

Abstract
We report studies of six individuals with marked elevations of cystathionine in plasma and/or urine. Studies of CTH, the gene that encodes cystathionine γ-lyase, revealed the presence among these individuals of either homozygous or compound heterozygous forms of a novel large deletion, p.Gly57_Gln196del, two novel missense mutations, c.589C>T (p.Arg197Cys) and c.932C>T (p.Thr311Ile), and one previously reported alteration, c.200C>T (p.Thr67Ile). Another novel missense mutation, c.185G>T (p.Arg62His), was found in heterozygous form in three mildly hypercystathioninemic members of a Taiwanese family. In one severely hypercystathioninemic individual no CTH mutation was found. Brief clinical histories of the cystathioninemic/cystathioninuric patients are presented. Most of the novel mutations were expressed and the CTH activities of the mutant proteins determined. The crystal structure of the human enzyme, hCTH, and the evidence available as to the effects of the mutations in question, as well as those of the previously reported p.Gln240Glu, on protein structure, enzymatic activity, and responsiveness to vitamin B6 administration are discussed. Among healthy Czech controls, 9.3% were homozygous for CTH c.1208G>T (p.Ser403Ile), previously found homozygously in 7.5% of Canadians for whom plasma total homocysteine (tHcy) had been measured. Compared to wild-type homozygotes, among the 55 Czech c.1208G>T (p.Ser403Ile) homozygotes a greater level of plasma cystathionine was found only after methionine loading. Three of the four individuals homozygous or compound heterozygous for inactivating CTH mutations had mild plasma tHcy elevations, perhaps indicating a cause-and-effect relationship. The experience with the present patients provides no evidence that severe loss of CTH activity is accompanied by adverse clinical effects.
Introduction
Cystathioninuria or hypercystathioninemia may be due to a variety of causes. Less marked elevations of urinary excretion and/or plasma concentrations occur with prematurity, deficiencies of vitamins B6, B12, or folic acid, neural tumors that produce cystathionine, a renal defect in the transport of this amino acid, or in genetically determined defects in the conversion of homocysteine to methionine. In the latter situation the excessive accumulation of homocysteine leads to an abnormally high rate of production of cystathionine by cystathionine β-synthase. These conditions have been reviewed in more detail elsewhere [1;2]. More marked cystathioninuria and hypercystathioninemia of genetic origin is caused by mutations that decrease the activity of cystathionine γ-lyase (CTH; EC 4.4.1.1)1, the enzyme that catalyzes the conversion of cystathionine to cysteine, ammonia, and 2-oxobutyrate. The initial patient in whom the metabolic findings strongly suggested a CTH defect was reported in 1959 by Harris and his colleagues [3], and by 1983 a survey of published cases listed briefly the metabolic and clinical findings in a total of 47 such cases with gross cystathioninuria thought to be of genetic origin [4]. However, among these cases the diagnosis had been confirmed in only ten by assay of CTH activity (using extracts of either liver or long-term lymphoid cell lines). Pyridoxal 5′-phosphate (PLP) is a cofactor for CTH. The effect of administration of oral pyridoxine (vitamin B6) on cystathionine excretion had been tested in 37 of the 47 individuals mentioned above. Most (33/37) showed marked decreases. The CTH activities in tissue extracts from B6-responsive individuals has been stimulated by the in vitro addition of PLP by between 1.3-fold and as much as 50-fold [4]. It was not until five years ago that the first report of identification of CTH mutations in cystathioninuric individuals was published. In four individuals two frame shift mutations, c.940_941delCT (p.Leu262ThrfsX20), and c.1220delC (p.Thr355IlefsX18) and two missense mutations, c.200C>T (p.Thr67Ile) and c.718C>G (p.Gln240Glu) were found, occurring in either homozygous or compound heterozygous forms [5]. The two missense CTH mutations have very recently been expressed in E coli and the mutant proteins purified and characterized with regard to their kinetic properties, the amounts of PLP they bind, and the effects of preincubation with PLP upon enzyme activity [6]. In the present paper we report clinical, metabolic, and molecular genetic studies of several additional cystathioninuric persons, among whose CTH genes three novel missense mutations, one of the previously reported alterations, and a novel large deletion have been identified. We have also further characterized in a large group of Czech individuals the effects of a CTH polymorphism previously studied in Canadian individuals and found to cause mild elevations of plasma total homocysteine (tHcy) [7].
Methods
Terminology
DNA and protein sequence variants are described as recommended by the Human Genome Variation Society (http://www.hgvs.org/mutnomen/recs-DNA.html and http://www.hgvs.org/mutnomen/recs-prot.html). The accession number for the CTH genomic sequence is AL354872. The single nucleotide polymorphism in CTH, termed in this paper c.1208G>T (p.Ser403Ile), was originally described as c.1364G>T (p.Ser403Ile) [7]. The nucleotide number has been changed here to agree with the current numbering convention. This polymorphism has also been termed SNP rs1021737 by Li and coauthors [8]
Immortalization of lymphocytes
This step was carried out by Dr William M. Sugden using plasmids derived from the Epstein-Barr virus (see http://www.warf.org/technologies.jsp?casecode=P93123US).
Cystathionine (Cysta), total cysteine (tCys), and total homocysteine (tHcy)
These compounds were assayed either by amino acid chromatography, high performance liquid chromatography [9] or by capillary gas chromatography-mass spectrometry [1; 10; 11]
Identification of genomic DNA variants in hCTH
In order to detect the full range of the mutations found among the present group of patients in was necessary to utilize several different methods. The reasons for this requirement amd the methods used are set forth in “Results and Discussion”. Amplification of coding regions and intron-exon boundaries of the CTH gene was done using primers that have been previously described [5]. The primer sequences were designed to anneal at a single temperature, however exon 10 required slight modification. For exons 1–9, 11, and 12, amplification conditions were 94 °C for 5 min, followed by 30 cycles comprised of 30 seconds each of denaturing at 94 °C, annealing at 57 °C, and extension at 72 °C. A final extension step was performed at 72 °C for 10 min. Genomic DNA (200 ng) was used for a 60 μL PCR containing 1X PCR Reaction Buffer (Invitrogen, Burlington, ON, Canada), 50 mM MgCl2, 0.2 mM dNTPs, 0.4 μM of each primer, and 1.0 U Taq polymerase (Invitrogen, Burlington, ON, Canada). For exon 10, the above reaction was used with the addition of 3% dimethylsulfoxide, and the annealing temperature was increased to 62 °C. Amplification products were run on 2% agarose gels and purified with the QIAquick gel extraction kit (QIAGEN, Valencia, CA). Purified PCR fragments were directly sequenced in both directions on an ABI 3730 Automated DNA Sequencer (Applied Biosystems, Mississauga, ON, Canada). DNA sequences were aligned with ABI Sequence Navigator software. Mutations were confirmed by restriction digestion of DNA from 100 Caucasian normal controls. For the c.589C>T (p.Arg197Cys) mutation in exon 6, control samples were PCR amplified and digested using HaeII (New England Biolabs, Pickering, ON, Canada) in NEB buffer 4. For the c.932C>T (p.Thr311Ile) mutation in exon 9, control samples were digested with BsrGI (New England Biolabs, Pickering, ON, Canada) in NEB buffer 2, and for the c.200C>T (Thr67Ile) mutation in exon 2 samples were PCR amplified and digested using BfaI (New England Biolabs, Pickering, ON, Canada) in NEB buffer 4. Samples were incubated overnight at 37°C. Mutations in exons 2 and 6 were analyzed by electrophoresis on 2.5% agarose, while exon 9 was analyzed on 8% polyacrylamide. Resultant bands were visualized by ethidium bromide staining and UV light exposure.
Detection of CTH intron 1–5 deletion, c.168+1917_589-1848del
This deletion was detected by PCR using primers # 700 and #701 (Supplemental Table 1). The forward primer is located in intron 1 (18843-19064); the reverse primer, in intron 5 (33671-33691). The annealing temperature is 60°C. 5% DMSO is used in the PCR reaction. The PCR product size from carriers of the deletion is 401 bp. Only carriers will have this product amplified (normal subjects have >14,000 bp of genomic DNA between those primers).
Identification of mutations using RT-PCR
Each RNA was made from one patient fibroblast roller bottle pellet using the PureLink Micro-to-Midi Total RNA Purification System (Invitrogen 12183-018, Carlsbad, CA). Following RNA purification, cDNA was made using the SuperScript First Strand kit (Invitrogen 12371-019; Carlsbad, CA). PCR was performed using the cDNA with a combination of the following primers: sense primers, #640 or #644; anti-sense primers, #641 or #645 (Supplemental Table 1). PCR was performed using either PfuTurbo DNA polymerase (Stratagene 600250, Cedar Creek, TX) or Taq DNA polymerase (New England Biolabs M0273S, Ipswich, MA). The PCR conditions used were 2 minutes at 94°C 1 cycle; 30 seconds at 94°C, 30 seconds at 52°C, 2 minutes at 72°C 35 cycles; 7 minutes at 72°C 1 cycle in the Stratagene Robocycler. After PCR, the product was purified using the Qiagen QIAquick PCR Purification kit (Qiagen Sciences 28104, MD) and/or by gel purification using the Qiagen QIAquick Gel Extraction kit (Qiagen Sciences 28704, MD). 50 ng of the purified PCR product along with 10 pmols of the primer used for PCR in a total of 17 μl was then sent for sequencing to the Colorado University Cancer Center DNA Sequencing & Analysis Core (CUCCC). The Vector NTI software (Invitrogen) was used to perform sequence alignments.
Genotyping of Czech controls
Three groups of individuals were selected from a larger group of healthy control subjects (described by Janošiková et al [12]), so that each group contained 55 members with genotypes c.1208 GG, GT, or TT. The DNAs of the 165 individuals so chosen were then screened for mutations c.168+1917_589-1848del, c.200C>T (p.Thr67Ile), c.589C>T (p.Arg197Cys), and c.932C>T (p.Thr311Ile) as described above.
Introduction of the mutations into two different expression plasmids
The construction of the vector pET22b+ (Novagen) expressing CTH in E.coli was described previously by Steegborn et al [13]. The vector pGEX6-P-1(GE Healthcare), which is used for expression of glutathione-S-transferase fusion proteins, was employed to prepare the GST-CTH fusion construct. Two complementary oligonucleotides, #621 and #622, with extensions coding for the restriction enzymes Apa I and XhoI, respectively, were synthesized (Supplemental Table 1). The CTH sequence was amplified from the template pET22b-CGL using these primers. The amplified DNA was digested with the two restriction enzymes and cloned into pGEX6-P-1. The vector pGEX6-P-1-CTH was then transformed in E.coli XL-1 cells.
Mutagenesis
Point mutations were introduced into the wild-type CTH that had been precloned into either expression vector pGEX 6-P-1 or pET22b+ by site-directed mutagenesis using the Quikchange™ kit (Stratagene) and primers containing the desired mutation. The DNA sequence was confirmed by sequencing at CUCCC.
Mutation primers
Primers for mutation c.200C>T were: #633 and #634; for mutation c.589C>T, #635 and #636; and for mutation c.932C>T, #637 and #638 (Supplemental Table 1).
Expression of recombinant human mutant CTH proteins in E. coli
The bacteria were cultured in 25 ml at 37°C in LB media supplemented with ampicillin at 100 μg/ml. CTH expression was induced with 1 mM IPTG when OD 600 of ~0.5 was reached and the expression was carried out for 3 hours.
Crude lysate preparation
The cells were harvested and resuspended in the lysis buffer containing 10 mM Tris-Cl, 150 mM NaCl, pH 8.0, 1 mM EDTA, 0.1 mM DTT, 1 μM PLP, and protease cocktail SIGMA (P8465) according to manufacturer’s suggestions. They were then sonicated at a 50% duty cycle, with an output 3–4 for 80 seconds in a 3000 Sonicator (Misonix, Inc.). The extracts were then spun at 14,000 g for 15 minutes. The pGEX 6-P-1 fusion protein product was cleaved with PreScission protease (GE Healthcare Life Sciences) overnight. Spin column (6-Tris Column; Biorad) was used to remove any free cysteine from the crude extract. Protein concentration was determined by the Lowry procedure [14].
Enzyme assays
The production of cysteine in crude extracts was assayed by a sensitive colorimetric reaction described by Gaitonde [15]. In this assay, an acid ninhydrin reagent reacts specifically with cysteine, which is a product of the CTH reaction, forming red color absorbing with a λmax of 560 nm. The standard assay was performed with 35 μl crude extract samples for 1 hour and contained 0.5 mg/ml BSA, 50 μM PLP, 1 mM DTT and 200 mM Bis-Tris Propane buffer pH 8.25 in 200 μl volume. The reaction was started by addition of the substrate, cystathionine. The reaction was terminated by removing a 50 μl aliquot and mixing it with 50 μl glacial acetic acid and 50 μl of the acidic ninhydrin reagent. After the contents were mixed thoroughly; the tube was closed and boiled for 10 min in a water bath. After boiling, the sample was cooled rapidly in an ice water bath and the contents were diluted by adding 850 μl of 95% EtOH. The A560 of the samples was measured and the time 0 blank was subtracted. One unit of activity is defined as the amount of CGL that catalyzes the formation of 1 μmol of cysteine in 1 hour at 37°C.
Crystallographic modeling of wild-type and mutant hCTH
Our analysis of hCTH mutations is based chiefly on the experimentally determined structure of hCTH-PLP presented in the Protein Data Bank (http://www.pdb.org/ (code 2NMP) [16] which best represents the physiological state of the enzyme. Our working model of the native CTH was formed from the C, D dimer taken from the 2NMP pdb file, the one better resolved in the experimental picture of the enzyme. Other important information was obtained from structures apo-hCTH (code 3ELP) and hCTH-PLP-PAG (code 3COG) [16] also determined by X-ray diffraction. For details see the relevant section in “Results and Discussion. Missing residues in terminal areas and hydrogens were added using programs PYMOL [17] and COOT [18] Minimization in the program AMBER-9 [19] was done under constraints that tethered all atoms of the main chain in their experimental positions well resolved in the diffraction experiment.
Patients
Many of the available metabolic and genetic data for the individuals covered in this paper (six with severe hypercystathioninemia and three members of a family with mild elevations of plasma cystathionine and only a heterozygous CTH mutation) are presented in Tables 1 and 2, with the subjects listed in descending order according to their most recent plasma cystathionine concentrations. The numbers designating these individuals are arbitrary, having been assigned by the laboratories in which initial genetic studies were performed. Detailed clinical histories are presented in Supplemental Material.
Table 1.
Chief clinical features of individuals with cystathionine elevations
| Patient (Gender) |
Cystathionine elevation found |
Age (yr) for last information |
Motor/ developmental retardation |
Mental capacity |
Other clinical abnormalities |
B6 responsive |
|
|---|---|---|---|---|---|---|---|
| Age | Reason investigated | ||||||
| 3183 (F) | 37 yr | mild hyperhomocysteinemia | 38 | no | excellent | venous thrombosis | no |
| 2930 (F) | 3 wk | routine newborn screen | 37 | no | normal | none; 4 normal children | yes |
| 2927 (F) | 2 yr | psychomotor retardation | 4 | yes | retarded | brain MRI abnormal | yes |
| 2929 (M) | 4 mo | born of father-daughter incest | 7 | yes | retarded | none | unknown |
| 2928 (F) | 2 yr | seizures; retardation | 25 | yes | retarded | mild hepatomegaly | yes |
| 3051 (F) | ?40–50 yr | chemical insensitivities | 63 | no | normal | episodic cognitive impairment | yes |
| S 15 child* (F) | 3 yr | mild hypermethioninemia | 7.5 | no | normal | none | no |
| Father S* (M) | 42 yr | family screen | 45 | no | normal | none | unknown |
| S 4 child*(F) | 13 yr | family screen | 17 | no | normal | none | unknown |
See also Table 3
Table 2.
Plasma concentrations of sulfur amino acids in individuals with different CTH genotypes
| Individual/ Nationality |
n | tHcy (μmol/l) |
Cysta (μmol/l) |
tCys (μmol/l) |
tCys/Cysta | CTH genotype cDNA |
Deduced amino acid change(s) |
|---|---|---|---|---|---|---|---|
| Homozygotes/compound heterozygotes for inactivating mutations | |||||||
| #3183 – Czech | 1 | 34.0 | 22.3 | 236 | 11 | * c.[169_588del]+[169_588del] | p.[Gly57_Gln196del]+ [Gly57_Gln196del] |
| #2930 – US | 1 | 18.2 | 11.8 | 246 | 21 | c.[932C>T]+[169_588del] | p.[Thr311Ile]+[Gly57_Gln196del]* |
| #2927 – Czech | 1 | 7.1 | (6)§ | 306 | 51 | c.[200C>T]+[200C>T] | p.[Thr67Ile]+[Thr67Ile] |
| #2929 – Czech | 1 | n.r.** | 5.4 | n.r. | n.r. | c.[589C>T]+[589C>T] | p.[Arg197Cys]+[Arg197Cys] |
| #2928 – Czech | 1 | 34.0 | 4.4 | 250 | 57 | c.[200C>T]+[169_588del] | p.[Thr67Ile]+[Gly57_Gln196del] |
| Individual with cystathioninuria of unknown origin | |||||||
| #3051 – US | 1 | 11.3 | 5.72 | 277 | 48 | No CTH mutation found | |
| Heterozygotes for inactivating mutations | |||||||
| Father of #2930 – US | 1 | 10.1 | 0.33 | 324 | 973 | c.[932C>T]+[=] | p.[Thr311Ile]+[=] |
| Mother of #2930 – US | 1 | 5.7 | 0.35 | 278 | 785 | c.[169_588del]+[=] | p.[Gly57_Gln196del]+[=] |
| Individual – Czech | 1 | 11.8 | 0.25 | 315 | 1251 | c.[169_588del]+[=] | p.[Gly57_Gln196del]+[=] |
| Individuals - Czechs (range) | 5‡ | 8.9 (7.1–12.5) | 0.50 (0.34–0.66) | 256 (234–320) | 601 (470–735) | c.[200C>T]+[=] | p.[Thr67Ile]+[=] |
| S family – Taiwanese (range) | 3‡‡ | 12.4 (5.9–20.0) | 1.29 (0.80–1.76) | 332 (214–443) | 263 | c.[185G>T]+[=] | p.[Arg62His]+[=] |
| Controls | |||||||
| Czechs (range) | 591† | 9.5 (4.4–47.6) | 0.17 (0.02–1.14) | 299 (142–499) | 1712 (233–14395) | ||
| Taiwanese (range) | 5‡‡ | 7.1 (4.5–10.8) | 0.18 (0.12–0.35) | 253 (210–336) | 1406 | ||
Abbreviations used and reference ranges (in parentheses): tHcy: total homocysteine (5.1–13.9 μM); Cysta: cystathionine (0.04–0.34 μM); tCys: total cysteine (203–369 μM; CTH: gene encoding cystathionine γ-lyase
The deletion in question (c.168+1917_589-1848del) starts in intron 1 and extends to intron 5, resulting in deletion of exons 2–5.
n.r. = not reported
Median values for these 5 individuals are listed.
Mean values for a father and two children from a Taiwanese family, each of whom is heterozygous for CTH p.Arg62His (see Table 3) are shown.
Czech controls were 591 apparently healthy individuals without a personal history of coronary artery disease, peripheral arterial disease, or stroke (described by Janosikova [12]). Median values for these controls are listed.
The concentration in parentheses was measured using an amino acid analyzer.
Taiwanese controls consist of the mother and 2 children of the S family who are not heterozygous for CTH p.Arg62His and 2 additional individuals. Mean values are listed.
Results and Discussion
In the following sections we discuss: (a) the identification of novel mutations and deletions in CTH among the patients reported upon in this paper; (b) the effects each of these novel mutations, as well as those previously characterized by Wang and Hegele [5] and Zhu et al [6] on enzyme activity (Fig. 2) and clinical responsiveness of persons carrying these mutations to dietary B6 supplementation; (c) the crystal structure of hCTH; and (d) the structural changes due to the mutations under discussion and their effects on the properties of the mutant proteins.
Figure 2.

Enzyme activities of wt and mutant human CTH expressed in E.coli. The enzymes were expressed and assayed as described in Methods.
(a) Identification of mutations and deletions and the genetic abnormalities discovered
PCR amplifications of the individual exons initially identified a previously described mutation, c.200C>T (p.Thr67Ile) in apparently homozygous form in patients #2927 and #2928 and two novel mutations: apparent homozygosity for c.589C>T (p.Arg197Cys) in patient #2929, and heterozygosity for c.932C>T (p.Thr311Ile) in patient #2930 (Table 2 and Supplemental Table 2). Since assay of the mutant enzyme carrying the p.Thr311Ile mutation yielded near normal activity (Fig. 2) we thought it unlikely that patient #2930 is in fact homozygous for the c.932C>T mutation as the genomic DNA sequencing suggested. Therefore, we decided to examine this patient’s RNA by performing RT-PCR. PCR amplification of this patient’s cDNA yielded two bands, one of the expected length of 1516 bp and a shorter one of ~1100 bp. DNA sequencing of the longer band confirmed the presence of the c.832C>T mutation while the shorter band had a sequence in which exons 2–5 were missing (Fig. 1A), suggesting that this patient has a large deletion at the genomic level or perhaps an unusual splicing mutation. In addition, using genomic DNA from patient #3183, repeated attempts to amplify exons 2 through 5 produced no products, strongly suggesting that a portion of the CTH gene was absent from the genome of this subject. We developed a strategy of walking primers to amplify genomic DNA regions spanning the breakpoint until we were able to sequence across the breakpoint. Patient #3183 was found to be homozygous for a 14.5 kb deletion spanning intron 1 to intron 5. The sequence for the deletion breakpoint is shown in Fig. 1B. The deletion eliminates exons 2 through 5 (see Fig. 1A) and results in a markedly truncated protein. We note that this deletion was found only because the RNA of patient #2930 had been examined. In the initial exon-by-exon sequencing heterozygote carriers of this deletion escaped detection because exons 2 to 5 yielded normal sequence from the other allele. These types of mutations have occasionally been causing problems in genomic DNA sequencing in other inherited diseases as well [26] (and unpublished results). In order to obtain accurate genotypes, patient DNA as well as RNA should be examined. Subsequently, specific primers were employed (see Materials and Methods) to directly screen for this deletion at a genomic level. All six patients and the parents of #2930 have been screened. The deletion was found in a homozygous form in patient #3183, in heterozygous form in patients #2930 and #2928, and in the mother of #2930 (Table 2 and Supplemental Table 2). It is interesting to note that an alignment of introns 1 and 5 of the CTH gene identified a region of ~85% similarity between these two introns (37 differences in 245 bp with no gaps introduced). Whether this extent of similarity accounts for the mechanism of recombination via unequal crossing over resulting in this deletion remains to be seen. In summary, coding changes in CTH have been found in five of the six severely cystathioninuric patients studied (Table 2 and Fig. 1). Human liver has been reported to contain two splice-variant forms of mRNA for CTH, a longer one with 85% amino acid homology to rat CTH, and a shorter one lacking 132 bases corresponding to exon 5. Enzyme activity is due to the longer mRNA, with expression of the shorter form being subject to post-transcriptional regulation [27;28]. We and others [13] have not observed a PCR product corresponding to the shorter mRNA. An enzyme missing 44 amino acid residues would result in a severe trimming of the PLP binding domain including several catalytically indispensable residues and would not be expected to be functional.
Figure 1.

CTH deletion. Panel A shows part of the cDNA sequence spanning the junction of exon 1 and 6 from patient #2930. The DNA was isolated from the shorter (approximately 1,100 bp) PCR band. Panel B shows details of the homozygous 14.5 kb deletion in patient #2930. The normal genomic structure of CTH is shown at the top of the panel with numbered horizontal bars corresponding to the exons. The electropherogram shows intron 1 genomic sequence directly adjacent to intron 5 genomic sequence, as indicated, with the transition break point indicated by the arrow. The predicted gene structure with exons 2–5 deleted is shown directly above the electropherogram. The precise number of deleted nucleotide base pairs (bp) is 14,448; the mutation name has been shortened to 14.5 kb deletion spanning CTH intron 1 to intron 5.
(b) Enzyme Activities of wild-type and mutant CTH enzymes
We have expressed wild-type human CTH and the three missense mutations in E.coli as recombinant proteins, using the pGEX system (Fig. 2). The expressions and activity measurements were repeated in the pET system with virtually identical results (data not shown), indicating that the activities of CTH mutants expressed in the presence or absence of a fusion partner were similar and thus the potential help of GST in folding of the mutant protein can be excluded.
(c) Structure of hCTH
Tetrameric structure
The molecular structure of hCTH in atomic resolution has been determined by X-ray diffraction as the apo-form (apo-hCTH), as a complex with cofactor PLP (hCTH-PLP) and also as the form inhibited by DL-propargylglycine (hCTH-PLP-PAG) [16]. All structures provide readily interpretable maps of electron density in stable parts of the protein, and, together, the three structures [16] provide a consensus model of hCTH needed for analysis of the effects mutations. In all three cases hCTH forms very similar tetrameric structures with virtual symmetry 222 (D21). Two active sites are formed at the interface between the A and B subunits, and another two active sites at the interface between the C and D subunits (Fig. 3). Thus, the tetramer is composed of two virtually stand-alone dimers, (A, B) and (C, D), mutually related by a pseudo-two-fold axis. The crystal structures of apo-hCTH, hCTH-PLP and hCTH-PLP-PAG show that the intra-dimer interface is relatively small and that a substantial part of it is formed by the substrate binding cleft. However, upon unbinding of PLP the cleft undergoes large conformational changes (see legend of Fig. 4 for details) leading to instability of stand-alone dimers. This finding emphasizes the importance of the stacking of the dimer into the more stable tetramer complexes (Fig. 3). The large and robust inter-dimer interface contains no residues involved in conformational changes during the enzyme reactions, and thus provides stability to the enzyme. Therefore, it is not surprising that the tetrameric structure of hCTH is evidently preserved (at least under non-extreme conditions) in all experimentally observed forms. These conclusions are all in full agreement with the experimental results of Sun et al who observed only tetramers or non-functional monomers under a variety of conditions [16].
Figure 3.
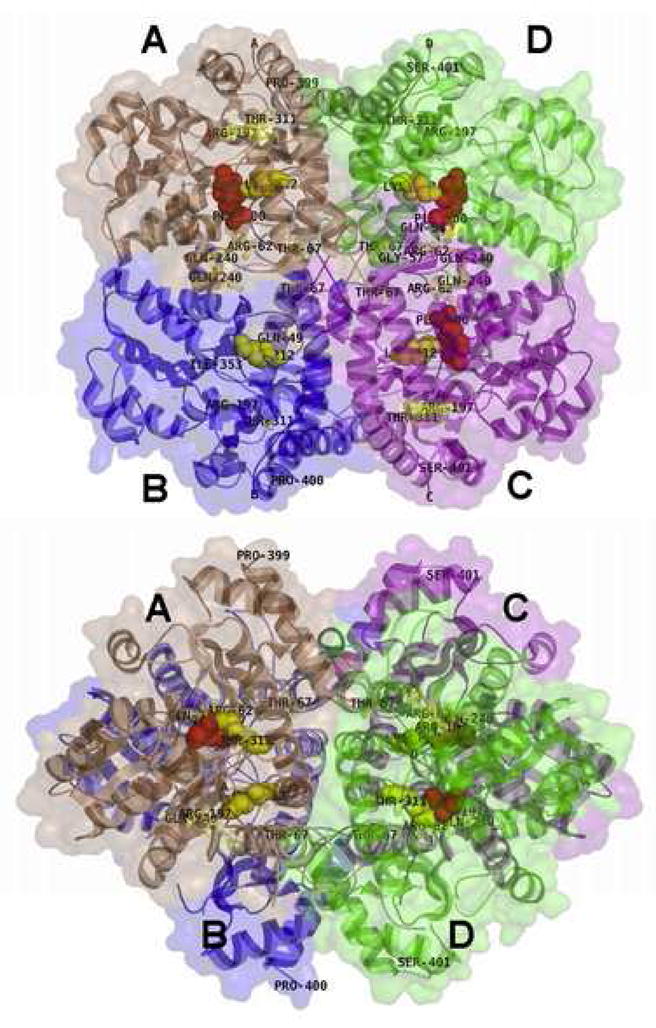
Front (a) and top (b) view of the hCTH-PLP tetramer (Protein Data Base code 2NMP). Two catalytic sites (red colored sticks) are shown on the interface between D and C subunits (green and cyan). The other two active sites lie at the interface between the A and B subunits (brown and blue), but, as detailed below, that of subunit B was not localized in electron density maps and is not shown. Mutated residues are indicated by yellow sticks. The interface between (A,B) and (D,C) dimers (vertical plane in both projections) serves to keep proper conformation of each dimer, stabilizing the face to face conformation of monomers in each dimer. The active sites in the refined models of subunits A and C are described with better accuracy than in the B and D subunits. In subunit D (green) 29 residues near the active site have several times higher displacement factors compared to those in subunits A and C. A substantial part of subunit B could not be localized in electron density maps, including the active site. Therefore no active site is shown for subunit B. In this paper, only the best resolved dimer CD and the active site with PLP in subunit C are discussed in detail. Drawn using the program PYMOL.
Figure 4.

Detailed view of the catalytic site formed between C and D subunits explaining the impact of mutated residues on stability of the active site of hCTH. The reaction site is formed upon PLP binding to Lys212 of the apo-form of hCTH. Loop Thr211-Met214 from subunit C (top right) moves from above, placing the phosphate group of PLP at its top, in contact with Tyr60 and Arg62 of subunit D. This is followed by flipping a second loop, Met110-Asn118, of subunit C (right-bottom) from below, placing Tyr114 parallel to PLP with its hydroxyl in contact with Arg62 of subunit D. This large reorganization leads to six strong, charge supported, hydrogen bonds. The figure shows also pathways from mutated residues to the catalytic site between cofactor PLP and Lys212 (top of the picture) and the importance of mutations p.T67I (top-left), p.R62H (center), and p.Q240E (left). Notice also that the active site is formed at the interface of two subunits, C (right side) and D (left side). Eight hydrogen bonds formed between these subunits fix the exact geometry of this site. Important stabilizing factors are the stacking of aromatic moieties (Tyr114 || PLP), the network of hydrogen bonds from the phosphate group, and the circle PLP-Lys212-Thr211-Arg60-PLP. The top-right corner of the figure shows a direct path from point mutation p.Thr311Ile to the reaction site between Lys212 and PLP. The nearby Cys310 might be the entry point for the effect of the p.Arg197Cys mutation if a putative S–S bond is formed between Cys197 and Cys310. For simplicity, water molecules present in the area are not shown in the figure. The lengths of important hydrogen bonds are given in angstrom units. Drawn using the program PYMOL [17].
Disordered ends of protein chains
A few residues at both the N and C terminals project into the solution, and thus are not localized in the final maps of electron density, undoubtedly because their conformations are not stabilized by any stable interactions with the bulk protein. The result is that the sequence SerMetGlnGluLysAspAlaSerSerGln- (1–10) at the N terminals of all four subunits is exposed to solvent without any definite conformation and are not present in the Protein Data Base [PDB] files. Similarly, at the C terminals, parts of the sequences GlySerHisSer (400–403), are not localized in the Protein Data Base files.
Active sites
The catalytic sites of hCTH-PLP are formed by the Lys212’s of the apo-enzyme binding PLP. Two loops, Thr211-Met214 and Met110-Asn118, on the surface of each subunit form a deep cavity with diameter of about 14 containing a reaction site with cofactor bound at the bottom. The ellipsoidal inlet to this cavity is quite narrow ~ 6×9 Å. A channel with diameter ~5 Å through which, during PLP binding and enzyme activity, water might pass runs from the bottom of the cavity to the opposite side of the enzyme, a total length of over 30 Å. The enzymatic reaction involves breaking the Lys212-PLP bond and thus is strongly influenced by the exact positions and orientations of residues in the cycle Thr60-Thr21-Lys212-PLP-Thr114.
Cofactor binding
In the ground state PLP is bound to the active site by Lys212, as confirmed by continuous density in the map of electron density of hCTH-PLP. The phosphate group of PLP from one subunit forms hydrogen bridges with Ser89, Gly90, and Leu91 of the same subunit (for clarity, not shown in Fig. 4), as well as with Tyr60 and Arg62 in the adjacent subunit (Fig. 4). Another important stabilization mechanism for PLP is that Tyr114 stacks parallel to the aromatic ring of the pyridoxal, forming a lid that locks the PLP in the active site cavity by π-π interactions between these two aromatic rings.
The crystal structures hCTH-PLP and hCTH-PLP-PAG indicate that the two active sites formed at the interface between C and D subunits are similar but not equivalent. The catalytic site with PLP in the C subunit has more stable geometry than the second catalytic site with PLP in the D subunit. Specifically, the PLP neighborhood in the C subunit has an average atomic displacement B-factor of ~ 24 Å2, whereas the neighborhood of PLP in the D subunit shows higher disorder (B-factors close to 64 Å2).
Binding of PLP (or both PLP and PAG) increases the structural stability of hCTH as shown by the facts that in the structures apo-hCTH, hCTH-PLP and hCTH-PLP-PAG, respectively, the mean atomic displacement factors, B, are 51, 25 and 17 Ǻ2, and the numbers of unidentified residues in tetramers decrease from 280 to 76 and 86 residues.
(d) Specific mutations
In the following sections we discuss, first, each of the CTH mutations identified among the present patients (doing so in the order in which these mutations are listed in Table 2); then, p.Gln240Glu, a mutation previously discovered by Wang and Hegele and studied by Zhu et al [5;6], but not found among our patients. In discussing these mutations it should be noted that experience has shown that point mutations often rebuild local interactions and local conformations, but very rarely cause significant shifts of atoms in the main chain. Therefore, we employed the tethering technique described under “Methods” so that in the structures of the mutant proteins to be discussed a majority of well resolved atoms from x-ray analyses stayed in the experimental electron density. Such an approach is expected to reveal trends of changes, but not definite structures of mutant enzymes.
p.Gly57_Gln196del
The large 14.5 kb deletion in the CTH gene leads to an in frame spliced RNA missing exons 2–5. Patient #3183, a homozygote for this large deletion, has excellent mental capability and, with the exception of a thrombotic episode during use of oral contraceptives, has generally enjoyed good health. This is noteworthy because inspection of the molecular structure shows that deletion of residues 57–196 should result in complete loss of hCTH activity due not only to removal of half of the catalytic site (left and bottom parts in Fig. 4), but also to the fact that the deletion leads to an altered quarternary structure. In agreement with complete loss of activity, patient #3183 had the highest plasma cystathionine among the patients studied.
In case of heterozygosity for p.Gly57_Gln196del, we assume that the mutated and differently folded subunit will not mix with wild-type subunits to form non-functional complexes, explaining the fact that little or no effect of such heterozygosity on plasma cystathionine was apparent in either the mother of patient #2930, or in the one p.Gly57_Gln196del heterozygous Czech identified by screening of healthy control subjects (Table 2); and suggesting that perhaps 50% of normal CTH activity may be sufficient to prevent abnormal elevation of cystathionine.
p.Thr311Ile
The effect of p.Thr311Ile, expressed in E. coli, on CTH activity was negligible (Fig. 2). p.Thr311Ile was not found among the 165 healthy Czechs genetically screened for this mutation, indicating it is not a common polymorphism. However, the very high plasma cystathionine of patient #2930, in whom this mutation is present together with p.Gly57_Gln196del, suggests that physiologically p.Thr 311Ile does not provide much catalytic activity. In the hCTH tetramer, Thr311 is situated at the inlet to the binding cavity and forms two strong hydrogen bonds to Tyr213, the amino acid next to Lys212, the principle residue in the reaction site (Fig. 4). Furthermore, nearby residues 211–208 are in close contact with the PLP (for clarity, not shown in Fig. 4). Replacement of Thr311 with Ile disrupts at least one of the H-bonds and may evoke a change of the Tyr213 side chain to a different conformer, accompanied by conformational changes in the quartet of residues, Lys212, PLP, Tyr60, and Thr211, critical for catalytic activity.
p.Thr67Ile
This mutation was first found by Wang and Hegele in both homozygous and compound heterozygous forms in two of the four cystathioninuric probands they studied, as well as in one of 120 alleles from normal control Europeans [5]. Three of the ten CTH alleles in the severely cystathioninemic/uric individuals reported upon here carried this mutation; and it was present in heterozygous form in 5 of the 165 control individuals genotyped in this regard in the Czech population (Table 2), thus having a prevalence of 5/(165 × 2) = 0.015 (i.e. 1.5%) and qualifying as a SNP by exceeding the level of 1%.
The p.Thr67Ile mutant protein had a catalytic activity of 13% of wild-type when expressed in E coli (Fig. 2), and Zhu et al reported a similarly decreased activity of 29% [6]. The latter workers found also that the mutant contained only about 19 % of the amount of PLP contained by wild-type protein and that preincubation with PLP restored catalytic activity to wild-type levels [6]. Functional effects of p.Thr67Ile are shown by the facts that plasma cystathionine was markedly elevated in patient #2927, a homozygote for this mutation, and mildly elevated in the 5 heterozygotes found in the Czech population (Table 2). During B6 administration there were marked decreases in both the urine and plasma cystathionine concentrations of patient #2927 (Supplemental Table 3), and in patient #2928 compound heterozygosity for p.Thr67Ile and p.Gly57_Gln196del was accompanied by B6 responsiveness. These effects may be due to the fact that Thr67 is in the chain leading to Arg62, the residue that joins Tyr114 and PLP to form a stable catalytic site (Figs. 3 & 4). Conformational change in this chain due to the p.Thr67Ile mutation may extend to Arg62 and reduce the stability of binding of PLP and thus, also, the stability of the entire catalytic site.
p.Arg197Cys
The data in Fig. 2 show that the loss of catalytic activity with this mutation, expressed in E. coli, is moderate, down to about 44% of wild-type. However, patient #2929, a homozygote for this mutation, clearly had a markedly elevated plasma cystathionine that was B6–responsive (Supplemental Table 3). Arg197 is far from the catalytic site and is not involved in any subunit-subunit interaction. However it forms a small, positively charged pocket at the outlet from the long, 5×5 Å wide channel that leads from the catalytic cavity to the opposite side of protein. Mutation to Cys197 changes the character of the pocket in question, and may reduce the capacity for water to escape via this channel, thereby influencing the kinetics of PLP binding and offering an explanation for the positive B6 effect. An alternative interpretation postulating formation of an S-S bond between Cys197 and Cys 310 (Fig 4) would require large conformational changes and would be energetically demanding.
p.Arg62His
Heterozygotes for this mutation have elevations of plasma cystathionine far more marked than were found in heterozygotes for the other CTH mutations studied (Tables 2 and 3). As detailed earlier in the discussion of the structure of hCTH, Arg62 is of central importance for correct formation of the active site and for cofactor binding because it binds the phosphate group of PLP and also locks Tyr114 parallel to PLP. Exchange of Arg62 for His62 shifts the geometry of the active site, decreases the strength of PLP binding, and makes this bond pH dependent under physiological conditions.
Table 3.
Taiwanese family S with heterozygous CTH p.[Arg62His] and MAT1A p.[ArgR264His] mutations
| Patient | Plasma Cysta μmol/l | plasma tCys μmol/l | tCys/Cysta Ratio | CTH genotype at 62 | plasma met μmol/l | MAT1A genotype at 264 |
|---|---|---|---|---|---|---|
| # 31*- father | 1.455 | 324 | 223 | p.[Arg62]+[Arg62His] | 126 | p.[Arg264]+[Arg264His] |
| # 4 – child | 1.761 | 281; 443** | 252; 310 | p.[Arg62]+[Arg62His] | 63; 123 | p.[Arg264]+[Arg264His] |
| #15 – child | 1.497 | 214; 399 | 267; 267 | P[Arg62]+[Arg62His] | 115; 248 | p.[Arg264]+[Arg264His] |
| # 32 – mother | 0.151 | 261 | 1728 | p.[Arg62]+[Arg62] | 26 | p.[Arg264]+[Arg264] |
| # 6 – child | 0.354 | 255; 336 | 949; 1232 | p.[Arg62]+[Arg62] | 121; 185 | p.[Arg264]+[Arg264His] |
| #33 – child | 0.199 | 238 | 1196 | p.[Arg62]+[Arg62] | 32 | not sequenced |
| Control | 0.12 | 237 | 1975 | not sequenced | 17 | not sequenced |
| Control | 0.183 | 210 | 1148 | not sequenced | 23 | not sequenced |
Numbers were assigned by the laboratory in which the hypermethioninemia was first studied [23].
If two values are listed, they are the results of assays of samples obtained at different times
p.Ser403Ile
p.Ser403Ile is a widespread polymorphism, with the TT genotype (producing p.[Ile403]+[Ile403]) accounting for 7.5% of a Canadian population studied (n = 496) [7], and 9.3 % of the 591 Czech controls for whom results are reported in this paper (Table 2). Fasting cystathionine levels were not significantly different between the GG, TG, or TT groups of Czechs. However, six-hours after challenging the transsulfuration pathway by oral administration of L-methionine, 100 mg/kg body weight, increased plasma cystathionine levels were observed in individuals with TT and GT genotypes compared to GG homozygotes (median plasma values 2.07 and 2.02 compared to 1.50 μmol/L, respectively (p<0.05 by Mann-Whitney test)), suggesting this polymorphism is not completely functionally neutral.
The TT genotype was found to cause a slight, but statistically significant, increase in the plasma tHcy of the Canadian group (13.3 ± 8.6 in the TT genotype compared to 11.4 ± 6.3 in GG+GT genotypes combined; p = 0.019) [7]. There was a similar trend among the Czech groups studied that fell just short of statistical significance (p = 0.051) (Table 2). However, a mechanism whereby this polymorphism might affect plasma tHcy remains to be defined.
The lack of a major effect of the p.Ser403Ile polymorphism on plasma cystathionine is understandable because it lies at the C terminus of the hCTH chains, distal to Ser401, the last residue localized in the electron density maps of the hCTH subunits, in the area exposed to solvent and without definite 3D structure (Fig 3). The amino acid in this position is likely to influence neither the tetramer structure, nor the conformation of the hCTH tetramer. In case of heterozygosity, one might expect the native and mutated subunits to form randomly mixed tetramers, and that the full activity of these heterotetramers will be preserved.
p.Gln240Glu
Zhu et al found that the catalytic activity of the p.Gln240Glu mutant protein was down about 70-fold, and that its PLP content was about 80-fold lower than that of the wild-type protein. Preincubation with PLP led to only partial restoration of catalytic activity to 10–13 % of wild-type [6]. As yet no human homozygous for p.Gln240Glu has been identified, and the effect of B6 administration on such a patient remains to be determined. The change of Gln240 to Glu influences conformational flexibility near the active site and may thus influence catalytic efficacy and the binding affinity for PLP. Fig. 4 shows that the Asn241 next to Gln240 has direct interaction with the side-chain of Arg62 responsible for PLP binding (compare with the p.Arg62His mutation described above). p.Gln240Glu can also influence the Tyr60 side-chain that helps form a suitable environment in the reaction site (Tyr60-Thr211-Lys212-PLP-Tyr114). Furthermore, Gln240 influences the binding tunnel via the chain Asn241…Arg119 -Tyr114, and may thus also affect the Tyr114 that locks the PLP in the correct position. The side-chain of Gln240 is bound to Arg237, Glu72 and Ala86 (not shown in Figure 4). Exchange of Gln240 for Glu makes this part of the structure more flexible and potentially allows release of stress in the areas just described. Thus the p.Gln240Glu mutation has the potential for three effects: change the substrate selectivity; modulate PLP binding; and change the local pK at the reaction site via Tyr60.
What are the clinical consequences of inactivating mutations in CTH?
Four of the patients discussed in this paper were found to have severe cystathioninuria during investigations prompted by medical problems; one, during studies of children born out of incest. Only patient #2930 and the three members of family S who are heterozygous for p.R62H were identified without what could be considered to be ascertainment bias. These latter four subjects are each free of adverse clinical effects at ages of as much as 36 and 44 years. Most strikingly, as stated earlier in her case report, patient #3183, a homozygote for the large CTH deletion, has excellent mental capability and, except for a thrombotic episode while on oral contraceptives, has generally had good health. No further siblings of the present patients are available who were found by family screening to have cystathioninuria. Such siblings would be helpful in determining whether or not cystathioninuria is associated with clinical effects. A previous literature review found that a wide assortment of clinical aberrations had been present in individuals with presumptive CTH deficiency. However, among the 26 CTH-deficient persons for whom ascertainment bias was expected to be minimal, only five had clinical difficulties that could have been related to the metabolic disorder, and for four of these either additional cystathioninuric sibs were normal, or sibs without severe cystathioninuria were similarly affected clinically [2].
The transsulfuration pathway by which the sulfur atom originating in methionine is transferred, via homocysteine and cystathionine, to cysteine is the major means by which mammals form cysteine. Thus, it is of interest that the patients reported upon in this paper who have impairments in their CTH activities all have normal plasma levels of tCys, including patient #3183 in whom even a small residual CTH activity is thought not to be present. It must be that intake of cyst(e)ine on normal diets provides a sufficient amount of this amino acid, thus presumably helping, also, to maintain glutathione, a compound for which cysteine is a metabolic precursor [29] or that cysteine is synthesized from serine and H2S by the serine sulfhydrase activity of CBS [2].
Two additional possible health risks for patients with CTH deficiency merit mention:
Three of the four homozygotes or compound heterozygotes for inactivating CTH mutations described here (patients #3183, #2930, and #2928) in whom plasma tHcy was assayed had elevations of plasma tHcy, suggesting a possible cause-and-effect relationship. It would be of interest to determine in a subject with B6-responsive hypercystathioninemia and mild hyperhomocysteinemia whether the elevated tHcy is mitigated by B6 administration. A causative relationship is supported by the recent report that mice homozygous for a deletion in CTH, have plasma tHcy values about 18 times those in age-matched wild-type mice [30]. However, the tHcy elevations observed in our patients were far milder, and may well not be severe enough to bring about any adverse effects.
It has been known since 1950 that CTH can catalyze the formation of H2S from cysteine [31]. Recent evidence suggests that H2S may function as both a neuromodulator or transmitter in brain [32] and a vasorelaxant [33], raising the question of whether in CTH deficiency a lack of H2S may have adverse effects on brain function or contribute to hypertension. Because H2S may be produced also by cystathionine β-synthase (CBS) [2; 34], and normal brain has CBS activity but is very poor in CTH activity [35], and because 3-mercaptopyruvate sulfurtransferase also produces H2S in the brain [36], CTH deficiency is unlikely to cause an abnormal lack of H2S in brain. In contrast, in portal vein, thoracic aorta, and other arteries H2S production is strongly or completely suppressed by DL-propargylglycine, an inhibitor of CTH, indicating that CTH may be responsible for generation of H2S in vascular tissues [33; 37]. With regard to hypertension, Yang et al found the mice mentioned above with homozygous deletions in their CTH genes not only have markedly reduced levels of H2S in serum, heart, aorta, and other tissues, but also develop mildly elevated blood pressures [30]. Li et al carried out a study of Northern Han Chinese to assess if there was a relationship between the p.Ser403Ile polymorphism and essential hypertension. No significant differences in the risks of hypertension were found among the p.Ser403Ile genotypes [8]. Most of the patients reported in this paper have more severe loss of CTH activity than that due to the Ser403Ile polymorphism. Blood pressures were 107/83 and 100/74 in the two hypercystathioninemic members of the S family (#4 and #15, respectively) in whom it was measured. Patient #2930 had normal blood pressure up to her last check-up at age 36 1/2 years. Hypertension has not been noted in any of the other patients described in this paper. However, because this parameter has not been checked systematically, a possible effect of CTH deficiency on human blood pressure remains to be more thoroughly investigated.
In summary, the present findings, taken together with those from the previous review [2], do not provide convincing evidence that CTH deficiency is a cause of adverse clinical consequences.
Supplementary Material
Acknowledgments
The authors thank Dr. William M. Sugden, Wisconsin Alumni Research Foundation for establishing the immortalized line of lymphocytes from patient #3051. The plasmid pET22b+hCTH was a generous gift from Dr. Markus Wahl. We are grateful to Dr. Harvey Levy for help in gathering patient information. This work was supported by grants from the American Heart Association (AHA 2-5-80663), from the NIH (HL065217-06A2 and AG09834) and Jerome Lejeune foundation to J.P.K, by grants from the Grant Agency of the Czech Republic 305/07/1073, GAAV CR IAA500500701, and MSMT T400500402 to J.H., and from the Wellcome Trust (reg. No 070255/Z/03/Z) and the Ministry of Education of the Czech Republic (MSM0021620806) to V.K. A generous gift from Ms. Margie McGlynn is also acknowledged.
Footnotes
The name for the same enzyme has also been abbreviated as “Cse”, “CGL” and “CTT”.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Stabler SP, Lindenbaum J, Savage DG, Allen RH. Elevation of serum cystathionine levels in patients with cobalamin and folate deficiency. Blood. 1993;81:3404–3413. [PubMed] [Google Scholar]
- 2.Mudd SH, Levy HL, Kraus JP. Disorders of transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 2007–2056. [Google Scholar]
- 3.Harris H, Penrose LS, Thomas DHH. Cystathioninuria. Ann Hum Genet. 1959;23:442–453. doi: 10.1111/j.1469-1809.1959.tb01485.x. [DOI] [PubMed] [Google Scholar]
- 4.Mudd SH, Levy HL. Disorders of transsulfuration. In: Stanbury JB, Wyngaarden JB, Frederickson DS, Goldstein JL, Brown MS, editors. The Metabolic Basis of Inherited Disease. McGraw-Hill Book Co.; New York: 1983. pp. 522–559. [Google Scholar]
- 5.Wang J, Hegele RA. Genomic basis of cystathioninuria (MIM 219500) revealed by multiple mutations in cystathionine gamma-lyase (CTH) Hum Genet. 2003;112:404–408. doi: 10.1007/s00439-003-0906-8. [DOI] [PubMed] [Google Scholar]
- 6.Zhu W, Lin A, Banerjee R. Kinetic properties of polymorphic variants and pathogenic mutants in human cystathionine γ-lyase. Biochemistry. 2008;47:6226–6232. doi: 10.1021/bi800351a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Huff AM, Spence JD, Hegele RA. Single nucleotide polymorphism in CTH associated with variation in plasma homocysteine concentration. Clin Genet. 2004;65:483–486. doi: 10.1111/j.1399-0004.2004.00250.x. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Zhao Q, Liu X-L, Wang L-Y, Lu X-F, Li H-F, Chen S-F, Huang J-F, Gu D-F. Relationship between cystathionine γ-lyase gene polymorphism and essential hypertension in Northern Chinese Han population. Chinese Medical Journal. 2008;121:716–720. [PubMed] [Google Scholar]
- 9.Krijt J, Vacková M, Kožich V. Measurement of homocysteine and other aminothiols in plasma: advantages of using tris(2-carboxyethyl)phosphine as reductant compared with tri-n-butylphosphine. Clin Chem. 2001;47:1821–1828. [PubMed] [Google Scholar]
- 10.Stabler SP, Marcell PD, Podell ER, Allen RH. Quantitation of total homocysteine, total cysteine, and methionine in normal serum and urine using capillary gas chromatography-mass spectrometry. Analyt Biochem. 1987;162:185–196. doi: 10.1016/0003-2697(87)90026-1. [DOI] [PubMed] [Google Scholar]
- 11.Stabler SP, Marcell PD, Podell ER, Allen RH, Savage DG, Lindenbaum J. Elevation of total homocysteine in the serum of patients with cobalamin or folate deficiency detected by capillary gas chromatography-mass spectrometry. J Clin Invest. 1988;81:466–474. doi: 10.1172/JCI113343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janosikova B, Pavlikova M, Kocmanova D, Vitova A, Vesela K, Krupkova L, Kahleova R, Krijt J, Kraml P, Hyanek J, Zvarova J, Andel M, Kozich V. Genetic variants of homocysteine metabolizing enzymes and the risk of coronary disease. Mol Genet Metab. 2003;79:167–175. doi: 10.1016/s1096-7192(03)00079-9. [DOI] [PubMed] [Google Scholar]
- 13.Steegborn C, Clausen T, Sondermann P, Jacob U, Worbs M, Marinkovic S, Huber R, Wahl MC. Kinetics and inhibition of recombinant human cystathionine γ-lyase Toward the rational control of transsulfuration. J Biol Chem. 1999;274:12675–12684. doi: 10.1074/jbc.274.18.12675. [DOI] [PubMed] [Google Scholar]
- 14.Lowry OH, Rosenrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 15.Gaitonde MK. A spectrophotometric method for the direct determination of cysteine in the presence of other naturally occurring amino acids. Biochem J. 1967;104:627–633. doi: 10.1042/bj1040627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Q, Collins R, Huang S, Holmberg-Schiavone L, Anand GS, Tan C-H, van-den-Berg S, Deng L-W, Moore PK, Karlberg T, Sivaraman J. Structural basis for the inhibition mechanism of human cystathionine-γ-lyase: an enzyme responsible for the production of H2S. J Biol Chem. 2008;M805459200 doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 17.DeLano WL. The PyMOL molecular graphics system. Delano Scientic; Palo Alto, CA: 2008. htp://www.pymol.org. [Google Scholar]
- 18.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozeli S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. AMBER 9. University of California; San Francisco: 2006. [Google Scholar]
- 20.Levy HL, Mudd SH, Uhlendorf BW, Madigan PM. Cystathioninuria and homocystinuria. Clin Chim Acta. 1975;58:51–59. doi: 10.1016/0009-8981(75)90484-2. [DOI] [PubMed] [Google Scholar]
- 21.Vargas JE, Mudd SH, Waisbren SE, Levy HL. Maternal γ-cystathionase deficiency: lack of teratogenic effects and of pregnancy complications. Am J Obstet Gynecol. 1999;181:753–755. doi: 10.1016/s0002-9378(99)70525-9. [DOI] [PubMed] [Google Scholar]
- 22.Hyanek J, Hoza J, Seemanova E. Primary cystathioninuria in an infant born out of incest. Acta Univ Carol Med Monogr. 1977:39–44. [PubMed] [Google Scholar]
- 23.Chien Y-H, Chiang S-C, Huang A, Hwu W-L. Spectrum of hypermethioninemia in neonatal screening. Early Hum Dev. 2005;81:529–533. doi: 10.1016/j.earlhumdev.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Chamberlin ME, Ubagai T, Mudd SH, Levy HL, Chou JY. Dominant inheritance of isolated hypermethioninemia is associated with a mutation in the human methionine adenosyltransferase 1A gene. Am J Hum Genet. 1997;60:540–546. [PMC free article] [PubMed] [Google Scholar]
- 25.Pérez-Mato I, Sánchez del Pino MM, Chamberlin MM, Mudd SH, Mato JM, Corrales FJ. Biochemical basis for the dominant inheritance of hypermethioninemia associated with the R264H mutation of MAT1A gene A monomeric methionine adenosyltransferase with tripolyphosphatase activity. J Biol Chem. 2001;276:13803–13809. doi: 10.1074/jbc.M009017200. [DOI] [PubMed] [Google Scholar]
- 26.Zschocke J, Quak E, Knauer A, Fritz B, Aslan M, Hoffman GF. Large heterozygous deletion masquerading as homozygous missense mutation: A pitfall in diagnostic mutation analysis. J Inherit Metab Dis. 1999;22:687–692. doi: 10.1023/a:1005527731397. [DOI] [PubMed] [Google Scholar]
- 27.Lu Y, O’Dowd BF, Orrego H, Israel Y. Cloning and nucleotide sequence of human liver cDNA encoding for cystathionine γ-lyase. Biochem Biophys Res Commun. 1992;189:749–758. doi: 10.1016/0006-291x(92)92265-y. [DOI] [PubMed] [Google Scholar]
- 28.Levonen A-L, Lapatto R, Saksela M, Raivio KO. Human cystathionine γ–lyase: developmental and in vitro expression of two isoforms. Biochem J. 2000;347:291–295. [PMC free article] [PubMed] [Google Scholar]
- 29.Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R. A functional transsuulfuration pathway in the brain links to glutathione homeostasis. J Biol Chem. 2006;281:35785–35793. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]
- 30.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ–lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Binkley F, Okeson D. Purification of the enzyme responsible for the cleavage of cystathionine. J Biol Chem. 1950;182:273–277. [Google Scholar]
- 32.Kimura H. Hydrogen sulfide as a neuromodulator. Mol Neurobiol. 2002;26:13–19. doi: 10.1385/MN:26:1:013. [DOI] [PubMed] [Google Scholar]
- 33.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhyration in liver and kidney of rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mudd SH, Finkelstein JD, Irreverre F, Laster L. Transsulfuration in mammals: Microassays and tissue distributions of three enzymes of the pathway. J Biol Chem. 1965;240:4382–4392. [PubMed] [Google Scholar]
- 36.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Oshii K, Kimura H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxidants & Redox Signaling. 2008 doi: 10.1089/ARS.2008.2253. [DOI] [PubMed] [Google Scholar]
- 37.Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.