. We analyzed all mate pairs with both ends mapped but with sequences on opposite strands (SOLiD mate-paired libraries create mate-paired tags in which both sequence reads are normally on the same strand). We look for multiple mate pairs to exhibit the same inverted orientation at the same coordinates in the genome and across multiple libraries. The supporting evidence of all inversions can be visually inspected using the SOLiD Alignment Browser (Supplemental Fig. S9).
We observe 91 inversions of which 22 are amongst the 90 inversions observed by Levy et al. (2007) and 37 are amongst the 72 inversions observed by Korbel et al. in NA18505 (Korbel et al. 2007). Some of the inversions will be missed by short reads due to an enrichment of repeats at the edges of the variations. A complex inversion and deletion is discovered on chromosome 4 (Supplemetnal Fig. S9), in which a homozygous deletion is nested within a heterozygous inversion, indicating a hotspot for variation in which the deletion occurred prior to the inversion. This genomic event is at the same location in which others have observed copy number changes (Redon et al. 2006) and where we have observed a high discordance in our SNP calling compared with the HapMap. The observed homozygous deletion near 190.845Mb of chromosome 4 corresponds to two annotated segmental duplications (chr4:190,844,046–190,845,646 and chr4:190,845,653–190,847,295).
There is potential with mate-paired reads to resolve the phase of these inversions by linking them with SNPs that are identified within the sequenced tags. Among the 68 heterozygous large (>400 bp) nonoverlapping inversions, 55 (81%) are detected with mate-paired reads that contain SNPs compared with the reference sequence, and 49 (72%) of them contain a heterozygous SNP. 41 of the 49 inversions that are in phase with a heterozygous SNP have more than one set of mate-paired reads confirming the phasing. These preliminary data suggest the potential for resolving the phase of all types of structural variations by linking them to heterozygous SNPs.
Copy number variations
Through the analysis of changes in depth of coverage in windows, we observe 565 CNVs in the size range of 2–937 kb in the autosomes. The distribution of size ranges is shown in Figure 7A. We identify 116 out of the 179 CNV events detected with the Affymetrix genotyping array 6.0 (McCarroll and Altshuler 2007). These CNVs are plotted in Figure 7B with the SOLiD-called copy numbers on the x-axis and CGH-called copy numbers in various colors. We demonstrate that there is good agreement between the copy numbers called by the different technologies. When the CNV regions are compared with the deletions detected using our mate-pair clusters, 102 out of 409 one-copy regions are supported by heterozygous deletions, 51 out of 78 zero-copy regions are supported by homozygous deletions, and there are no one-copy regions overlapping with homozygous deletions or zero-copy regions overlapping with heterozygous deletions.
Figure 7.

Copy number variations detected with SOLiD mate-paired reads in NA18507. (A) The size distribution of CNVs detected with SOLiD mate-paired reads. (B) Overlap of copy numbers computed from normalized SOLiD coverage and from Affymetrix array CGH (aCGH) (McCarroll and Altshuler 2007). Colors indicate CNV calls from aCGH. On the top of the figure are the numbers of SOLiD CNV calls that overlap with aCGH data at each copy number.
Evaluation of sequencing strategies for genetic variation discovery
Whole-genome human sequencing with massively parallel platforms is poised to become the workhorse of population and medical genetic studies. However, little information exists on how to better design these studies and balance cost versus benefit of different sequencing strategies. Thus, we investigated the fraction of genetic variation discovery at relatively low levels of average sequence coverage: 2×, 4×, 8×, and 10×. Figure 8 shows the percent of the genome that meets the coverage requirements for SNPs and intra-read indels at various levels of average sequence coverage (panel A) as well as the actual number of these variants that are detected (panel B). Significant homozygous SNP discovery can be achieved at 2× with a significant increase at 4×, but only a modest increase at 8×. Determining heterozygote status requires more coverage, but at 8×, 84.3% of the genome is accessible to heterozygous SNP detection. The identification of small indels under the sequence read is fairly incomplete at these low coverage depths due to more stringent mapping requirements.
Figure 8.

Theoretical and actual detection of SNPs and indels at various levels of average sequence coverage. (A) The upper bound on the number of SNPs and intra-read indels that can be detected at various levels of coverage. This is calculated by assessing how much of the genome meets the coverage requirements for each type of variant, 2× coverage for homozygous SNPs, 4× coverage for heterozygous SNPs, and 6× coverage without considering the 3 bp on each end of the reads for intra-read indels. For small indels, two split reads are required to make a call, but due to the more restrictive manner of these calls, only about one in three reads (as found in simulations) can be used for this. (B) The actual number of SNPs and intra-read indels detected at various levels of average sequence coverage. (C) The number of insertions and deletions ≥200 bp detected between mate-paired reads at various average levels of sequence coverage.
To understand the benefits of deeper sequencing in identifying structural variants, we assessed the indels detected between mate-paired reads from sets of one, two, three, and four slides of long mate-paired data that amount to 2.2×, 4.0×, 5.6×, and 8.4× average sequence coverage, respectively. Figure 8C illustrates that the number of insertions and deletions rises steadily from 2.2× to 8.4× average coverage and indicates that further sequencing will enable more variants to be detected. While the full data set in this study is from 8.4× average sequence coverage of long mate-paired data, 37%, 61%, and 72% of the deletions ≥200 bp in the 8.4× data set are detected at 2.2×, 4.0×, and 5.6× average sequence coverage, respectively. The advantage of deeper sequencing is even larger for insertions in which 16%, 49%, and 68% of the insertions ≥200 bp in the 8.4× data set are detected at 2.2×, 4.0×, and 5.6× average sequence coverage, respectively.
In regard to the combination of mate-pair libraries and the choice of insert size, we investigated the contributions of multiple insert sizes to the power to detect and resolve large indels by simulation and comparison with our empirical data. Supplemental Figure S10 shows the theoretical and observed probability of detecting an insertion or a deletion with two different insert-sized libraries independently (green and blue lines) and combined (red line). Our results imply that while a larger insert size increases detection, the probability of resolving a breakpoint increases with a combined library approach such that the probability of detecting a break point with a 600-bp and a 2841-bp library is higher than using either library exclusively. Higher-resolution break point mapping is anticipated to greatly facilitate any PCR-based validation of next-generation results.
Diversity amongst human genomes
We compared the SNPs and structural variations identified in NA18507 to those found in the Venter (Levy et al. 2007), Watson (Wheeler et al. 2008), and YH (Wang et al. 2008) genomes. Supplemental Figure S11 demonstrates that >20% of the SNPs found in NA18507 are in each of the other genomes, while 20%–40% of the SNPs identified in each genome are unique to that genome. Fewer insertions, deletions, and inversions are in common amongst the four genomes, and a higher proportion of them are unique to the genome in which they are identified. While it is noteworthy to compare these data, it must be understood in the context of what is still yet to be uncovered in each genome. The percent of the NA18507 variants that are in common with the other genomes is a lower bound, while the number of variants that are unique to each genome is an upper bound. These values will certainly shift as more variation is uncovered in each of the genomes. Since it is likely that we have identified most of the SNPs that are present in NA18507, while structural variations are typically more difficult to identify than SNPs with current sequencing technologies and have not yet all been identified, it will be exciting to discover whether the tendency for structural variations to be more distinct to single genomes than SNPs will hold as more of the variations in each genome are revealed, or whether this is an artifact of the current state of detection.
Functional consequences of genetic variation in the Yoruba individual genome
To assess disease-relevant variations present in the NA18507 sequence as described previously in the literature, we used the disease variants as described in the Online Mendelian Inheritance in Man (OMIM) database (Hamosh et al. 2005; McKusick 2007), a database of gene–disease relationships. Here, we investigate only the amino acid allele variants from the OMIM database—a list of 9239 variants of amino acid and terminator mutations that we can position uniquely and with confidence onto the genomic sequence as described in the Methods. We then compare the NA18507 sequence variants with this list encompassing 2161 human genes found in OMIM as of August 2008. Within these 9239 mutations, Supplemental Table S4 shows the list of OMIM variants for which this individual is a carrier of the disease-related allele in the homozygous or heterozygous states. Based on the annotations in OMIM and after reviewing the corresponding literature, we further filtered the homozygous and heterozygous alleles into high, medium, and low reliability for annotation (Supplemental Table S5). In total, NA18507 carries five disease-relevant OMIM alleles with stronger evidence in the homozygous form. For all five of these, the sequence quality is sufficient to trust the zygosity assignment. We found an additional 10 for which the annotations in OMIM were inconclusive (“medium” reliability). The remaining three overlaps had contradictory or disease-irrelevant annotations (“low” reliability). Furthermore, NA18507 carries 49 OMIM alleles in the heterozygous state.
In reviewing the disease associations for the homozygous alleles, each association is with a common, multifactorial disease, including susceptibility to obesity, drug addiction, atopy, thrombocytosis, and bladder cancer, as well as the trait of slow acetylation. As expected, from this initial analysis it seems none of the many Mendelian disease-causing variants listed in OMIM are found in the NA18507 genome sequence in the homozygous state. In addition, in Supplemental Table S6, we list all variations in NA18507 that generate an in-frame stop codon in homozygous and heterozygous states and overlap a known SNP in dbSNP.
We studied the impact of large insertions and deletions (detected by the inter-read approach) on gene integrity, by looking for breakpoint regions that overlap with gene regions, defined as the transcription start site to the end of the mRNA transcript including all exons and introns of the gene. We detected 2477 potential gene disruption events (44.3% of the 5590 large indels) that fall within gene boundaries in the NA18507 genome sequence, disrupting a total number of 2015 unique human genes, including some that are disrupted by multiple events. Amongst these 2015 disrupted genes, 303 (15.0%) are contained in a curated collection of 3600 human disease genes (∼15% of all human genes) assembled using a previously published collection of 923 human disease genes (Jimenez-Sanchez et al. 2001), all genes listed in OMIM and in the Human Genome Mutation Database (HGMD v7.1; Stenson et al. 2003) as of August 2008, and further extended by a comprehensive and rigorous literature review (Supplemental Table S7). The number of observed events should be treated as an upper bound of the total number of gene overlaps since the boundaries for the large indels are wider than the actual events, so the structural variation is either occurring within the gene or in close proximity to it. In summary, we can see a trend for disruption events to cluster around genes, but no clear preference to cluster around disease genes. Further analysis of these disruption events along with an evaluation of whether an exon is disrupted is warranted.
We identify a number of gene pairs that appear to be fused by a structural variation. Based on the empirical distribution of clone lengths, and the positioning of the discordant mate-paired reads, we compute a confidence estimate on the gene fusion event (Bashir et al. 2008). Table 1 reports five events, with the probability of genomic fusion equal to one, that are supported by at least three distinct pairs of reads. Interestingly, all but one of the predicted fusions involve tandem duplicated genes. The distance between the pairs of duplicated fused genes ranges from 16 bp to 368 kb. Two of the gene pairs have been previously observed as chimeras in the literature: APOBEC3A–APOBEC3B (Kidd et al. 2007) and EMR2–CD97 (Chiu et al. 2008). The gene pair CTRB1–CTRB2 is caused by an inversion in which six of the mate-paired reads that detect the inversion also contain a SNP compared with the reference sequence and of which three of them contain a heterozygous SNP, providing the potential to resolve the phase of this fusion.
Table 1.
Predicted gene fusions created by structural variation events

Signature of purifying selection in the pattern of nonsynonymous mutations
We discovered 6131 nonsynonymous SNPs that are pre-annotated using PolyPhen for the expected degree of damage they will cause to the protein (Ramensky et al. 2002). 4912 of these nonsynonymous SNPs are annotated as “benign” (79.6%), 765 are “possibly damaging” (12.4%), and 454 are “probably damaging” (7.4%). This compares with the proportions of these categories among all 76,434 nonsynonymous SNPs in the PolyPhen database. Of annotated SNPs, 66.2% are “benign,” 18.9% are “possibly damaging,” and 14.9% are “probably damaging.” The nonsynonymous SNPs in this Yoruba sample are significantly less damaging than the full collection of dbSNP nonsynonymous SNPs (P < 10−5). The homozygous state is significantly underrepresented for “probably damaging” and “possibly damaging” alleles as compared with “benign” variants in this individual genome (Supplemental Table S8).
We investigated whether damaging SNPs were over- or underrepresented in certain protein classes. There are 986 proteins with annotated function in the PANTHER protein classification database (Thomas et al. 2003; http://www.pantherdb.org) containing possibly or probably damaging nsSNPs. When comparing with the distribution of proteins in PANTHER categories of the human proteome, we identify protein families significantly underrepresented for damaging SNPs (binomial test, P < 0.05 with Bonferroni correction), including nucleic acid binding proteins (P = 0.00012), ligases (P = 0.0053), transferases (P = 0.0063), transcription factors (P = 0.0086), and kinases (P = 0.084). Categories overrepresented for damaging SNPs include receptors (P = 0.0013) (especially G protein-coupled receptors, P = 10−9), extracellular matrix glycoproteins (P = 0.009), cell adhesion molecules (P = 0.03), and cytoskeletal protein (P = 0.07); as well as biological functions of genes including sensory perception (P = 10−11), specifically olfaction (P = 10−17), G protein-mediated signaling (P = 0.00057), and cell adhesion-mediated signaling (P = 0.054).
Discussion
Benefits of 2BE for error correction and SNP discovery
For human sequencing it is advantageous to have a substitution error rate that is substantially lower than the anticipated substitution polymorphism rate (10−3), such that any single read can be trusted for homozygous SNP detection (Venter et al. 2001). In absence of this level of accuracy one must build confidence based on the overlap of aligned reads. Due to Poisson sampling limitations of shotgun sequencing, this overlap usually requires at minimum threefold more reads per allele (Lander and Waterman 1988), and thus there is distinct value in higher accuracy read generation to maximize polymorphism detection per Gb of sequence generated.
The 2BE method introduced here provides in theory a 37.5-fold gain in sensitivity for detecting real SNPs next to raw measurement noise (see Supplemental material “Error Correction” for details and further discussion). We evaluated the raw sequencing error and the remaining error after 2BE correction for all of the runs in this study, and we indeed observe a >99.9% average accuracy (Supplemental Table S1). This gain in accuracy is reflected in the ability to call SNPs and in particular heterozygotes at relatively modest levels of average coverage (Figs. 3, 8).
One possible drawback to color space is that it requires two mismatches per SNP in a given read length. As a result, a 50-bp read with five SNPs is difficult to align to the human reference, as it consumes 10 color mismatches. Unless the alignment tools treat pairs of valid adjacent mismatches as a single mismatch, such highly polymorphic sequences can exhibit a reference bias with strict reference matching algorithms. Therefore, mate pairs are preferred when aligning to polymorphic regions such as the major histocompatibility complex (MHC) of the human genome, since one read can act as an anchor and more relaxed or gapped alignments can be performed with the polymorphic tag. Alternatively, four-frame dynamic programming alignment implemented in tools such as SHRiMP (Lee et al. 2008) can be utilized. A second traversable concern is de novo assembly with color space, discussed in the Supplemental Information “Error Correction.”
On the sequencing of hundreds to thousands of human genomes with short reads
Variation discovery is clearly coverage-dependent, asymptotic, and, in the case of SNPs, very sensitive to read error rate. An alternative for projects considering the sequencing of hundreds of genomes where cost is still a driving factor is to sequence these at a low sequence coverage (e.g., 4×–6×) at the expense of partial genetic variation discovery. For structural variation discovery by discordant mate-paired clones, the relevant parameter is clearly clone coverage, which is easier to achieve with larger inserts at equivalent sequencing cost. We have demonstrated the number of homozygous and heterozygous SNPs as well as intra- and inter-read indels that are detected at a variety of coverage levels and how these compare to what is detected with our full data set of ∼18× sequence coverage. We show that it is possible to physically phase clusters of alleles interconnected by mate-paired reads, providing a significant amount of information that should improve complete haplotype resolution by statistical methods. This information can be used as a guide when planning experiments to understand how much of the genome will be accessible to each type of variant given the average level of sequence coverage.
We believe these data support the recent priorities in building a more comprehensive human reference sequence that better captures these forms of variation, which whole-genome sequence assemblers may have condensed or undersurveyed in regions of the reference that were haploid-derived (BACs and PACs) (Gresham and Kruglyak 2008). Algorithms can improve variant detection substantially if they are informed of regions of common CNVs or structural variations.
Comparison with other personal genomes
It is tempting to compare the individual African genome reported here and the two previously reported genomes of European descent in terms of the patterns of genetic variation. Our expectation is to find more genetic variants in a heterozygous state in the African versus European genomes (Li et al. 2008). However, there are several factors that complicate this comparison. First, the levels of coverage between these studies vary significantly, in part due to the cost, but also due to length of the reads underlying sequencing technologies: 500–1000 bp in Levy et al. (2007), 200 bp in Wheeler et al. (2008), and 25–50 bp here. Clearly, shorter reads have more difficulty spanning different types of repeats in the human genome and could be more susceptible to chemistry biases due to local sequence composition (e.g., GC content). Some of these limitations can be overcome by deeper coverage (Levy et al. [2007] and Wheeler et al. [2008] 7×, Bentley et al. [2008] 30×, this study 18×), which at the same time renders the comparison challenging. In addition, the different sequencing platforms used in these studies have different error patterns and rates (0.01% for Sanger [Shendure and Ji 2008], 3% for pyrosequencing [Quinlan and Marth 2007], 0.1% for SOLiD, 1%–2% for Illumina [Hillier et al. 2008]), error rates for all platforms continue to improve, and these numbers represent an ephemeral comparison, which together with the shotgun sampling statistics and coverage biases results in different demands for calling SNPs and genotypes.
Furthermore, the power to detect other types of genetic variation varies widely between studies due to the different sequencing strategies used: In Venter, indels were detected by read overlap or comparisons to the reference genome that restricted discovery to the 1–100 bp range; in Wheeler, reads were aligned to the reference genome and thus restricted discovery of indels to much shorter than the read length; and in our case, we utilized read overlap with the reference in addition to mate-paired mapping analysis to infer larger inter-read indels, which is coverage-dependent and thus gave us power on the 100- to 100,000-bp range. Therefore, differences between these studies are more likely reflective of the power of the different methods involved rather than population of origin differences and simply reflect the ranges of unexplored variation by former studies.
On the signatures of natural selection and demography in a single genome
A question of considerable interest that could be addressed with data from individual genomes is the number of deleterious mutations per human genome. Based on population genetic theory, it has been suggested in the past that the number of lethal mutations per individual should be extraordinarily low (Morton et al. 1956). Lohmueller et al. (2008) recently addressed this question based on a sequencing survey of the exons of over 20,000 genes in 20 European and 19 African-American individuals. Their results suggest that the number of potentially damaging mutations per individual (as predicted by PolyPhen) is much higher than expected, on the order of several hundreds, and that the population of European descent has more probably damaging mutations than that of African descent, presumably due to the population bottleneck suffered by the former. Our results are consistent with these findings.
We demonstrate that in the genome of this Yoruba individual, over 50 mutations previously implicated with disease, as well as over 1500 putatively deleterious mutations (as predicted by PolyPhen) and 2000 possible gene disruptions events (300 in genes previously implicated with disease) are present. Potentially damaging SNPs are underrepresented in exons of genes with molecular functions that are essential for cell survival, but are overrepresented in exons of genes undergoing rapid evolution in human populations. Many of these biological functions, most notably olfaction and immunity, have previously been demonstrated to be overrepresented among gene variants that differ in frequency among human populations, suggesting at minimum a relaxation of purifying selection and perhaps directional selection and/or an increase in mutation rate. Our observation that both SNPs and indels tend to reside outside of exons, and that both disease susceptibility alleles and putatively deleterious mutations are present in a homozygous state less than expected, is consistent with the operation of purifying selection.
Conclusions
With the rapid improvements in next-generation sequencing, at the time of this writing, the data for this study could be generated in just one to two 30- to 50-Gb runs from a SOLiD instrument at an estimated reagent cost of under $30,000. The time to analyze such large data sets is not keeping pace with these increases in data generation, and we anticipate much pioneering work ahead on whole-genome sequence analysis. We have placed many of the provisional analysis tools as open source tools on the web and encourage critique and improvement to this software (http://solidsoftwaretools.com).
Previous studies have considered mostly SNPs as sources of deleterious mutations; however, it is becoming clear that structural variation can have functional implication in gene integrity and function (Kidd et al. 2008). Here, we are able to complete the landscape of potentially deleterious variation by considering insertion and deletion events that damage genes and/or elicit potential gene fusions. These results suggest that it is important to consider structural variation in determining the potential disease alleles in genome and population studies.
It seems that even in a single human genome, the signatures of natural selection and human demography are ever present, and that the exploration of personal genomes has significant potential to be of importance in healthcare and personalized medicine. Our studies provide guidance for future exploration of human genetic variation with ultra-high-throughput short-read sequencing technologies such as SOLiD and confirm that accuracy is an important factor that interplays with throughput in determining the cost-effectiveness of the new sequencing methods in whole human resequencing. As with the initial sequencing of the human genome, it appears that longer-range mate pairs continue to provide structure and phasing information of significant value to understand personal genomes.
Methods
DNA sample
We sequenced the genome of the Yoruba sample NA18507, obtained from the Coriell Institute. This sample has been previously consented for genomic research by the International HapMap Project (Frazer et al. 2007).
Sequencing
We sequenced the genomic DNA of the sample using a combination of mate-paired libraries and fragment libraries with the Applied Biosystems SOLiD System analyzer according to the manufacturers' instructions. Two different methods for generating mate-paired libraries were utilized: TypeIII-generated libraries and nick translation libraries.
TypeIII libraries
Briefly, using the method described by Smith et al. (2006), we generated mate-paired libraries with the TypeIII restriction endonuclease EcoP15I (Applied Biosystems SOLiD Library Preparation Guide).
Nick translation libraries
To generate paired 50-base tags, the EcoP15I cap adaptors were left dephosphorylated so that circularization of the target DNA left a nick on the 3′ ends of the internal adaptor. These nicks were bidirectionally extended into the insert DNA using a timed nick translation reaction. Tags were liberated with S1 nuclease, end repaired with the Epicentre Endit kit, and varied in size from 50 to 75 bp per tag. All libraries were primer ligated with T4 DNA ligase (Ambion) and utilized identical adaptors (P1 and P2, Applied Biosystems SOLiD Library Oligos #4392456) for emulsion PCR (Applied Biosystems Long Mate Pair Library Protocol).
Fragment libraries
Additionally, we generated sheared “fragment” libraries that were sequenced as unidirectional reads. Briefly, fragment libraries were generated by shearing genomic DNA to a 60- to 90-bp range using various shearing methods (DNase I, Nebulization, and adaptive focused acoustic bombardment with a Covaris S2) and end repairing the DNA.
Emulsion PCR was performed according to Dressman et al. (2003) with a few minor modifications (Supplement Emulsion Methods). Since limited dilution of DNA is utilized to produce clonal bead amplification, 70%–80% of the beads in any given emulsion are unamplified beads. An enrichment step is performed to select for the templated beads and provide a higher number of sequence-generating features per run. Enrichment of amplified beads was performed as previously described (Shendure et al. 2005) with a few modifications (supplement enrichment). Once emulsions are broken, the beads are enriched, end modified, and deposited on a microscope slide ready for SOLiD sequencing (supplement end modification and deposition).
Ligation sequencing is performed in five different frames of sequencing. As a result, five different 5′-phosphorylated primers that are each offset by one base with respect to each other are used. The detection probes have a cleavable phosphorothiolate linkage fixed between the fifth and sixth base such that sequencing with one primer generates partial dinucleotide information in 5-base increments. Primer 1 will survey dinucleotides 1,2 and 6,7 and 11,12 and so on to bases 46 and 47. Primer 2 will survey dinucleotides 0,1 and 5,6 and 10,11…45,46. Primers 3, 4, and 5 will be nested more than two bases into the known adaptor sequence and thus do not require their first ligation cycle to be imaged (Supplemental Fig. S1).
SOLiD sequence alignment
The ABI SOLiD alignment tool mapreads, translates the reference sequence to dibase encoding (color space), and aligns the reads in color space. The program guarantees finding all alignments between a read and the reference sequence with up to M mismatches (a user-specified parameter). It uses multiple spaced seeds (discontinuous word patterns) to achieve a rapid running time (Z Zhang, AP Blanchard, HE Peckham, J Ni, FM De La Vega, A Siddiqui, KJ McKernan, and E Spier, in prep.). Reads that align in only one location in the color space reference with up to the given number of mismatches are referred to as uniquely aligned.
Representation of reads in terms of GC content
The percent GC content of the human genome is calculated for each 250-bp window. The average coverage depths of the windows are grouped by associated GC content and ranked within the groups. The mean of each group defines a Poisson distribution, and the tenth and 90th centiles are compared with those of actual data.
SNP identification
Single nucleotide polymorphisms are initially identified using the SOLiD Consensus Calling algorithm. SNPs are called by a consensus of valid adjacent two-base encoded mismatches. The confidence of each base call is determined by the type of call in color space, the position in the read, and the 6-mer base space context in which the base call occurs, and this confidence is used to weight the contribution of each set of adjacent base calls to the consensus call. The SNPs are further filtered to eliminate all variants with coverage greater than three times the mean, variants amongst three SNPs called in a 10-bp window, and variants within 15 bp of an intra-read indel that we identify in this analysis.
We also present SNPs called with another SOLiD SNP detection algorithm called diBayes. diBayes is a Bayesian algorithm that includes color space error detection (FCL Hyland, S Tang, O Sakarya, X Xing, and FM De La Vega, in prep.), an error model that uses probe and positional errors as well as color quality values, and the prior probability of population heterozygosity, in a framework similar to that of PolyBayes (Marth et al. 1999).
SNP validation
To confirm our findings, we randomly selected 333 of our novel autosomal SNP calls (those not found in dbSNP 129) for validation using the SNPlex Genotyping System (Tobler et al. 2005). There were 34 assay failures, which left a total of 299 successful assays in eight multiplex pools. The genotyping was performed as described (Tobler et al. 2005) on a panel of 46 Coriell DNA samples (22 African-American and 24 Caucasian samples) to populate genotype clusters and the Yoruban DNA sample NA10859.
Identification of small insertions and deletions under the sequencing tag
Indels can be detected within the actual sequencing reads (intra-read indels) or by observing the expected clone sizes to stretch or compress (inter-read indels). Intra-read indels have the benefit of single-base resolution of the variation while inter-read indels are less precise on the coordinate of the event. We surveyed indels with one end anchored (OEA) mate pairs (Kidd et al. 2008) in order to increase the accuracy since the search range for the unanchored tag is drastically reduced compared with searching the entire human genome.
Small insertions (up to 3 bp) and deletions (up to 11 bp)
Using mate-paired libraries, we realign our OEA pairs using the anchored pair as a seed and perform a more aggressive alignment with the other tag in a several-kilobase window (depending on the insert size of the library) from the anchored mate. Using the unanchored tag we align both ends of the read until the maximum number of two mismatches for 2 × 25-mers or five mismatches for 2 × 50-mers occur. Disallowing for indels one or two bases of either end of the read (not including the first base of the read), we identify if we are able to piece together both ends only allowing for a single gap of up to 3 bp inserted (present in read but not in reference) or up to 11 bp deleted. Furthermore, we identify a gapped alignment if the above joining can be done with the fewest (up to a maximum of two for 2 × 25 and five for 2 × 50) number of mismatches and identify the location of it by where this joining occurs. The algorithm considers only unanchored tags in which only this gapped alignment can be found and condenses two or more nonredundant alignments allowing up to four candidates to be within five bases and unlimited candidates within two bases of another. It also requires that three-quarters of the supporting reads have a consistent indel size and that, for cases with only two supporting nonredundant reads, the indels are on average greater than 9.1 bases from the end of the read. Ambiguity of this location is common but is reduced by checking the color space compatibility of the sequences that the gap traverses.
We use fragment libraries to provide additional evidence to identify indel candidates. We use the same algorithm with the fragment data, except that in this case the first and last 20 bases of the read are matched against the genome allowing for one mismatch and the window locations are 40 bases upstream to 80 bases downstream from the match location. Only fragment reads that do not align ungapped and align uniquely to the genome with a gap are considered for indel detection. Using the gapped alignment procedure described above with OEA pairs, we find gapped alignments by disallowing evidences that are within nine bases for insertions and 12 bases for deletions of the ends of the reads and allowing a maximum of three mismatches in the read.
Medium insertions (up to 19 bp) and deletions (up to 500 bp)
We are able to take advantage of the longer reads in the 2 × 50 mate pairs to search for intra-read insertions up to 19 bp and deletions up to 500 bp. With these longer tag lengths, we disallow evidences that have gapped deletions within 19 bases of either end of the read and align both ends of the read until the maximum number of five mismatches occurs. For gapped insertions, we disallow 13 bases, but allow up to five mismatches for sizes four to 14, and three mismatches for sizes 15–19. For medium insertions, we condense these results to call candidates in the same manner as small indels. For medium deletions, however, we do not require that the indel size is the same in three-quarters of the supporting molecules.
Large structural variation inference
To search for larger insertions or deletions (inter-read indels) from 100 bp to 100 kb we evaluate the average pairing distance across the genome and look for pairs that significantly deviate from the expected insert size (Tuzun et al. 2005). We harness the power of high clone coverage to enable us to detect smaller indels (<1 kb) with a high level of statistical significance that have been previously undetectable with mate-paired distances. A look-up table is created in which the amount that the clones must be deviated to achieve one standard deviation of significance is the standard error at each level of clone coverage. SE = SD/ 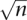 in which SD is the standard deviation of all of the normal clones in the library and n is the number of clones. This produces an asymptotic curve in which the minimum size of detectable indels at a given level of significance drops rapidly as the clone coverage increases. We use the look-up table to determine the significance of the deviation in average insert size at each position in the genome. Regions of the genome that are significantly deviated are selected as candidate indels, and hierarchical clustering is used to segregate the clones into groups in which the difference in the sizes of all clones in a group is less than the range of normal insert sizes of the given libraries. All clusters with less than two clones are removed and the candidates are assessed to determine if there is a homozygous or heterozygous population of deviated insert sizes. Any candidate indels with more than two populations with at least two clones in each are removed from consideration. All clones deviated by ≥100 kb are discarded.
in which SD is the standard deviation of all of the normal clones in the library and n is the number of clones. This produces an asymptotic curve in which the minimum size of detectable indels at a given level of significance drops rapidly as the clone coverage increases. We use the look-up table to determine the significance of the deviation in average insert size at each position in the genome. Regions of the genome that are significantly deviated are selected as candidate indels, and hierarchical clustering is used to segregate the clones into groups in which the difference in the sizes of all clones in a group is less than the range of normal insert sizes of the given libraries. All clusters with less than two clones are removed and the candidates are assessed to determine if there is a homozygous or heterozygous population of deviated insert sizes. Any candidate indels with more than two populations with at least two clones in each are removed from consideration. All clones deviated by ≥100 kb are discarded.
Clones from various libraries with various insert sizes contribute to a single indel call by combining the probabilities associated with the clones from each library (supplement inter-read insertions and deletions). We limited the study of these structural variations to the longer tag mate-paired libraries. The clones used to detect the large indels are limited to those in which both tags place uniquely against the reference sequence allowing up to five mismatches per 50-bp tag and in which the number of mismatches in both tags sum to five or less.
Inversions
An inversion is defined by its two breakpoints. The number of mate pairs with one flipped end supporting the occurrence of the starting or ending inversion breakpoint is counted for each base pair. The genomic ranges corresponding to local peaks of these counts, if above a score threshold, are called as candidate breakpoint ranges. To define an inversion, its starting and ending breakpoints are paired up only if they are the reciprocal nearest neighbor of each other in the correct order. The score for such an inversion is the harmonic mean of its two breakpoints. Finally, each breakpoint range is scanned for coverage of normal mate pairs to identify a sub-range with the lowest normal mate-paired coverage as the most probable location of a breakpoint and to differentiate homozygous inversions from heterozygous ones.
Copy number variations
Copy number variations are analyzed using a hidden Markov model on variable-length windows. The mappability of the genome is calculated for a given tag length and number of mismatches by matching a uniformly fragmented genome to itself. Subsequently, the per-base count of sequence coverage is summed by sliding windows (the sizes of which are varied to keep the sum of the corresponding mappable coverage constant). GC normalization is calculated using the empirical distribution of coverage as a function of GC content, normalizing observed coverage to adjust for GC content. A hidden Markov model is used for segmentation, and a final set of filters is used to remove CNVs at segments with low mappability or small size.
SNP annotation analysis
Amino acid variants, found in the allele variant list in each OMIM entry, were transferred onto the reference genome using a battery of computational methods described elsewhere (DK Hendrix, F Salas, AR MacBride, and MG Reese, in prep.). In short, OMIM alleles were aligned to a translated genomic sequence, reverse-translated and transcribed, and each mutation was located as a single base change in the genomic sequence using Golden Path coordinates (Kent et al. 2002), creating a whole-genome map of OMIM in genomic coordinates. The results of this pipeline are 9239 variants of amino acid and terminator mutations within OMIM that we can position uniquely and with confidence onto the genomic sequence.
PolyPhen is a tool to predict the possible impact of an amino acid substitution on the structure and function of a human protein, using sequence, phylogenetic, and structural information characterizing the substitution (Ramensky et al. 2002). PolyPhen mapped dbSNP SNPs to the protein identifier from the SWALL database (SWISS-PROT). We compared the nonsynonymous SNPs we detected against the predicted impact of 76,434 nonsynonymous SNPs in dbSNP build 126. We found 2892 nonsynonymous SNPs in this Yoruba sample that have a PolyPhen annotation.
Prediction and validation of gene fusions
The gene fusion prediction was performed as described in Bashir et al. (2008) using mate-paired reads that match the human genome with zero mismatches and in only one location. The mate-paired data used in this analysis consist of all of the 2 × 25 data sets (see Supplemental Table S1) as well as a small sample of 2 × 50 mate-paired data that are available at the NCBI Short Read Archive via the accession no. SRA000272 under the slide name Florence_20080201_1. The constraints imposed by multiple, spanning reads were used to reduce uncertainty in breakpoint location, and the probability of any particular breakpoint was evaluated by additionally considering the insert size distribution of each mapped read. Six of the predicted breakpoints (four of which resulted from inversions, one from a deletion, and one from an undefined rearrangement) have a breakpoint probability equal to one and are supported by at least three pairs of reads. Five of these breakpoints (the four inversions and one deletion) were selected for further validation.
The regions predicted to contain the fusion genes were PCR-amplified from genomic DNA and then sequenced by conventional Sanger sequencing (Agencourt Bioscience) (supplement Sanger confirmation of gene fusions). Assembly of these sequences and alignment to the human genome (hg18) either identified the precise fusion point, or, in cases of high sequence homology between the fused genes, localized the breakpoint to within 100 bp. We Sanger sequenced both fused and reference alleles to validate possible heterozygous rearrangements. Four of the five predicted gene fusions have been confirmed by Sanger sequencing.
All analysis algorithms are freely available in open source format at http://solidsoftwaretools.com. The variants identified in this study are available at http://solidsoftwaretools.com/gf/project/yoruban.
Acknowledgments
We thank Jason Warner, Lynne Apone, Ali Aslam, Deyra Rodriguez, and Chunlin Xiao for preliminary feasibility studies for the project; Ryan Koehler for early algorithmic development for CNV analysis; Brian Tuch for a ns-SNP annotation script; Charles Scafe for data mining; and Aaron Kitzmiller for making the variant lists publicly available. We thank Eugene Spier and Michael Wenz for valuable advice and Roger Canales for assistance in bioinformatics tool release. This work was supported in part by NIH grants HG004120 to E.E.E., HG002993 to M.G.R., and NIH R01 HG004962-01 to V.B. and A.B.
Footnotes
[Supplemental material is available online at http://www.genome.org. The data from this paper have been submitted to the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA000272. The variants identified in this study are available at http://solidsoftwaretools.com/gf/project/yoruban.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.091868.109.
References
- Bashir A, Volik S, Collins C, Bafna V, Raphael BJ. Evaluation of paired-end sequencing strategies for detection of genome rearrangements in cancer. PLoS Comput Biol. 2008;4:e1000051. doi: 10.1371/journal.pcbi.1000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braslavsky I, Hebert B, Kartalov E, Quake SR. Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci. 2003;100:3960–3964. doi: 10.1073/pnas.0230489100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 2000;18:630–634. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu PL, Ng BH, Chang GW, Gordon S, Lin HH. Putative alternative trans-splicing of leukocyte adhesion-GPCR pre-mRNAs generates functional chimeric receptors. FEBS Lett. 2008;582:792–798. doi: 10.1016/j.febslet.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci. 2003;100:8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Gresham D, Kruglyak L. Rise of the machines. PLoS Genet. 2008;4:e1000134. doi: 10.1371/journal.pgen.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- Ju J, Kim DH, Bi L, Meng Q, Bai X, Li Z, Li X, Marma MS, Shi S, Wu J, et al. Four-color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators. Proc Natl Acad Sci. 2006;103:19635–19640. doi: 10.1073/pnas.0609513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. DNA sequencing. A plan to capture human diversity in 1000 genomes. Science. 2008;319:395. doi: 10.1126/science.319.5862.395. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007;3:e63. doi: 10.1371/journal.pgen.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Waterman MS. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics. 1988;2:231–239. doi: 10.1016/0888-7543(88)90007-9. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lee S, Cheran E, Brudno M. A robust framework for detecting structural variations in a genome. Bioinformatics. 2008;24:i59–i67. doi: 10.1093/bioinformatics/btn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Indap AR, Schmidt S, Boyko AR, Hernandez RD, Hubisz MJ, Sninsky JJ, White TJ, Sunyaev SR, Nielsen R, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth GT, Korf I, Yandell MD, Yeh RT, Gu Z, Zakeri H, Stitziel NO, Hillier L, Kwok PY, Gish WR. A general approach to single-nucleotide polymorphism discovery. Nat Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39:S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick VA. National Library of Medicine; Bethesda, MD: 2007. Online Mendelian Inheritance in Man, OMIM (TM). McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/omim/. [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NE, Crow JF, Muller HJ. An estimate of the mutational damage in man from data on consanguineous marriages. Proc Natl Acad Sci. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Marth GT. Primer-site SNPs mask mutations. Nat Methods. 2007;4:192. doi: 10.1038/nmeth0307-192. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaghi M, Nygren M, Lundeberg J, Nyren P. Analyses of secondary structures in DNA by pyrosequencing. Anal Biochem. 1999;267:65–71. doi: 10.1006/abio.1998.2978. [DOI] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- Smith D, Malek J, Mckernan K. Methods for producing a paired tag from a nucleic acid sequence and methods of use thereof. EP Patent 1,682,680. 2006. http://www.wipo.int/pctdb/en/wo.jsp?wo=2005042781.
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler AR, Short S, Andersen MR, Paner TM, Briggs JC, Lambert SM, Wu PP, Wang Y, Spoonde AY, Koehler RT, et al. The SNPlex genotyping system: A flexible and scalable platform for SNP genotyping. J Biomol Tech. 2005;16:398–406. [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–1063. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Guo Y, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch RA, Lazaruk K, Haque KA, Hyland F, Xiao N, Wronka L, Burdett L, Chanock SJ, Ingber D, De La Vega FM, et al. Validation of the performance of a comprehensive genotyping assay panel of single nucleotide polymorphisms in drug metabolism enzyme genes. Hum Mutat. 2008;29:750–756. doi: 10.1002/humu.20703. [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]