

Summary
Glutamyl-prolyl tRNA synthetase (EPRS) is a component of the heterotetrameric GAIT (Gamma-interferon Activated Inhibitor of Translation) complex that binds 3′UTR GAIT elements in multiple interferon-gamma (IFN-γ)-inducible mRNAs and suppresses their translation. Here we elucidate the specific EPRS phosphorylation events that regulate GAIT-mediated gene silencing. IFN-γ induces sequential phosphorylation of Ser886 and Ser999 in the non-catalytic linker connecting the synthetase cores. Phosphorylation of both sites is essential for EPRS release from the parent tRNA multisynthetase complex. Ser886 phosphorylation is required for the interaction of NSAP1, which blocks EPRS binding to target mRNAs. The same phosphorylation event induces subsequent binding of ribosomal protein L13a and GAPDH, and restores mRNA binding. Finally, Ser999 phosphorylation directs the formation of a functional GAIT complex that binds initiation factor eIF4G and represses translation. Thus, two-site phosphorylation provides structural and functional pliability to EPRS, and choreographs the repertoire of activities that regulates inflammatory gene expression.
Introduction
The canonical function of aminoacyl-tRNA synthetases (AARS) is to decipher the genetic code by accurate ligation of amino acids to their cognate tRNAs (Ibba and Söll, 2000; Ribas de Pouplana and Schimmel, 2001). The AARS are ancient and ubiquitous enzymes with catalytic cores conserved in bacteria, archaea, and eukarya. The 20 AARS are divided into two classes of 10 enzymes each, distinguished by structures and signature sequences in their catalytic domains. During evolution, certain AARS acquired structural characteristics and functions beyond those required for protein synthesis (Park et al., 2005a). For example, many chordate AARS are distinguished from their bacterial counterparts by a greater degree of complexity exemplified by noncanonical functions unrelated to aminoacylation, by intracellular organization into large complexes, and by non-catalytic appended domains (Robinson et al., 2000; Shiba, 2002). These features of chordate AARS appear to be interrelated, e.g., the appendages facilitate interactions between AARS and other proteins (or inter-AARS interactions), and they may contribute to AARS noncanonical activities (Ko et al., 2002).
Via their noncanonical activities, several AARS may be important regulators of diverse cellular processes, including several related to inflammation. For example, mast cell LysRS exerts transcriptional control via its catalytic product Ap4A (Lee et al., 2004b), and upon secretion triggers a proinflammatory response (Park et al., 2005b). GlnRS prevents apoptosis by inhibiting the kinase activity of apoptosis signal-regulated kinase 1 (Ko et al., 2001). Proteolytic fragments of TyrRS and TrpRS exhibit opposing, cytokine-like activities that regulate angiogenesis (Tzima and Schimmel, 2006). In humans and other chordates, GluRS and ProRS are linked to form a single bifunctional protein, GluProRS (or EPRS) (Jeong et al., 2000). Human EPRS exhibits a noncanonical function as a post-transcriptional regulator of inflammatory gene expression (Sampath et al., 2004). In some cases, domains that are neither a part of the enzymatic core nor present in bacterial homologues contribute to noncanonical functions (Jia et al., 2008; Shiba, 2002). These domains are usually appended to the N or C terminus, and include EF1Bγ-like domains, an endothelial monocyte-activating polypeptide II-like domain, and a helix-turn-helix domain termed the WHEP-TRS (after three AARS containing them, i.e., Trp(W)RS, His(H)RS, and GluPro(EP)RS); GlyRS and MetRS also contain appended WHEP domains. Interestingly, no other proteins contain this domain, and all 5 WHEP domain-bearing proteins express noncanonical functions (Park et al., 2005a; Sampath et al., 2004; Shiba, 2002). In mammalian cells, 9 of the 20 AARS activities are organized in a cytosolic, 1.5 mDa tRNA multisynthetase complex (MSC) (Lee et al., 2004a; Robinson et al., 2000). The complex also contains three AARS-interacting multifunctional proteins (AIMP) that lack synthetase activity, AIMP1/p43, AIMP2/p38, and AIMP3/p18, which may serve as a scaffold for the complex, but also express additional cellular functions (Park et al., 2005a; Robinson et al., 2000). The function of the MSC remains unclear, but it may facilitate channeling of aminoacylated tRNAs to the ribosome during protein biosynthesis (Kyriacou and Deutscher, 2008). In addition, the complex can serve as a depot for AARS and non-AARS proteins released to perform noncanonical activities in a stimulus- or context-dependent manner (Ray et al., 2007).
Human EPRS is a 172 kDa monomeric protein that displays all of the characteristics that differentiate eukaryotic AARS from their bacterial counterparts. The protein contains two distinct appended domains, namely, an N-terminus EF1Bγ-like domain and a linker domain containing three tandem WHEP repeats that connects the catalytic domains. EPRS resides exclusively in the MSC, but in monocytic cells it is released upon interferon (IFN)-γ activation to join three other proteins to form the cytosolic IFN-Gamma-Activated Inhibitor of Translation (GAIT) complex (Sampath et al., 2004). The GAIT complex binds a defined RNA element (GAIT element), consisting of a stem-loop with an internal bulge in the 3′ untranslated region (UTR) of target transcripts, e.g., vascular endothelial growth factor (VEGF)-A, ceruloplasmin (Cp), death-associated protein kinase, zipper-interacting protein kinase, several chemokines and their receptors, and silences their translation (Mukhopadhyay et al., 2008; Ray and Fox, 2007; Ray et al., 2009; Sampath et al., 2003; Vyas et al., 2009). The GAIT complex forms in two stages. After about 2 h of IFN-γ stimulation, EPRS is phosphorylated and released from its MSC residence to interact with NS1-associated protein (NSAP1) and form the inactive pre-GAIT complex. About 14-16 h later, ribosomal protein L13a and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) join the pre-GAIT complex to form the active, 4-protein complex that binds GAIT element-bearing mRNAs and suppresses their translation by intercepting the 43S ribosomal subunit binding site on eukaryotic initiation factor 4G (eIF4G) (Kapasi et al., 2007; Sampath et al., 2003). As the sole target mRNA-binding protein, EPRS has a central role in GAIT system function (Sampath et al., 2004). The two upstream WHEP repeats are essential and sufficient for high-affinity binding to the GAIT element (Jia et al., 2008). RNA-binding activity of EPRS is both negatively and positively regulated. Coincident with its release from the MSC, NSAP1 binds EPRS in a domain partially overlapping the RNA-binding domain, and efficiently inhibits binding to target transcripts. Upon subsequent joining of L13a and GAPDH to the pre-GAIT complex, RNA binding and translational silencing is restored.
Phosphorylation is a near-universal regulatory mechanism applied to both global and transcript-selective translational control (Dever, 2002; Gebauer and Hentze, 2004; Proud, 2007). Two key GAIT complex constituents, EPRS and L13a, are phosphorylated in response to IFN-γ (Mazumder et al., 2003; Mukhopadhyay et al., 2008; Sampath et al., 2004). EPRS phosphorylation is required for its release from the MSC and for formation of the functional GAIT complex (Jia et al., 2008; Sampath et al., 2004). However, the specific site(s) of phosphorylation, and GAIT pathway processes controlled by EPRS phosphorylation are unknown. Here we show IFN-γ-inducible, 2-site phosphorylation of EPRS at Ser886 and Ser999 in the linker domain choreographs the specific events required for noncanonical EPRS activity: they are essential for induced release of EPRS from the MSC, for negative and positive regulation of EPRS binding to target mRNAs, for phospho-L13a binding to eIF4G, and ultimately, for translational silencing of inflammatory gene expression.
Results
Multisite Ser Phosphorylation is Essential for GAIT Complex Assembly
To investigate EPRS phosphorylation in vivo, cytosolic lysates from IFN-γ-treated U937 cells were detected with anti-EPRS antibody after prolonged resolution by SDS-PAGE. IFN-γ treatment for 1 h caused a small electrophoretic mobility shift of EPRS that markedly increased after 2 h, indicative of multisite phosphorylation (Fig. 1A, top panel). Approximately half of the cellular EPRS exhibited reduced electrophoretic mobility, which was reversed by phosphatase treatment. Immunoprecipitation with anti-EPRS antibody and detection with anti-pSer antibody confirmed multisite phosphorylation of one or more Ser residues (Fig. 1A, bottom panel). EPRS has two catalytic synthetase domains, ERS and PRS, joined by a linker containing 3 tandem WHEP repeats (R1, R2, and R3), each exhibiting a helix-turn-helix structure (Cahuzac et al., 2000; Jeong et al., 2000) (Fig. 1B, top). To determine the specific domain(s) phosphorylated by IFN-γ, E. coli-expressed and purified, His-tagged polypeptides were phosphorylated by lysates from IFN-γ-treated cells. The results revealed highly specific phosphorylation of the linker domain (Fig. 1B, bottom, left). Domain immunoprecipitation with anti-His antibody and detection with anti-pSer antibody confirmed specific linker phosphorylation (not shown). Maximal phosphorylation of linker was observed with lysates from cells treated with IFN-γ for 4 h (Fig. 1C).
Figure 1. Inducible, Multisite Serine Phosphorylation in EPRS Linker Domain.
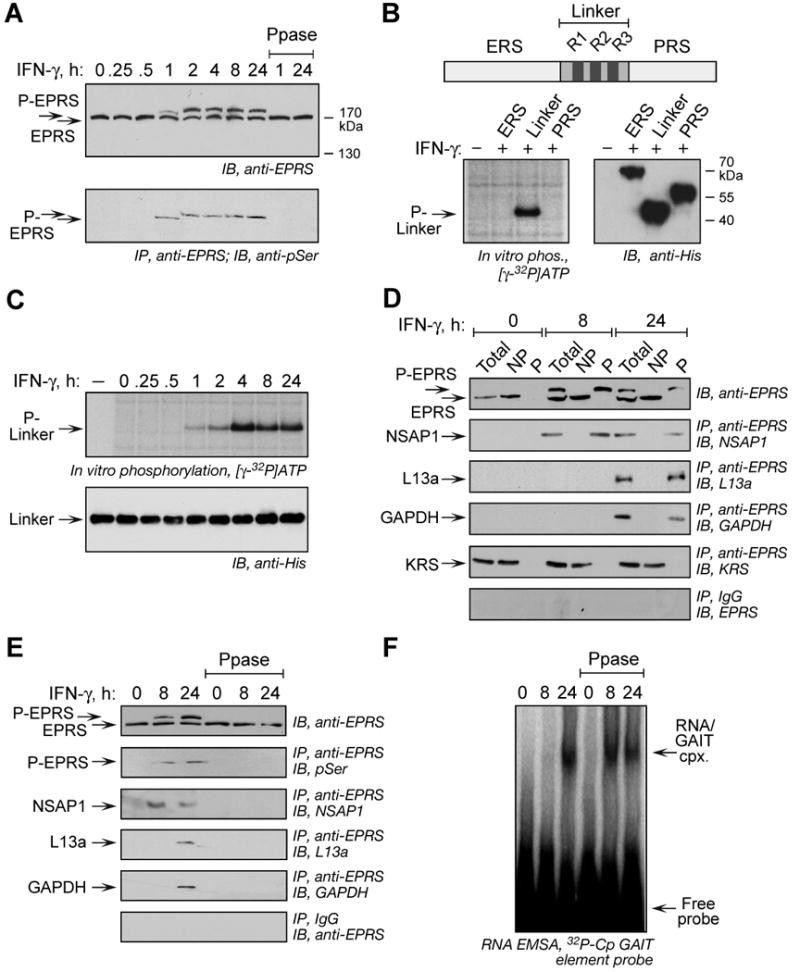
(A) Multisite EPRS phosphorylation. Phosphorylated EPRS (P-EPRS) in lysates from IFN-γ-treated U937 cells was resolved by SDS-PAGE (4.5% polyacrylamide) and detected by immunoblot with anti-EPRS antibody. Two samples were incubated with shrimp alkaline phosphatase (PPase) before immunoblot analysis. The same lysates were immunoprecipitated with anti-EPRS antibody and probed with anti-pSer antibody.
(B) The non-catalytic EPRS linker domain is phosphorylated. Schematic of major human EPRS domains. In vitro phosphorylation of His-tagged EPRS domains by lysates of 24-h, IFN-γ-treated U937 cells. All domains detected by immunoblot with anti-His antibody.
(C) Lysates from IFN-γ treated cells were used to in vitro phosphorylate recombinant, His-tagged EPRS linker. Linker was detected by immunoblot with anti-His antibody.
(D) EPRS phosphorylation is required for release from MSC and interaction with GAIT proteins. Lysates from IFN-γ treated U937 cells were applied to phosphoprotein affinity column to isolate P-EPRS and unmodified EPRS. Total, non-phosphorylated (NP), and phosphorylated (P) fractions were assessed by immunoblot with anti-EPRS antibody. The same fractions were immunoprecipitated with anti-EPRS and detected with antibodies against GAIT constituent proteins and the MSC constituent KRS. As a control, fractions were immunoprecipitated with rabbit IgG and immunoblotted with anti-EPRS.
(E) Dephosphorylation of EPRS disrupts the pre-GAIT and GAIT complexes. Lysates from IFN-γ-treated U937 cells were incubated with shrimp alkaline phosphatase and dephosphorylation detected with anti-EPRS antibody, and by immunoprecipitation with anti-EPRS and immunoblot with anti-phosphoSer. Precipitates were probed with antibodies against all GAIT proteins.
(F) EPRS phosphorylation is not required for binding to GAIT element. EPRS binding to GAIT element was determined by RNA EMSA using 32P-labeled Cp GAIT element RNA probe in control and phosphatase-treated lysates of IFN-γ-activated cells.
To evaluate the role of phosphorylation in interaction with other GAIT constituents, unmodified and phosphorylated EPRS in cell lysates were separated by phosphoprotein enrichment chromatography (Fig. 1D, top panel). Phosphorylated, nonphosphorylated, and total EPRS fractions were immunoprecipitated with anti-EPRS antibody and probed with antibodies against GAIT constituents. All GAIT proteins interacted specifically with phospho-EPRS; NSAP1 bound EPRS after 8 h whereas binding of L13a and GAPDH to EPRS required 24 h of IFN-γ stimulation (Fig. 1D, panels 2-4). LysRS (KRS), a constituent of the MSC, interacted specifically with nonphosphorylated EPRS (Fig. 1D, panel 5). These results show that only phosphorylated EPRS leaves the MSC and interacts with other GAIT proteins. Phosphatase treatment efficiently dephosphorylated EPRS, and completely disrupted the pre-GAIT and GAIT complexes formed after 8 and 24 h (Fig. 1E). The role of EPRS phosphorylation in binding GAIT RNA was examined by EMSA with 32P-labeled Cp GAIT RNA element. As shown previously (Sampath et al., 2003), the GAIT complex binds the GAIT element only after 24-h IFN-γ treatment of cells (Fig. 1F). Phosphatase treatment of the 24-h lysate did not disrupt this interaction. Interestingly, disruption of the pre-GAIT complex by phosphatase treatment of the 8-h lysate allows GAIT ribonucleoprotein complex formation, consistent with our previous observation that binding of NSAP1 to phosphorylated EPRS inhibits the interaction of EPRS with target RNA (Jia et al., 2008). Together these results indicate EPRS phosphorylation is essential for GAIT complex assembly, but not for GAIT element binding.
IFN-γ Induces Phosphorylation of EPRS at Ser886 and Ser999
A proteomic approach was used to identify specific EPRS phosphorylation sites. Full-length, phosphorylated EPRS was isolated from IFN-γ-treated cells (Fig. 2A, left), and subjected to microsequence analysis by liquid chromatography (LC)-tandem mass spectrometry (MS). Database searches comparing collision-induced dissociation (CID) spectra with human EPRS (GI: 62241042) revealed 72 peptides with 57% coverage, and two phosphopeptides, 871EYIPGQPPLSQSSDSSPTR889 (Fig. 2A, middle) and 992NQGGGLSSSGAGEGQGPK1009 (Fig. 2A, right). Detailed spectra analysis revealed Ser886 and Ser999 (or possibly Ser1000) as the phosphorylation sites; assignment of the Ser999 site was tentative due to low abundance of signature fragment ions.
Figure 2. IFN-γ Induces Phosphorylation of Ser886 and Ser999 in EPRS Linker.
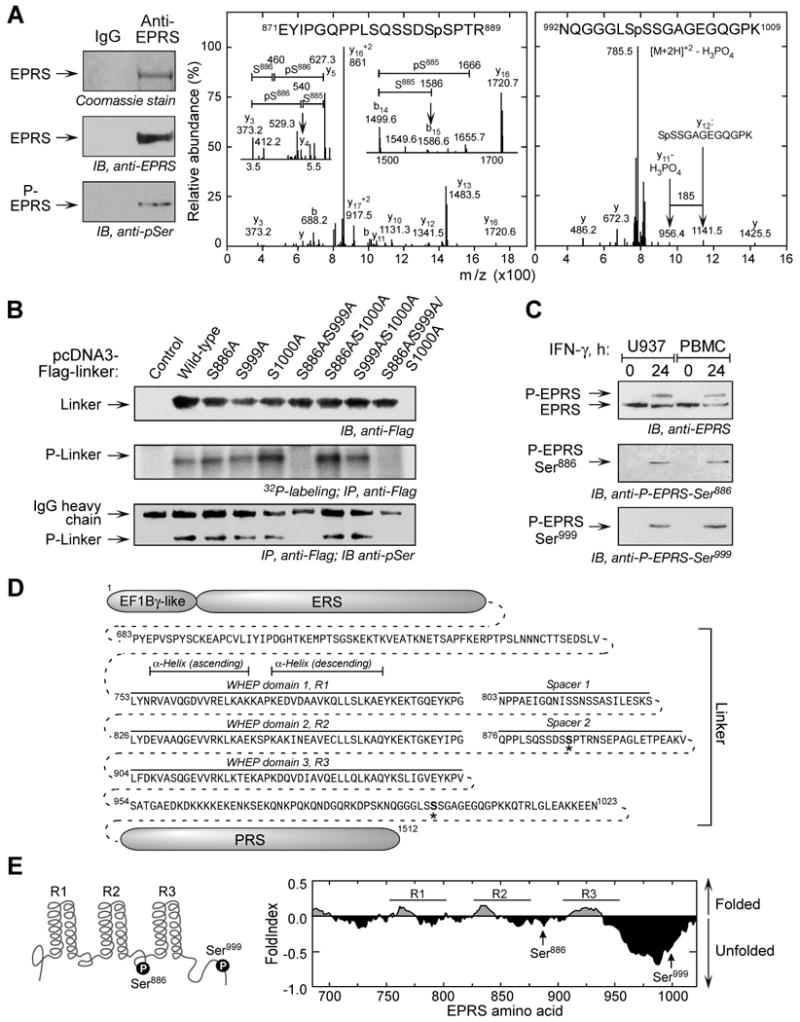
(A) Mass spectrometric identification of phosphorylation sites. Phospho-EPRS was isolated from 24-h, IFN-γ-treated U937 cells by phosphoprotein affinity column and immunoprecipitation with anti-EPRS antibody. Phosphorylation was shown by immunoblot with anti-pSer antibody (left). Excised, Coomassie-stained P-EPRS was subjected to in-gel tryptic digest, and CID spectra obtained from LC-tandem MS identified two phospho-peptides. CID spectra of 871EYIPGQPPLSQSSDSSPTR889 were dominated by fragmentation at several Pro residues, including Pro874 forming the y16+2 ion and Pro877 to give y13 ion (center). Other sequence-specific y and b ions limited the phospho-site to Ser885 or Ser886. Detailed inspection indicated a low-abundance y4 (m/z 540) and b15 (m/z 1586) ions (insets). The latter indicated lack of phosphorylation at Ser885, and the 167 Da mass difference between y4 and y3 ions indicated Ser886 as the phosphorylation site. The fragmentation spectra of 992NQGGGLSSSGAGEGQGPK1009 were dominated by [M+2H]+2-H3PO4 ion at m/z 785.5 (right). Low-abundance, sequence-specific y-ions were on both sides of three potential phosphorylation sites in 998SSS1000. Ser999 was identified as the likely phosphorylation site by the y11 ion at m/z 956.4, which differs from y12 ion at m/z 1141.5 by 185 Da, consistent with the loss of unmodified Ser998 (87 Da) and H3PO4 (98 Da). Also, MS3 analysis of the ion at m/z 785.5 showed fragment ions indicating Ser999 as the dehydroalanine residue resulting from loss of H3PO4 of a phosphoserine residue (not shown).
(B) Verification of phosphorylation sites. N-terminus-Flag-tagged, wild-type and mutant linkers in pcDNA3 vector were transiently transfected into U937 cells, and expression was determined by immunoblot. Cells were incubated for 4 h with 32P-orthophosphate and IFN-γ. Lysates were immunoprecipitated with anti-Flag antibody, and phosphorylation determined by autoradiography, and by detection with anti-pSer antibody.
(C) Phospho-specific EPRS antibodies confirm inducible, 2-site phosphorylation. Phospho-specific antibodies raised against peptides containing pSer886 and pSer999 were used to probe lysates from IFN-γ-treated U937 cells and peripheral blood mononuclear cells (PBMC).
(D) Sequence and domains of human EPRS linker. The N-terminus EF1Bγ-like domain and ERS domain are connected to the C-terminus PRS domain by a linker consisting of three WHEP repeats, R1, R2, and R3, separated by two spacers. Phosphorylation sites indicated by “*”.
(E) Schematic of two phosphorylation sites in EPRS linker domain (left). Predicted folding probability of EPRS linker polypeptide sequence determined by FoldIndex using a window size of 51 (right).
To verify phosphorylation at Ser886, and distinguish between the Ser999 and Ser1000 sites, pcDNA3-Flag-linker constructs harboring Ser-to-Ala mutations were expressed in U937 cells (Fig. 2B, top panel). IFN-γ-treated cells were labeled with 32P-orthophosphate and immunoprecipitated with anti-Flag antibody (Fig. 2B, middle panel). IFN-γ induced phosphorylation of all linker polypeptides except for those containing mutations at both Ser886 and Ser999, specifying them as EPRS phosphorylation sites. Immunoblotting with anti-pSer antibody verified both sites (Fig. 2B, bottom panel). Finally, EPRS phosphorylation in U937 cells was detected by immunoblot with phosphospecific antibodies generated against peptides containing phosphorylated Ser886 and Ser999 (Fig. 2C). Similarly, IFN-γ-dependent phosphorylation at both sites was detected in human peripheral blood mononuclear cells (PBMC). Both phosphosites are in the linker domain, known to be critical for interaction with other GAIT components (Jia et al., 2008). Ser886 is in the spacer between the 2nd (R2) and 3rd (R3) WHEP repeats domains (Fig. 2D, E, right); NMR spectroscopy indicates the phospho-site is within an unstructured loop connecting highly compact, α-helical WHEP domains (Jeong et al., 2000). Ser999 is in the post-WHEP flanking domain just upstream of the PRS domain. Structural data is not available for this site, but FoldIndex, an algorithm that predicts folding probability of protein domains, indicates that the Ser999 site is in an extremely unfolded domain (Fig. 2E, right), comparable to proteins completely unfolded in their native state (Prilusky et al., 2005).
Distinct Kinases Coordinate Sequential Phosphorylation at Ser886 and Ser999
The two-step shift in EPRS electrophoretic mobility suggests sequential phosphorylation events (Fig. 1A). Using phospho-specific antibodies, phosphorylation of Ser886 and Ser999 were detected after 1 and 2 h of IFN-γ treatment, respectively (Fig. 3A). To confirm the order and kinetics of phosphorylation, lysates from cells treated with IFN-γ for up to 24 h were used to in vitro phosphorylate purified, His-tagged EPRS linker polypeptides containing mutations ensuring site-specific phosphorylation (Fig. 3B). The Ser886 phospho-acceptor (Ser999-to-Ala, S999A mutant) is phosphorylated by lysates from cells treated with IFN-γ for 1 h or more (Fig. 3B, top), whereas phosphorylation of the Ser999 phospho-acceptor (Ser886-to-Ala, S886A mutant) required lysates from cells treated for at least 2 h (Fig. 3B, middle). Maximal phosphorylation of all the linker substrates was observed by 4 h. These results confirmed the sequential EPRS phosphorylation first at Ser886, then at Ser999. Moreover, they show that the phosphorylation events can occur independently, i.e., phosphorylation of Ser999 does not require phosphorylation of Ser886 as a priming event.
Figure 3. Ordered Phosphorylation of Ser886 and Ser999 by two Ser/Thr kinases.
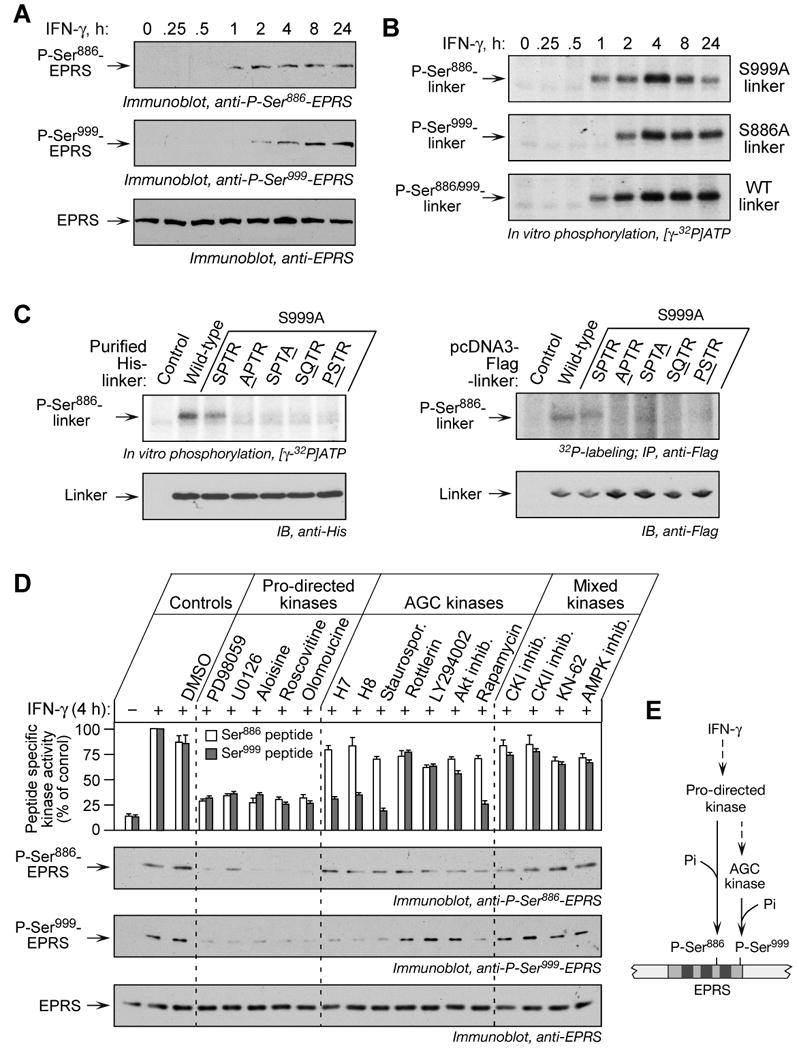
(A) Ser886 phosphorylation precedes Ser999 phosphorylation. Lysates from U937 cells treated with IFN-γ were resolved by SDS-PAGE (12% polyacrylamide) and analyzed by immunoblot with phospho-specific antibodies, and with anti-EPRS antibody.
(B) Ser886 and Ser999 phosphorylation events are independent. Lysates of IFN-γ-treated U937 cells were used to in vitro phosphorylate His-tagged wild-type and mutant linkers.
(C) Identification of the kinase recognition motif for Ser886 phosphorylation. Lysates of 4-h, IFN-γ-treated cells were used to in vitro phosphorylate purified His-tagged wild-type and linkers with mutations in key motif amino acids; equal linker amount was detected with anti-His antibody (left panels). The pcDNA3-Flag-linker DNAs containing motif mutations were transiently expressed in U937 cells and labeled with 32P-orthophosphate in presence of IFN-γ for 4 h. Lysates were immunoprecipitated with anti-Flag antibody, and 32P incorporation determined by autoradiography (right panels).
(D) Distinct kinases phosphorylate Ser886 and Ser999. U937 cells were incubated with IFN-γ for 0.5 h and then with kinase inhibitors for an additional 3.5 h. Cells incubated with or without IFN-γ and dimethyl sulfoxide (DMSO, 0.5%) served as controls. Pro-directed kinase group inhibitors were PD98059 (10 μM), U0126 (10 μM), aloisine (5 μM), roscovitine (10 μM), and olomoucine (50 μM). AGC kinase group inhibitors were H-7 (10 μM), H-8 (10 μM), staurosporine (Staurospor. 20 nM), rottlerin (100 μM), LY294002 (2.5 μM), Akt inhibitor-VIII (2.5 μM), rapamycin (10 nM). Other inhibitors were CKI inhibitor (100 μM), CKII inhibitor (100 μM), KN-62 (5 μM), and AMPK inhibitor (2 μM). Lysate kinase activity was determined as phosphorylation of Ser886 (white) and Ser999 (grey) phospho-acceptor peptides, and expressed as % of control lysates (mean ± SEM, top panel). Lysates were resolved on SDS-PAGE (12% polyacrylamide) and analyzed by immunoblot with indicated antibodies (panels 2-4).
(E) Schematic of kinase pathway phosphorylating EPRS Ser886 and Ser999.
The independent, temporally discrete phosphorylation of Ser886 and Ser999 suggests the involvement of distinct proximal kinases. To further test this possibility, we investigated kinase recognition motifs near the EPRS phospho-acceptor sites. Scansite Motif Scanner and PhosphoMotif Finder, complemented by literature search, revealed EPRS sequence 886SPTR as a potential “S/T-P-X-K/R” (X, any amino acid) kinase recognition motif for Ser886 phosphorylation (Amanchy et al., 2007; Obenauer et al., 2003; Songyang et al., 1996). Motifs near Ser999 were not identified. We tested the “S/T-P-X-K/R” motif requirement by in vitro phosphorylation of His-tagged EPRS linkers with mutations near the Ser886 phospho-acceptor site, in the context of S999A (to prevent Ser999 phosphorylation). As expected, wild-type and S999A mutant linkers were phosphorylated (Fig. 3C, left). However, 886SPTR mutation to APTR, SPTA, SQTR, and PSTR abolished Ser886 phosphorylation. Similar results were obtained in 32P-labeled cells expressing pcDNA3-Flag-linker constructs harboring the same mutations in the motif (Fig. 3C, right). These studies establish 886SPTR as an essential kinase recognition site for Ser886 phosphorylation, and the absence of a similar motif surrounding Ser999 supports EPRS phosphorylation by distinct proximal kinases.
The “S/T-P-X-K/R” motif is the target of multiple Pro-directed kinases, e.g., cyclin-dependent kinases, mitogen-activated protein kinases, and others, and belongs to the CMGC kinase group (Manning et al., 2002; Songyang et al., 1996). Several AGC group kinases (e.g., PKA, PKC etc.,) and casein kinases (CK) can phosphorylate AARSs in vitro (Pendergast and Traugh, 1985; Van Dang and Traugh, 1989; Venema and Traugh, 1991). The effect of group-specific pharmacological inhibitors was investigated. All inhibitors targeting Pro-directed kinases significantly inhibited IFN-γ-stimulated phosphorylation of both Ser886 and Ser999 14-mer peptide substrates (Fig. 3D, top panel); similar results were obtained after immunoblotting with EPRS phospho-specific antibodies (Fig. 3D, panels 2-4). Several AGC group kinase inhibitors, inhibited phosphorylation of Ser999, but not Ser886. Among the PKC inhibitors, only staurosporine inhibited the phosphorylation. Significant inhibition with H7, H8, and rapamycin suggest a member of the cyclic-nucleotide dependent protein kinase (e.g., PKA, PKG, etc.) or ribosomal S6 kinase families might mediate Ser999 phosphorylation. Akt inhibitor-VIII and the PI3K inhibitor LY294002 showed negligible inhibition of Ser999 phosphorylation. Inhibitors of CKI, CKII, and CAMK group members did not inhibit phosphorylation of either site. The temporal difference in phosphorylation, the requirement of the 886SPTR motif for Ser886 phosphorylation, and the specific inhibition of Ser999 phosphorylation by AGC kinase group inhibitors show that the two phosphorylation events are mediated by distinct Ser/Thr kinases. However, inhibition of both phosphorylation events by inhibitors of Pro-directed kinases suggests the Ser886 kinase may be an upstream activator of the Ser999 kinase, consistent with the observed delay in Ser999 phosphorylation (Fig. 3E).
Dual Ser Phosphorylation is Required for EPRS Exit from the MSC
Previous studies showed that IFN-γ induces EPRS release from the MSC (Jia et al., 2008; Sampath et al., 2004). The role of phosphorylation in inducing EPRS release was investigated using roscovitine, an inhibitor of Pro-directed kinases, which suppressed phosphorylation of Ser886, and as a consequence, Ser999 (Fig. 4A). Immunoprecipitation with anti-EPRS antibody and immunoblot with antibodies against two MSC constituent proteins, KRS and AIMP3/p18 showed that the inhibitor prevented EPRS release from the MSC. Likewise, immunoblot with antibody against NSAP1 showed EPRS failed to form the pre-GAIT complex with NSAP1. To evaluate the role of specific phosphorylation events in release, full-length, Flag-tagged EPRS containing phosphorylation-defective (Ser-to-Ala) mutations was expressed in cells. Cycloheximide was added after 18 h of transfection to permit accumulation of newly synthesized EPRS in the MSC. Immunoblot with anti-Flag antibody showed comparable expression of all proteins (Fig. 4B). Immunoprecipitation with anti-Flag antibody and immunoblot with antibodies against MSC components or anti-NSAP1 revealed incorporation of transfected, wild-type EPRS into the MSC, and essentially near-complete release upon stimulation with IFN-γ. However, none of the Ser-to-Ala mutants exhibited IFN-γ-dependent translocation from the MSC to the pre-GAIT complex indicating phosphorylation of both Ser residues is required for EPRS release. To further investigate the role of phosphorylation, cells were transfected with single or double phosphorylation mimetics (Ser-to-Asp) with the expectation that only the double-mutant would fail to incorporate in the MSC. Surprisingly, along with S886D/S999D, S999D mutant EPRS was also absent from the MSC suggesting Ser999 phosphorylation prevented EPRS incorporation into the MSC during assembly (Fig. 4C). The S886D mutant that was incorporated into the MSC was released upon second-site phosphorylation after IFN-γ treatment (Fig. 4D). Together these studies show that phosphorylation of both Ser886 and Ser999 are essential for EPRS release from the MSC; however, their roles are not identical because Ser999, but not Ser886, prevents EPRS incorporation into the MSC, and it may have a distinct role in regulating the stable dissociation of EPRS from the MSC.
Figure 4. EPRS Phosphorylation is Required for Release from MSC.
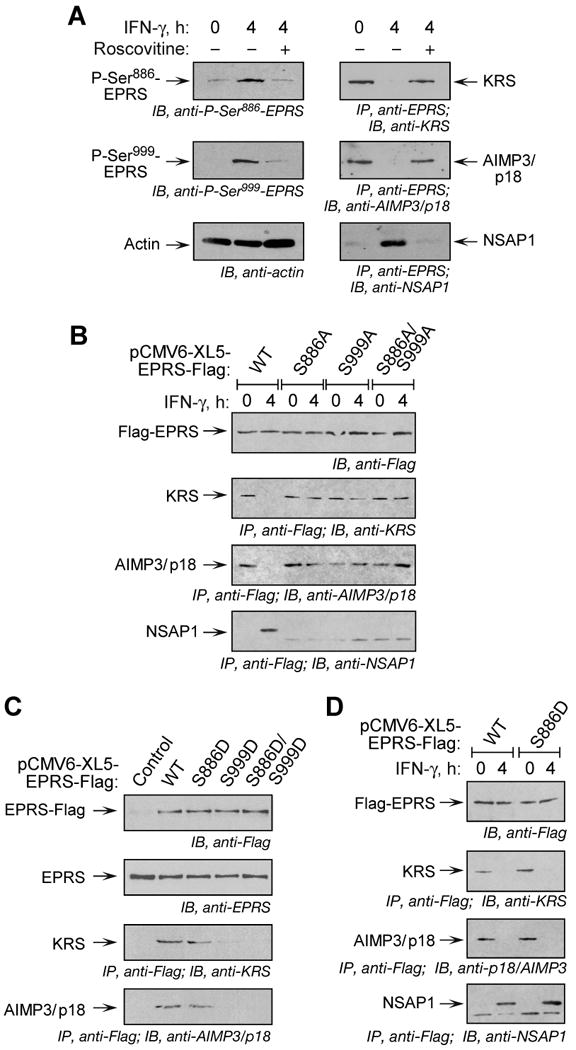
(A) Inhibition of phosphorylation blocks EPRS release from MSC. U937 cells were treated with IFN-γ and roscovitine or DSMO as in Fig. 3D. Lysates were immunoblotted with antibodies indicated. Same lysates were immunoprecipitated with anti-EPRS antibody and immunoblotted with antibodies against MSC components, KRS and AIMP3/p18, or against the pre-GAIT complex protein, NSAP1.
(B) Both Ser phosphorylation events are required for EPRS release from MSC. Flag-tagged, full-length, wild-type (WT) or phosphorylation-defective (Ser-to-Ala, S-A) EPRS mutants were cloned into pCMV6-XL5 vector and expressed in U937 cells by transient transfection. After 18 h, cycloheximide (50 μg/ml, 30 min) was added to block new synthesis, and then incubated with IFN-γ for 4 h. Lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with antibodies indicated.
(C) Incorporation of EPRS phospho-mimetics into MSC. Flag-tagged, full-length, WT and phospho-mimetic (Ser-to-Asp, S-D) mutant EPRS was expressed in U937 cells by transient transfection as in (B) and measured by immunoblot with anti-Flag antibody. Endogenous EPRS was detected by anti-EPRS antibody. Lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with antibodies indicated.
(D) Release of S886D phospho-mimetic establishes dual-site phosphorylation-dependent release of EPRS from MSC. Cells expressing Flag-tagged, full-length EPRS were incubated with IFN-γ for 4 h. Lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with antibodies indicated.
Differential Interaction of GAIT Proteins with Phosphorylated Ser886 and Ser999
All interactions of GAIT complex constituents with EPRS require a phosphorylated linker domain, which in previous studies was phosphorylated by incubation with cytosolic lysates from IFN-γ-treated cells (Jia et al., 2008). In-cell binding of GAIT proteins with EPRS linker harboring phospho-defective (Fig. 5A, middle row) and phospho-mimetic (Fig. 5A, bottom row) mutations was determined since, unlike full-length-EPRS, the linker does not accumulate in the MSC and thus is highly accessible (not shown). After IFN-γ treatment, NSAP1 interacted with wild-type and S999A mutant linker, but failed to bind the S886A mutant (Fig. 5A, left column). Also, NSAP1 interacted with the S886D mutant even in the absence of stimulation by IFN-γ. These results indicate Ser886 phosphorylation is essential for NSAP1 binding. The presence of Ser886 in the spacer connecting WHEP repeats R2 and R3 sheds light on previous results showing NSAP1 binding to this EPRS region (Jia et al., 2008).
Figure 5. Role of Specific EPRS-Ser Phosphorylation in GAIT Complex Assembly.
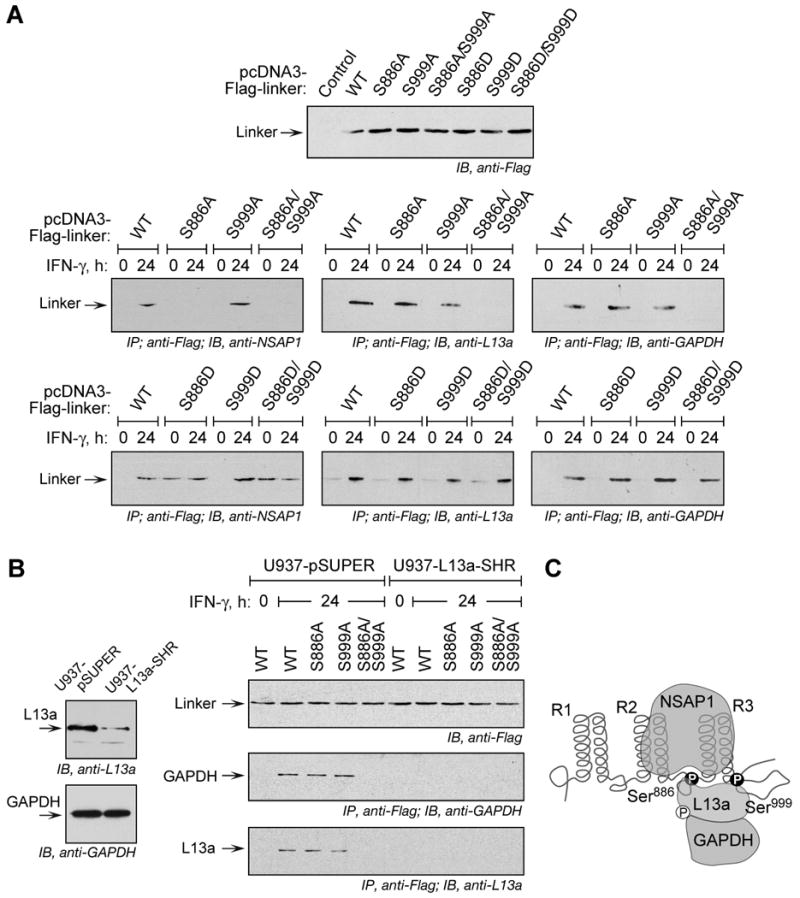
(A) Phosphorylation of Ser886 is essential for EPRS interaction with NSAP1, and phosphorylation of either Ser886 or Ser999 is essential for EPRS interaction with L13a and GAPDH. Flag-tagged wild type (WT), phospho-defective (Ser-to-Ala, S-A) and phospho-mimetic (Ser-to-Asp, S-D) mutant linkers were expressed by transient transfection and determined by immunoblot with anti-Flag antibody. After IFN-γ treatment, lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with antibodies against NSAP-1, L13a, and GAPDH.
(B) GAPDH interaction with phospho-EPRS requires L13a. Immunoblot showing depletion of L13a, and unaltered expression of GAPDH, in U937 cells with stable knockdown of L13a (U937-L13a-SHR) and vector control cells (U937-pSUPER) (left). Cells were transiently transfected with Flag-tagged WT and phosphorylation-defective EPRS linker constructs (right). Expression was determined by immunoblot with anti-Flag antibody. The cells were incubated with IFN-γ and lysates immunoprecipitated with anti-Flag antibody followed by immunoblot with anti-GAPDH or anti-L13a antibodies.
(C) Schematic depicting interaction of NSAP1, L13a, and GAPDH to EPRS linker phosphorylated at Ser886 and Ser999.
L13a interacted with EPRS linker bearing either Ser886- or Ser999-to-Ala mutation; however, the double mutation abrogated binding (Fig. 5A, middle column). L13a did not bind any phospho-mimetic EPRS mutants in unstimulated cells, consistent with a requirement for IFN-γ-induced phosphorylation of L13a for GAIT complex formation (Jia et al., 2008; Mukhopadhyay et al., 2008). Essentially identical results were seen when binding of GAPDH to EPRS linker was determined (Fig. 5A, right column) indicating L13a and GAPDH can bind either phospho-site. In addition, the covariant binding of L13a and GAPDH to EPRS linker, and the inability of GAPDH to bind phospho-mimetic EPRS linker in the absence of IFN-γ, suggests GAPDH binding to EPRS requires L13a. To test this, binding studies were done in L13a-knockdown cells; GAPDH expression was unaltered in these cells (Fig. 5B, left). In control cells, GAPDH binds all EPRS linkers that bind L13a; however, binding is completely abrogated in L13a-knockdown cells (Fig. 5B, right). These results suggest a series of interactions in which GAPDH binds phospho-EPRS indirectly via phospho-L13a that acts as a connecting bridge (Fig. 5C).
Dual Phosphorylation of EPRS Facilitates Negative and Positive Regulation of RNA-binding and Translational Control
EPRS is the only GAIT constituent that directly binds target mRNA (Sampath et al., 2004). EPRS binding to mRNA in cells is blocked by the negative regulator NSAP1, and restored by subsequent binding of phospho-L13a and GAPDH (Jia et al., 2008). The role of specific EPRS phosphorylation events on RNA binding was investigated by surface plasmon resonance (SPR) spectrometry. Biotinylated Cp GAIT element RNA was immobilized on the sensor chip and binding activity measured using solutions containing modified forms of EPRS linker by themselves, or in the presence of NSAP1 or NSAP1, GAPDH, and phospho-L13a. High-affinity binding of non-phosphorylated, wild-type linker to the RNA was not blocked by pre-incubation with NSAP1 (Fig. 6A, upper right). However, NSAP1 blocked binding of purified, phosphorylated wild-type linker (Fig. 6A, center left). Addition of GAPDH and phospho-L13a to NSAP1 restored binding of the linker to the RNA element, consistent with previous results showing an induced conformational change (Jia et al., 2008). Binding studies were done with EPRS linker containing the S886D mutation within the NSAP1 binding domain. In this case, NSAP1 blocked linker interaction with the RNA element, and binding was restored by pre-incubation with GAPDH and phospho-L13a (Fig. 6A, center right). The S999D mutant linker showed binding behavior identical to the wild-type, and the double-mutant was similar to the S886D mutant (Fig. 6A, bottom). Thus, Ser886 phosphorylation is essential and sufficient for negative regulation of EPRS binding to RNA by NSAP1, and for positive regulation by GAPDH and phospho-L13a. However, these studies do not shed light on the specific role of Ser999 phosphorylation in GAIT complex function.
Figure 6. Role of EPRS Linker Phosphorylation in Binding the GAIT RNA Element and GAIT-mediated Translational Silencing.
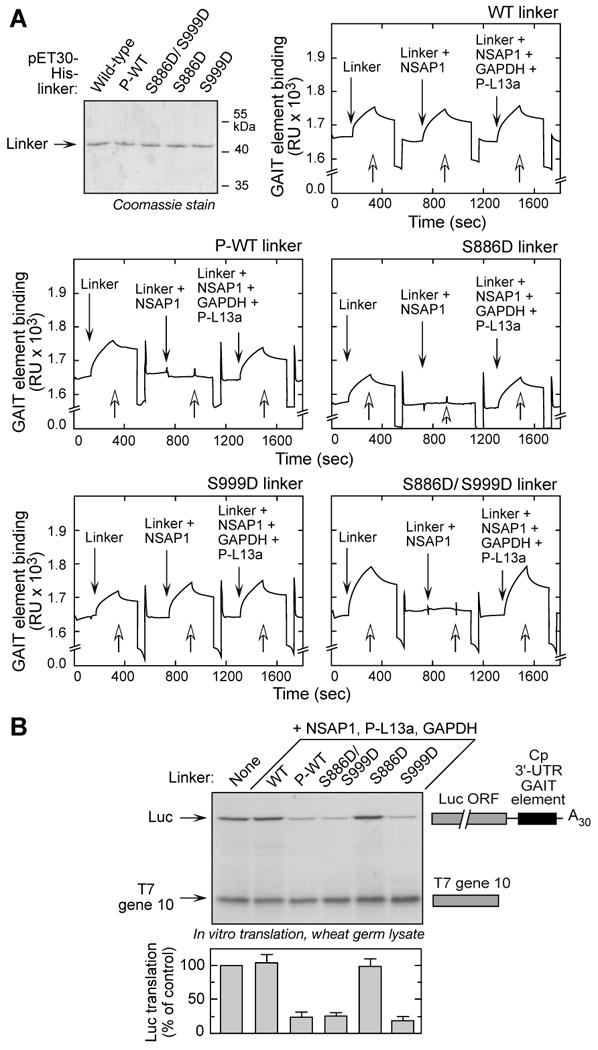
(A) Ser886 phosphorylation is sufficient for reconstituting binding to GAIT RNA element. N-terminus, His-tagged wild-type (WT) and phospho-mimetic (Ser-to-Asp, S-D) mutant linkers were expressed in E. coli and purified by Ni-affinity chromatography. Phosphorylated, wild-type (P-WT) linker was obtained by in vitro phosphorylation of purified protein with lysates of 4-h, IFN-γ-treated U937 cells, and repurification by Ni-affinity column. Biotinylated Cp GAIT RNA element was immobilized on a streptavidin sensor chip, and binding of linker to RNA was determined by SPR and expressed as resonance units (RU). After removal of bound protein with wash buffer (open arrows), binding of linkers preincubated with NSAP1 was measured, then linkers preincubated with NSAP1, GAPDH, and phospho-L13a (P-L13a) (each at 10-fold molar excess).
(B) Ser999 phosphorylation is critical for GAIT complex activation. His-tagged, wild-type (WT), phosphorylated-WT (P-WT), and phospho-mimetic linker proteins were preincubated with His-tagged NSAP1, GAPDH, and phosphorylated, His-tagged L13a (P-L13a), and then added to wheat germ extract for in vitro translation of luciferase (Luc)-Cp-GAIT element reporter in presence of 35S-labeled Met. T7 gene 10 RNA was co-translated as a specificity control. Luc expression was quantified by densitometry, normalized by T7 gene 10 translation, and expressed as % of control (mean ± SEM, n = 3 experiments).
We investigated the role of Ser886 and Ser999 phosphorylation in reconstituting GAIT-mediated translational silencing activity. A luciferase reporter transcript bearing the Cp GAIT element (Luc-Cp-GAIT) was translated in vitro in a wheat germ extract in the presence of EPRS linker, NSAP1, GAPDH, and phospho-L13a. Translational silencing was observed when the wild-type linker was phosphorylated, but not when unmodified (Fig. 6B). Unexpectedly, the linker bearing the single S999D mutation fully reconstituted translational silencing activity in the presence of the other GAIT components; the double-mutant was also effective, but the single S886D mutant was inactive. These results also show that the noncanonical, GAIT-mediated functions of EPRS do not require either of the synthetase domains, and that complete reconstitution requires only the phosphorylated linker domain.
Ser999 Phosphorylation is Essential for L13a Interaction with eIF4G
To investigate the specific requirement for Ser999 phosphorylation for functional reconstitution of the GAIT complex, we took advantage of previous studies of the molecular mechanism of GAIT-mediated translational suppression (Kapasi et al., 2007). Phospho-L13a binds eIF4G at or near the site that binds the eIF3e subunit of the eIF3 complex, which in turn blocks recruitment of the eIF3-containing 43S complex to GAIT element-bearing mRNA, and represses translation-initiation (Kapasi et al., 2007; LeFebvre et al., 2006). We tested the hypothesis that Ser999 phosphorylation is required for interaction of the GAIT complex with eIF4G, and thus for competitive inhibition of eIF3e binding to eIF4G. GAIT activity was reconstituted with EPRS linker containing Ser-to-Asp mutants along with purified GAIT complex proteins in the presence of GAIT element-bearing reporter RNA and wheat germ extract (to supply eIF4G, eIF3e, and other translation factors). The reaction mixture was treated with RNaseA/T1 (to prevent confounding, RNA-dependent interactions during the pull-down), and then subjected to co-immunoprecipitation with anti-EPRS and anti-eIF4G antibodies. In the co-immunoprecipitates with anti-EPRS antibody, in vitro phosphorylated EPRS linker and the double phospho-mimetic bound all GAIT proteins and eIF4G, whereas wild-type linker bound none of these proteins (Fig. 7A). The S999D mutant bound eIF4G and the GAIT proteins (with the expected exception of NSAP1). However, the S886D mutant did not bind eIF4G despite interaction with all GAIT complex proteins. eIF3e was not detected in any anti-EPRS antibody co-immunoprecipitates, indicating it does not interact directly with GAIT proteins, and that the translational-repression-competent GAIT complex prevents eIF3e binding to eIF4G. To determine if GAIT complex interaction with eIF4G blocks recruitment of the 43S ribosomal complex under conditions of translational repression only, we investigated the interaction of eIF3e with eIF4G (and with GAIT proteins), by co-immunoprecipitation with anti-eIF4G antibody. Reconstitution of the GAIT complex with phosphorylated wild-type, double, and single S999D phospho-mimetic mutant linkers significantly reduced the eIF3e–eIF4G interaction (Fig. 7A, bottom 4 panels). In contrast, GAIT complex reconstituted with unphosphorylated or single S886D mutant linkers showed efficient binding of eIF3e to eIF4G. These results suggest that Ser999 phosphorylation is required for interaction of the GAIT complex with eIF4G, which prevents the recruitment of 43S ribosomal complex and facilitates translational inhibition of GAIT element-bearing mRNA.
Figure 7. Ser999 Phosphorylation is Required for L13a-mediated Interaction of GAIT Complex with eIF4G.
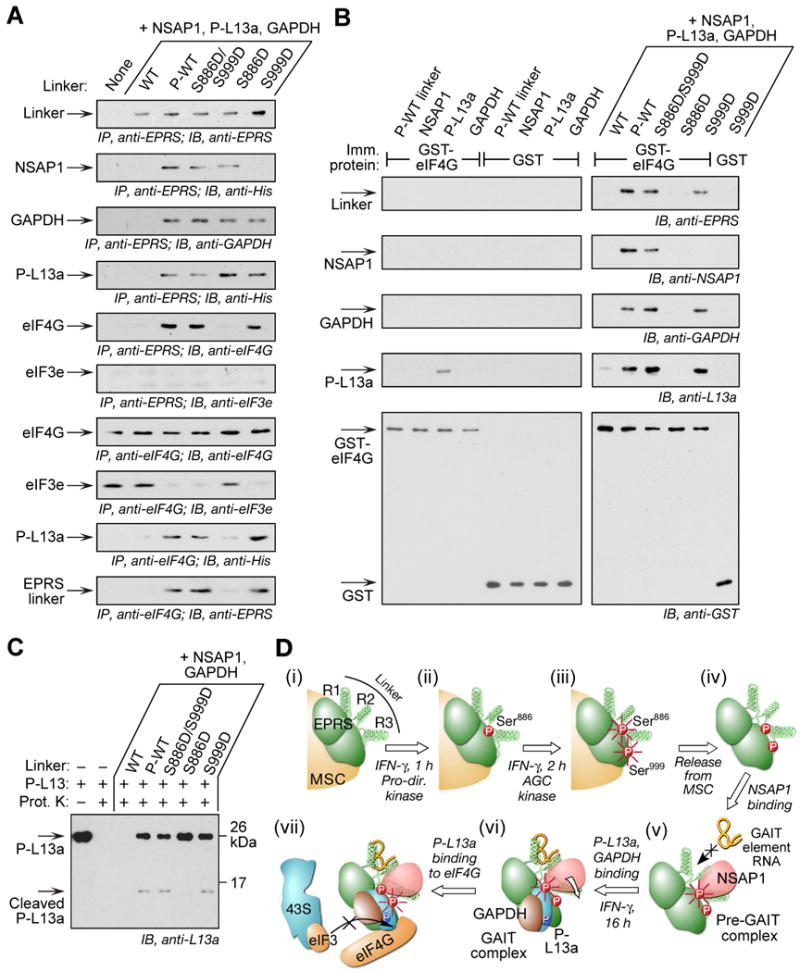
(A) Ser999 phosphorylation is required for GAIT complex interaction with eIF4G during in vitro translation. In vitro translation of GAIT element-bearing reporter in wheat germ extract as in Figure 7B. The reaction mixture was immunoprecipitated with anti-EPRS and eIF4G antibodies after digestion with RNase A/T1, and immunoblotted with antibodies indicated.
(B) Ser999 phosphorylation facilitates P-L13a-mediated interaction of GAIT complex with recombinant eIF4G. Individual (left panels) or combined (right panels) GAIT complex proteins were incubated with GST-eIF4G or GST immobilized to glutathione-agarose beads. After washing, the interaction was analyzed by immunoblotting with antibodies against GAIT components and eIF4G.
(C) EPRS Ser999 phosphorylation alters protease susceptibility of L13a. P-L13a (150 ng) was preincubated with GAIT components including mutant EPRS linkers and then treated with 10 ng proteinase (Prot.) K for 15 min at 30°C. Untreated and protease-treated P-L13a served as controls. Cleavage was determined by immunoblot with anti-L13a antibody.
(D) Schematic illustrating function of 2-site phosphorylation of EPRS
(i) EPRS resides in the MSC.
(ii) IFN-γ induces phosphorylation of Ser886 by a proline-directed kinase.
(iii) IFN-γ induces phosphorylation of Ser999 by an AGC kinase group member. Phosphorylation of both Ser886 and Ser999 (highlighted with radial lines) induces EPRS release from the MSC.
(iv) Diphosphorylated EPRS translocates from the MSC to the cytoplasm.
(v) Phosphorylation of EPRS Ser886 is required for NSAP1 binding that blocks binding of EPRS to the GAIT RNA element.
(vi) EPRS Ser886 phosphorylation is required for binding of phospho-L13a and GAPDH that induces a conformational shift and permits GAIT RNA element binding to EPRS.
(vii) Phosphorylation of EPRS Ser999 is required for translational silencing activity of GAIT complex by permitting interaction of phospho-L13a with eIF4G, and blocking the binding of eIF3 of the 43S ribosomal complex to eIF4G.
Phospho-L13a binds eIF4G directly (Kapasi et al., 2007); however, potential interaction of other GAIT proteins with eIF4G has not been investigated. We determined binding of each GAIT protein to an immobilized chimera of GST with eIF4G containing the essential eIF3 binding site (GST-eIF4G). Only phospho-L13a bound eIF4G (Fig. 7B, left panels). Similarly, binding of reconstituted GAIT complex to eIF4G was measured in the presence of EPRS linker phospho-mimetics. As before, binding to eIF4G was observed when the complex was reconstituted with phosphorylated wild-type, S999D, and S886D/S999D mutant linkers, but not with S886D (Fig. 7B, right panels). The absence of NSAP1 in the GAIT complex reconstituted with S999D linker did not prevent binding to eIF4G, consistent with its primary role as negative regulator. Phospho-L13a interacted with eIF4G much more robustly when reconstituted with phospho-EPRS in the GAIT complex than when free (in the context of binding-incompetent, wild-type linker). This result suggests the GAIT complex might influence the conformation of L13a, possibly by exposing a binding site, to facilitate its interaction with eIF4G. Moreover, the failure of the EPRS S886D-containing GAIT complex proteins to bind eIF4G suggests phosphorylation of Ser999 might be critical for establishing the binding-efficient conformation of L13a.
To evaluate the influence of EPRS linker mutations on L13a conformation, phospho-L13a by itself and in reconstituted GAIT complexes were subjected to limited proteolysis with proteinase K, and L13a cleavage determined. Phospho-L13a alone or in the presence of reconstitution-defective, wild-type linker were completely digested (Fig. 7C). When phospho-L13a was reconstituted with phosphorylated wild-type linker, or S999D or S886D/S999D EPRS linker mutants, L13a was largely protected from limited proteolysis with the exception of a single 14-15 kDa cleavage product. Notably, this product was absent when L13a was reconstituted with GAIT complex containing S886D linker. These results suggest that L13a is almost completely ensconced within the GAIT complex with limited accessibility to protease. The differential protease susceptibility between the functional (P-wild-type, S999D, S886D/S999D) and non-functional (S886D) EPRS linkers verifies their conformational differences. We speculate that the greater protease susceptibility of the functional linkers may be due to an extended conformation facilitating the interaction of L13a with eIF4G.
Discussion
We have shown that IFN-γ-induced, two-kinase-mediated, dual-site phosphorylation of EPRS at Ser886 and Ser999 in the non-catalytic linker domain control multiple events required for GAIT-mediated gene silencing activity (Fig. 7D). This includes EPRS escape from the MSC, interaction with negative and positive regulators of binding to the GAIT RNA element, binding to eIF4G, and translational silencing of target transcripts. We have also elucidated the specific role of each phosphorylation site in these events. Phosphorylation at both Ser886 and Ser999 are required for EPRS exit from its normal residence in the MSC. Released phospho-EPRS interacts with NSAP1 to form an inactive pre-GAIT complex that does not bind the GAIT element; this interaction requires EPRS phosphorylation at Ser886. After about 14-16 h, phospho-L13a and GAPDH bind EPRS inducing a conformational shift that displaces NSAP1 (Jia et al., 2008) to form the active GAIT complex that binds the GAIT element in the 3′UTR of target mRNAs. EPRS phosphorylation at Ser886 is sufficient for this interaction. Although the L13a/GAPDH complex can bind EPRS phosphorylated at either site, phosphorylation at Ser999 is essential for reconstitution of a functional GAIT complex as it permits their interaction with eIF4G of the translation initiation complex, and inhibits translation of GAIT element-bearing mRNAs by blocking the binding of eIF3 of the preinitiation complex to eIF4G
Global phosphoproteomic analysis has shown phosphorylation of several AARS, including EPRS, but in no case is the function known (Beausoleil et al., 2004; Olsen et al., 2006; Rikova et al., 2007) The concept that AARS phosphorylation may regulate aminoacylation rate was first explored in female rats fed estradiol to increase overall protein synthesis (Berg, 1977). Phosphorylation of multiple AARS was detected, but relatively small increases or decreases in catalytic activity were measured. Minor effects on aminoacylation activities and translation-elongation rate have been confirmed by others (Clemens, 1990; Venema and Traugh, 1991), suggesting that post-translational modification may regulate AARS noncanonical functions. Also, phosphorylation may contribute to AARS pathological activity as in the case of phosphorylated HisRS which is a major antigen in myositis, a human autoimmune disease (Mathews and Bernstein, 1983). The phosphorylation state of AARS in the MSC remains controversial with several reports showing phosphorylated MSC components (Damuni et al., 1982; Pendergast et al., 1987), but another failing to detect any phosphorylated protein under basal conditions (Mirande et al., 1985). Immunological stimulation of mast cells leads to the interaction of the MSC component LysRS with microphthalmia-associated transcription factor and upstream stimulatory factor-2, which enhances Ap4A production and transcriptional activity (Lee and Razin, 2005). The source of LysRS was not determined in these experiments; LysRS free in the cytoplasm, from de novo synthesis, or released from the MSC, are possible sources. Based on the observation that phosphorylation increases Ap4A production by ThrRS and SerRS (Van Dang and Traugh, 1989), phosphorylation may likewise be involved in LysRS activation (Lee and Razin, 2005). Ultraviolet irradiation induces specific phosphorylation of AIMP2/p38, an MSC component thought to function as a scaffold for the synthetases (Han et al., 2008). Like EPRS, phospho-AIMP2/p38 is released from the parental MSC to perform a noncanonical activity, in this case, interacting with nuclear p53 and preventing its degradation. These studies, together with ours on EPRS, suggest an underlying principle, namely, agonist-induced phosphorylation of AARS components induce their specific release from the MSC, and are critical for their consequent noncanonical function. We have generalized this principle as the “depot hypothesis” in which proteins in macromolecular complexes, including the ribosome, are subject to stimulus-inducible release to perform auxiliary, moonlighting functions unrelated to their role in the parent complex (Ray et al., 2007). To our knowledge, EPRS is the only AARS (or non-AARS component of the MSC) known to undergo inducible, multisite phosphorylation.
Protein phosphorylation is a major intracellular mechanism regulating diverse protein interactions important for signal transduction and for assembly of macromolecular complexes, among other functions. Multisite protein phosphorylation (i.e., phosphorylation of two or more sites of a single protein), by multiplying the potential input stimuli and output responses, can confer an array of additional benefits (Cohen, 2000; Holmberg et al., 2002). In the simplest case, phosphorylation of each individual site generates the same response, but concurrent phosphorylation of multiple sites can yield a well-defined threshold or a steeper or stronger output response (Ferrell, 1996; Nash et al., 2001). In some cases, phosphorylation of individual sites yields multiple distinct responses, for example, growth factor-stimulated receptors exhibit several autophosphorylation sites that specifically interact with distinct adapter or signaling proteins to transduce a simple input into a highly complex response. In other cases, disparate inputs induce phosphorylation at distinct sites to generate an integrated response. The classic example is p53, containing at least a dozen phosphorylation sites that bind distinct proteins which act combinatorially to give a concerted response to a variable, context-dependent group of inputs (Meek, 1998). In some cases, phosphorylation can exert either positive or negative regulation, e.g., site-specific phosphorylation of Bim can increase or decrease the sensitivity of the cell to apoptotic stimuli (Hübner et al., 2008).
As yet there is no evidence that EPRS integrates signals from multiple stimuli, instead, it choreographs the time-dependent behavior of the GAIT complex in response to IFN-γ. EPRS phosphorylation is unique among multisite phospho-targets in that sequential phosphorylation of two sites by a single stimulus, induces at least five distinct responses involving interactions with three GAIT complex proteins and an RNA structural element, and also facilitates the interaction of L13a with eIF4G. In addition to contributing to attractive forces involved in protein-protein interaction, EPRS phosphorylation can also reduce such interactions. For example, phosphorylation could facilitate EPRS release by decreasing its affinity for interacting partners within the parent MSC. Electron microscopy indicates EPRS is localized at the MSC exterior (Wolfe et al., 2005), which could minimize disruption of the remaining complex upon release. The near-simultaneous release of EPRS from the MSC and EPRS interaction with NSAP1 (Sampath et al., 2004) suggests that NSAP1 may have a role in the release process, by contributing to the conformational shift or by preventing EPRS return to the MSC. However, our finding that EPRS containing a S999D mutation, which does not bind NSAP1 and does not associate with the MSC, suggests that NSAP1 may not have a critical role in the release mechanism, and that the phosphorylation events by themselves are sufficient. Our results show that Ser886 phosphorylation is sufficient for binding all other GAIT proteins, but is not sufficient for inhibition of translation of target RNAs, which requires Ser999 phosphorylation. The latter phosphorylation is required to optimize the interaction of phospho-L13a with eIF4G in the translation-initiation complex, previously shown to block recruitment of the 43S preinitiation complex and thereby suppress translation-initiation (Kapasi et al., 2007).
The specific molecular mechanisms underlying the induced functions of phospho-EPRS are, for the most part, unknown. Phosphorylation can introduce an altered recognition site, induce an allosteric conformational change, or disrupt repulsive and attracting forces by alteration of the energy landscape (Groban et al., 2006; Sprang et al., 1988). The latter bulk electrostatic mechanism is unlikely since the addition of only two negatively charged groups is not sufficient to offset most biologically significant binding energies (Serber and Ferrell, 2007). Förster resonance energy transfer (FRET) studies suggest the EPRS linker is highly dynamic as binding of L13a and GAPDH induces a conformational shift that exposes the binding site for target RNA (Jia et al., 2008). Also, the substantial electrophoretic mobility shift induced by phosphorylation, suggests that phosphorylation-induced conformational changes may be responsible for some, or all, of the GAIT-related functions of EPRS (Li et al., 1998). The presence of both phosphorylation sites in unfolded/unstructured domains may be a key to the mechanism underlying some or all of the GAIT-related functions of EPRS. The high proportion of protein sequence encoding unstructured domains, and their persistence though evolution, has led to their recent recognition as “hotspots” for post-translational modification with critical regulatory functions (Wright and Dyson, 1999). In some cases, including one involving multisite phosphorylation (Pufall et al., 2005), phosphorylation of an unstructured domain results in increased structure in that domain, and also increased affinity for target molecules. According to one proposed thermodynamic mechanism these processes are intimately related: the entropic penalty associated with the folding transition is offset by the enthalpy of binding (Parker et al., 1999). Future structural studies will resolve the possibility that 2-site phosphorylation of unstructured domains within the EPRS linker results in gains-of-structure coincident with the observed gains-of-function.
Experimental Procedures
Reagents
Recombinant His-tagged NSAP1 and phosphorylated, His-tagged L13a were prepared as described (Jia et al., 2008), and GAPDH was purchased from Sigma (St. Louis, MO). Purified GST and GST-eIF4G were obtained from Pierce (Rockford, IL) and Abnova (Taipei, Taiwan), respectively. Kinase inhibitors were from EMD Biosciences (Darmstadt, Germany). Affinity-purified polyclonal phosphospecific antibodies against Ser886 and Ser999 phosphorylation sites were generated using 880SQSSDS(pS)PTRNSE892 and 991KNQGGGLS(pS)SGAGE1004 peptides, respectively (Open Biosystems, Huntsville, AL). Anti-EPRS (Leu753 to Thr956) and anti-L13a antibodies were obtained as described (Mazumder et al., 2003; Ray and Fox, 2007).
Cell Culture
Human U937 monocytic cells (ATCC, Rockville, MD) were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS). Cells (1×107) were treated with human IFN-γ (500 units/ml, R&D Systems) for up to 24 h (Mazumder et al., 2003). Stably knocked-down L13a-U937 cells were obtained as described (Ray and Fox, 2007). Cell lysates were prepared in Phosphosafe extraction buffer (Novagen) containing protease inhibitor cocktail (Roche).
Plasmids, Site-directed Mutagenesis, and Recombinant Protein Expression
The EPRS domains, ERS (Met1 to Val687), PRS (Lys1019 to Tyr1512), and the connecting linker (Pro683 to Asn1023) were cloned in pET-30 expression vector and expressed as described (Jia et al., 2008). pcDNA3-N-terminus-Flag-tagged linker was generated using pET30-EPRS linker as template and cloning between EcoRI and NotI restriction sites. Full-length human EPRS cDNA cloned in pCMV6-XL5 was obtained from Origene (Rockville, MD) and recloned with an in-frame, Flag-tag downstream of the open-reading frame. Specific mutations at Ser886, Ser999, and Ser1000 to Ala or Asp were introduced into the full-length EPRS or linkers using primers with the desired mutation and GeneTailor Site-Directed Mutagenesis System (Invitrogen). Mutations to alter the 886SPTR kinase recognition motif were introduced by the same method.
Phosphoprotein Enrichment
Phosphorylated proteins from control and IFN-γ-treated U937 cells were isolated using phosphoprotein enrichment kit (BD Biosciences). Cells were lysed in buffer A, and centrifuged at 10,000 g for 15 min. Supernatant (3 mg protein) was loaded onto phosphoprotein affinity column pre-equilibrated with buffer A, and incubated for 30 min. Phospho-proteins were eluted with buffer B, and exchanged with Phosphosafe extraction buffer containing protease and phosphatase inhibitors.
Immunoprecipitation and Co-immunoprecipitation Assays
To isolate phosphorylated EPRS from U937 cells, the phospho-enriched cytosolic fraction was incubated with anti-EPRS antibody cross-linked to protein A-Sepharose beads in immunoprecipitation (IP) buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, EDTA-free complete protease inhibitor, and phosphatase inhibitor cocktail). The beads were washed in detergent-containing wash buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, and 0.1% Triton X-100) and twice in detergent-free wash buffer. Flag-tagged, non-phosphorylated or phosphorylated linkers expressed in U937 cells were immunoprecipitated using anti-Flag M2 agarose (Sigma). For protein-protein interaction studies by co-immunoprecipitation, lysates were incubated with antibody in detergent-free IP buffer followed by several washes with detergent-free buffer.
Mass Spectrometry
Coomassie-stained, phosphorylated EPRS was in-gel digested with trypsin and analyzed by capillary column LC-tandem MS analysis to map peptides, and identify phosphopeptides. The experiments were done using LTQ-linear ion trap MS and LCQ-Decaion trap MS systems (ThermoFinnigan, San Jose, CA) equipped with nanospray ionization sources. Data were acquired in data-dependent mode to simultaneously record full-scan mass and CID spectra. For peptide mapping, the CID spectra were compared to the sequence of human EPRS using Sequest (ThermoFinnigan). For identification of phosphopeptides and specific phosphorylation sites, CID spectra were searched for peptides that contain pSer or pThr or pTyr modification by a combination of database searches, by plotting neutral loss chromatograms to show characteristic loss of phosphate group, and by selected reaction-monitoring experiments.
Cell Transfection
Plasmid DNA suspended in 100 μl of nucleofector solution V (Amaxa, Köln, Germany) was nucleofected into U937 cells. Transfected cells were immediately transferred to OptiMEM media for 6 h, and then to RPMI 1640 containing 10% FBS for 18 h.
32P-labeling
U937 cells were transfected with Flag-tagged plasmid DNA encoding wild-type or mutant linker. The cells were incubated with IFN-γ for 30 min, and then labeled with [32P]orthophosphate (500 μci, Perkin-Elmer, Boston, MA) for 4 h in phosphate-free medium. The 32P-labeled cells were lysed in Phosphosafe buffer (Novagen, Darmstadt) and labeled proteins immunoprecipitated with anti-Flag antibody as described above.
RNA EMSA
32P-labeled Cp GAIT element was prepared by oligonucleotide-directed transcription using MEGAshortscript system (Ambion) and custom-synthesized RNA probe template (Invitrogen) as described (Sampath et al., 2003).
Reconstitution of GAIT Complex Function
Capped, poly(A)-tailed Luc-Cp GAIT and T7 gene 10 reporter RNAs were prepared as described (Jia et al., 2008). The GAIT complex was reconstituted in vitro by incubating 5 pmol each of EPRS linker (wild-type or mutant) and GAIT component proteins (His-tagged NSAP1, GAPDH, and phosphorylated, His-tagged L13a). Gel-purified reporter RNAs (200 ng each) were incubated with in vitro reconstituted GAIT complex in the presence of 35S-labeled Met in wheat germ extract. To determine interaction of GAIT complex proteins with eIF4G and eIF3e, the in vitro translation reaction mixture was scaled up 10-fold in the presence of a 100-fold excess of reconstituted GAIT complex, and unlabeled complete amino acids. The reaction mixture was digested with RNase A/T1, co-immunoprecipitated with anti-EPRS or anti-eIF4G antibodies cross-linked to protein A-Sepharose, and analyzed by immunoblot.
In Vitro Phosphorylation
Cell lysates (1 μg protein) and purified, recombinant His-tagged ERS, linker, or PRS substrates (0.5 μg) were pre-incubated for 5 min in kinase assay buffer (50 mM Tris-HCl, (pH 7.6), 1 mM dithiothreitol, 10 mM MgCl2, and 1 mM CaCl2) containing phosphatase inhibitor cocktail. The reaction was initiated by incubation with 5 μCi [γ-32P]ATP (Perkin-Elmer) for 15 min, and terminated using SDS gel-loading dye and heat-denaturation. Phosphorylation was determined by SDS-PAGE and autoradiography. To assay kinase activity using peptide substrates, synthetic Ser886 (881QRRDRSPTRNREPA) and Ser999 (993QRGGLHSSGAGEGQ) peptides (50 μM) were phosphorylated with [γ-32P]ATP (1 μCi); Arg and His substitutions were made to facilitate peptide binding to phosphocellulose (Casnellie, 1991). Equal volumes were spotted onto P81-phosphocellulose squares, washed, and radioactivity measured by scintillation counting.
Analysis of Protein-RNA Binding by SPR
Binding of purified, recombinant His-tagged linker proteins to GAIT RNA element was determined by SPR in a Biacore 3000 system as described (Jia et al., 2008). Biotinylated GAIT RNA element was immobilized on a streptavidin sensor chip in buffer containing 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA and 0.005% v/v surfactant P20. The flow rate for the analyte was 30 μl/min in the same buffer with 5 mM MgCl2. Binding was analyzed by Biaevaluation software.
Protein-protein Interaction by GST pull-down
Purified GST or GST-eIF4G were incubated with glutathione-agarose beads for 1 h in buffer containing 20 mM Tris-HCl pH 7.6, 0.1 mM EDTA, and 100 mM NaCl. Equimolar amounts of GAIT components and immobilized proteins were incubated for 2 h in the same buffer. After washing, the samples were heat-denatured in SDS gel-loading dye and the interaction detected by immunoblotting.
Acknowledgments
This work was supported by N.I.H. grants P01 HL29582, P01 HL76491, R01 HL94441, and R01 GM86430 (to P.L.F.), and by A.H.A., Ohio Valley Affiliate Postdoctoral Fellowships (to A.A. and J.J.). None of the authors have any financial conflict of interest with the information in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A. A curated compendium of phosphorylation motifs. Nat Biotechnol. 2007;25:285–286. doi: 10.1038/nbt0307-285. [DOI] [PubMed] [Google Scholar]
- Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A. 2004;101:12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg BH. The early influence of 17-β-oestradiol on 17 aminoacyl-tRNA synthetases of mouse uterus and liver. Phosphorylation as a regulation mechanism. Biochim Biophys Acta. 1977;479:152–171. doi: 10.1016/0005-2787(77)90136-8. [DOI] [PubMed] [Google Scholar]
- Cahuzac B, Berthonneau E, Birlirakis N, Guittet E, Mirande M. A recurrent RNA-binding domain is appended to eukaryotic aminoacyl-tRNA synthetases. EMBO J. 2000;19:445–452. doi: 10.1093/emboj/19.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casnellie JE. Assay of protein kinases using peptides with basic residues for phosphocellulose binding. Methods Enzymol. 1991;200:115–120. doi: 10.1016/0076-6879(91)00133-h. [DOI] [PubMed] [Google Scholar]
- Clemens MJ. Does protein phosphorylation play a role in translational control by eukaryotic aminoacyl-tRNA synthetases? Trends Biochem Sci. 1990;15:172–175. doi: 10.1016/0968-0004(90)90153-3. [DOI] [PubMed] [Google Scholar]
- Cohen P. The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- Damuni Z, Caudwell FB, Cohen P. Regulation of the aminoacyl-tRNA synthetase complex of rat liver by phosphorylation/dephosphorylation in vitro and in vivo. Eur J Biochem. 1982;129:57–65. doi: 10.1111/j.1432-1033.1982.tb07020.x. [DOI] [PubMed] [Google Scholar]
- Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21:460–466. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groban ES, Narayanan A, Jacobson MP. Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput Biol. 2006;2:e32. doi: 10.1371/journal.pcbi.0020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, Park BJ, Park SG, Oh YS, Choi SJ, Lee SW, Hwang SK, Chang SH, Cho MH, Kim S. AIMP2/p38, the scaffold for the multi-tRNA synthetase complex, responds to genotoxic stresses via p53. Proc Natl Acad Sci U S A. 2008;105:11206–11211. doi: 10.1073/pnas.0800297105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg CI, Tran SE, Eriksson JE, Sistonen L. Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem Sci. 2002;27:619–627. doi: 10.1016/s0968-0004(02)02207-7. [DOI] [PubMed] [Google Scholar]
- Hübner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–425. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Söll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- Jeong EJ, Hwang GS, Kim KH, Kim MJ, Kim S, Kim KS. Structural analysis of multifunctional peptide motifs in human bifunctional tRNA synthetase: Identification of RNA-binding residues and functional implications for tandem repeats. Biochemistry. 2000;39:15775–15782. doi: 10.1021/bi001393h. [DOI] [PubMed] [Google Scholar]
- Jia J, Arif A, Ray PS, Fox PL. WHEP domains direct noncanonical function of glutamyl-prolyl tRNA synthetase in translational control of gene expression. Mol Cell. 2008;29:679–690. doi: 10.1016/j.molcel.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapasi P, Chaudhuri S, Vyas K, Baus D, Komar AA, Fox PL, Merrick WC, Mazumder B. L13a blocks 48S assembly: Role of a general initiation factor in mRNA-specific translational control. Mol Cell. 2007;25:113–126. doi: 10.1016/j.molcel.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko YG, Kim EY, Kim T, Park H, Park HS, Choi EJ, Kim S. Glutamine-dependent antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276:6030–6036. doi: 10.1074/jbc.M006189200. [DOI] [PubMed] [Google Scholar]
- Ko YG, Park H, Kim S. Novel regulatory interactions and activities of mammalian tRNA synthetases. Proteomics. 2002;2:1304–1310. doi: 10.1002/1615-9861(200209)2:9<1304::AID-PROT1304>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Kyriacou SV, Deutscher MP. An important role for the multienzyme aminoacyl-tRNA synthetase complex in mammalian translation and cell growth. Mol Cell. 2008;29:419–427. doi: 10.1016/j.molcel.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Cho BH, Park SG, Kim S. Aminoacyl-tRNA synthetase complexes: beyond translation. J Cell Sci. 2004a;117:3725–3734. doi: 10.1242/jcs.01342. [DOI] [PubMed] [Google Scholar]
- Lee YN, Nechushtan H, Figov N, Razin E. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcεRI-activated mast cells. Immunity. 2004b;20:145–151. doi: 10.1016/s1074-7613(04)00020-2. [DOI] [PubMed] [Google Scholar]
- Lee YN, Razin E. Nonconventional involvement of LysRS in the molecular mechanism of USF2 transcriptional activity in FcεRI-activated mast cells. Mol Cell Biol. 2005;25:8904–8912. doi: 10.1128/MCB.25.20.8904-8912.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeFebvre AK, Korneeva NL, Trutschl M, Cvek U, Duzan RD, Bradley CA, Hershey JW, Rhoads RE. Translation initiation factor eIF4G-1 binds to eIF3 through the eIF3e subunit. J Biol Chem. 2006;281:22917–22932. doi: 10.1074/jbc.M605418200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Cornea RL, Autry JM, Jones LR, Thomas DD. Phosphorylation-induced structural change in phospholamban and its mutants, detected by intrinsic fluorescence. Biochemistry. 1998;37:7869–7877. doi: 10.1021/bi9801053. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Mathews MB, Bernstein RM. Myositis autoantibody inhibits histidyl-tRNA synthetase: a model for autoimmunity. Nature. 1983;304:177–179. doi: 10.1038/304177a0. [DOI] [PubMed] [Google Scholar]
- Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto P, Fox PL. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell. 2003;115:187–198. doi: 10.1016/s0092-8674(03)00773-6. [DOI] [PubMed] [Google Scholar]
- Meek DW. Multisite phosphorylation and the integration of stress signals at p53. Cell Signal. 1998;10:159–166. doi: 10.1016/s0898-6568(97)00119-8. [DOI] [PubMed] [Google Scholar]
- Mirande M, Le Corre D, Waller JP. A complex from cultured Chinese hamster ovary cells containing nine aminoacyl-tRNA synthetases. Thermolabile leucyl-tRNA synthetase from the tsH1 mutant cell line is an integral component of this complex. Eur J Biochem. 1985;147:281–289. doi: 10.1111/j.1432-1033.1985.tb08748.x. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R, Ray PS, Arif A, Brady AK, Kinter M, Fox PL. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol Cell. 2008;32:371–382. doi: 10.1016/j.molcel.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Park SG, Ewalt KL, Kim S. Functional expansion of aminoacyl-tRNA synthetases and their interacting factors: new perspectives on housekeepers. Trends Biochem Sci. 2005a;30:569–574. doi: 10.1016/j.tibs.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Park SG, Kim HJ, Min YH, Choi EC, Shin YK, Park BJ, Lee SW, Kim S. Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc Natl Acad Sci U S A. 2005b;102:6356–6361. doi: 10.1073/pnas.0500226102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker D, Rivera M, Zor T, Henrion-Caude A, Radhakrishnan I, Kumar A, Shapiro LH, Wright PE, Montminy M, Brindle PK. Role of secondary structure in discrimination between constitutive and inducible activators. Mol Cell Biol. 1999;19:5601–5607. doi: 10.1128/mcb.19.8.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast AM, Traugh JA. Alteration of aminoacyl-tRNA synthetase activities by phosphorylation with casein kinase I. J Biol Chem. 1985;260:11769–11774. [PubMed] [Google Scholar]
- Pendergast AM, Venema RC, Traugh JA. Regulation of phosphorylation of aminoacyl-tRNA synthetases in the high molecular weight core complex in reticulocytes. J Biol Chem. 1987;262:5939–5942. [PubMed] [Google Scholar]
- Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J. 2007;403:217–234. doi: 10.1042/BJ20070024. [DOI] [PubMed] [Google Scholar]
- Pufall MA, Lee GM, Nelson ML, Kang HS, Velyvis A, Kay LE, McIntosh LP, Graves BJ. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–145. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- Ray PS, Arif A, Fox PL. Macromolecular complexes as depots for releasable regulatory proteins. Trends Biochem Sci. 2007;32:158–164. doi: 10.1016/j.tibs.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Ray PS, Fox PL. A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. EMBO J. 2007;26:3360–3372. doi: 10.1038/sj.emboj.7601774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457:915–919. doi: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas de Pouplana L, Schimmel P. Aminoacyl-tRNA synthetases: potential markers of genetic code development. Trends Biochem Sci. 2001;26:591–596. doi: 10.1016/s0968-0004(01)01932-6. [DOI] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Robinson JC, Kerjan P, Mirande M. Macromolecular assemblage of aminoacyl-tRNA synthetases: Quantitative analysis of protein-protein interactions and mechanism of complex assembly. J Mol Biol. 2000;304:983–994. doi: 10.1006/jmbi.2000.4242. [DOI] [PubMed] [Google Scholar]
- Sampath P, Mazumder B, Seshadri V, Fox PL. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol Cell Biol. 2003;23:1509–1519. doi: 10.1128/MCB.23.5.1509-1519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, Fox PL. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004;119:195–208. doi: 10.1016/j.cell.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Serber Z, Ferrell JE., Jr Tuning bulk electrostatics to regulate protein function. Cell. 2007;128:441–444. doi: 10.1016/j.cell.2007.01.018. [DOI] [PubMed] [Google Scholar]
- Shiba K. Intron positions delineate the evolutionary path of a pervasively appended peptide in five human aminoacyl-tRNA synthetases. J Mol Evol. 2002;55:727–733. doi: 10.1007/s00239-002-2368-3. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Lu KP, Kwon YT, Tsai LH, Filhol O, Cochet C, Brickey DA, Soderling TR, Bartleson C, Graves DJ, et al. A structural basis for substrate specificities of protein Ser/Thr kinases: Primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol Cell Biol. 1996;16:6486–6493. doi: 10.1128/mcb.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprang SR, Acharya KR, Goldsmith EJ, Stuart DI, Varvill K, Fletterick RJ, Madsen NB, Johnson LN. Structural changes in glycogen phosphorylase induced by phosphorylation. Nature. 1988;336:215–221. doi: 10.1038/336215a0. [DOI] [PubMed] [Google Scholar]
- Tzima E, Schimmel P. Inhibition of tumor angiogenesis by a natural fragment of a tRNA synthetase. Trends Biochem Sci. 2006;31:7–10. doi: 10.1016/j.tibs.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Van Dang CV, Traugh JA. Phosphorylation of threonyl- and seryl-tRNA synthetase by cAMP-dependent protein kinase. A possible role in the regulation of P1, P4-bis(5′-adenosyl)-tetraphosphate (Ap4A) synthesis. J Biol Chem. 1989;264:5861–5865. [PubMed] [Google Scholar]
- Venema RC, Traugh JA. Protein kinase C phosphorylates glutamyl-tRNA synthetase in rabbit reticulocytes stimulated by tumor promoting phorbol esters. J Biol Chem. 1991;266:5298–5302. [PubMed] [Google Scholar]
- Vyas K, Chaudhuri S, Leaman DW, Komar AA, Musiyenko A, Barik S, Mazumder B. Genome-wide polysome profiling reveals an inflammation-responsive post-transcriptional operon in IFN-γ-activated monocytes. Mol Cell Biol. 2009;29:458–470. doi: 10.1128/MCB.00824-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe CL, Warrington JA, Treadwell L, Norcum MT. A three-dimensional working model of the multienzyme complex of aminoacyl-tRNA synthetases based on electron microscopic placements of tRNA and proteins. J Biol Chem. 2005;280:38870–38878. doi: 10.1074/jbc.M502759200. [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]