

Abstract
Tissue-nonspecific alkaline phosphatase (TNAP) plays a central role in regulating extracellular matrix calcification during bone formation and growth. High throughput screening (HTS) for small molecule TNAP inhibitors led to the identification of hits in the sub-micromolar potency range. We report the design, synthesis and in vitro evaluation of a series of pyrazole derivatives of a screening hit which are potent TNAP inhibitors exhibiting IC50 values as low as 5 nM. A representative of the series was characterized in kinetic studies and determined to have a mode of inhibition not previously observed for TNAP inhibitors.
The alkaline phosphatase isozyme family in mammals is comprised of two groups, the tissue-specific alkaline phosphatases (placental, intestinal and germ cell) and tissue-nonspecific alkaline phosphatase (TNAP).1 The major function of TNAP in bone tissue is the degradation of extracellular inorganic pyrophosphate (PPi), a potent inhibitor of calcification, to inorganic phosphate. In this way a controlled steady state level of PPi, is maintained, thus sustaining normal bone mineralization. Increased expression of TNAP accelerates calcification in bovine vascular smooth muscle cells (VSMCs),2 and macrophages can induce a calcifying phenotype in human VSMCs by activating TNAP in the presence of IFNγ and 1,25(OH)2D3.3 Small molecule inhibitors of TNAP therefore have the potential to probe the causative mechanisms, or treat the pathology, of diseases caused by medial calcification such as idiopathic infantile arterial calcification, end-stage renal disease and diabetes. 4-6 Until now, levamisole and theophilline were the only available inhibitors of TNAP with Ki values of 16 and 82 μM, respectively.7 We recently reported the discovery of novel potent and selective small molecule inhibitors of TNAP using high-throughput screening (HTS).8 Herein we report our efforts on the hit-to-lead optimization of a pyrazole TNAP inhibitor screening hit with micromolar potency to provide novel derivatives with low nanomolar in vitro potency and excellent selectivity for TNAP. The structures and IC50 data for compounds were deposited to PubChem under AID 1056 (http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1056).
High throughput screening (HTS) of 66,000 compounds using a luminescence-based assay9,10 (see PubChem link to AID 1056 for details) developed in the Burnham Center for Chemical Genomics (BCCG) led to the identification of the pyrazole derivative CID-646303 (1 in Figure 1). Preliminary hit follow up was accomplished by performing similarity searches on databases of commercially available analogues. In this initial phase, 50 commercial analogues were identified, purchased and tested for their ability to inhibit TNAP. This allowed us to define some important features of the structure-activity relationships (SAR). For example, the potency in this series was improved from IC50 = 0.98 μM for the lead pyrazole 1 to IC50 = 0.50 μM for the 2,4-dichlorophenyl ester derivative 2 (Figure 1). Furthermore, conversion of the tricyclic derivative 3, with an IC50 value of 1.33 μM, to the pyrrolidine amide analogue 4 led to a 3-fold improvement in potency (IC50 = 0.50 μM). Encouraged by these results we designed and synthesized two focused libraries of substituted pyrazole amide analogues. In order to optimize the potency of the hit structure the pyrazole acid scaffold 8 was selected as the key synthon for the preparation of amide analogues (Scheme 1).
Figure 1.

Initial hit from screening and commercial analogues.
Scheme 1.
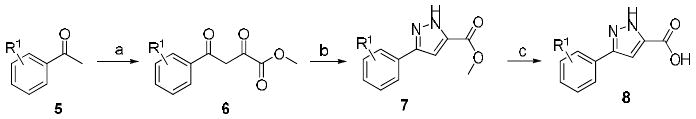
Reagents and conditions: (a) (i) NaOMe, Et2O, dimethyl oxalate, 25 °C, 4 -12h, (ii) AcOH (75-90%); (b) N2H2, AcOH, 100 °C, 12 h (50-85%) (c) LiOH, THF, MeOH, reflux (90-95%).
The synthetic chemistry used for the preparation of the pyrazole acid scaffolds is shown in Scheme 1. Reaction of acetophenone derivatives 5 with sodium methoxide and dimethyl oxalate yielded the 1,3-diketone derivatives 6 in excellent yields (75–90%). Compound 6 was then reacted with hydrazine to give the corresponding pyrazole ester 7. Saponification of the methyl ester provided access to the pyrazole acids 8.
The synthetic chemistry used for hit optimization is shown in Scheme 2. The pyrazole acid 8 was treated with HOBT, EDC and DIEA to produce the amides 911 or the desired hydrazide derivative 10.
Scheme 2.

Reagents and conditions: (a) EDC, HOBT, DMF, DIEA, NH2X (85-95%).
In light of the preliminary data generated from the HTS hits and commercial analogues our goal was to determine the key components of the SAR required for potency. For the focused library synthesis we selected a 2,4-dichloro and 2,4-dichloro-5-fluoro substitution pattern for the phenyl ring based on the initial SAR data. In the first library, twenty six compounds were synthesized and tested in the in vitro assay. This led to the identification of four analogues with potency values of 100 nM or better (Table 1). The incorporation of a hydroxyl group on the amide generally increased potency (9a and 9j). In all cases the 2,4-dichloro analogues were more potent than the corresponding 2,4-dichloro-5-fluoro analogues (Table 1). A second generation set of pyrazoles consisting of a library of twenty eight compounds were synthesized next (Table 2). In this series we found that branching of the amides generally decreased potency in the in vitro assay, especially when the chain length was greater than three carbon atoms. We also observed that amides with chain lengths of three carbons or less were the most active (Tables 1 and 2).
Table 1.
Summary of in vitro data from first focused library.
 | |||
|---|---|---|---|
| Compound | R1 | RNH2 | IC50(μM) |
| 9a | H | NH2CH2CH2OH | 0.044 |
| 9b | H | 0.051 | |
| 9c | H | 0.119 | |
| 9d | H | 0.398 | |
| 9e | H | 0.828 | |
| 9f | H | 0.488 | |
| 9g | H |  |
0.375 |
| 9h | H | 0.574 | |
| 9i | H |  |
1.450 |
| 9j | F | NH2CH2CH2OH | 0.100 |
| 9k | F | 0.324 | |
| 9l | F | 4.850 | |
| 10 | H | NH2NH2 | 0.044 |
Table 2.
Summary of in vitro data from second focused library.
 | |||
|---|---|---|---|
| Compound | R1 | RNH2 | IC50 (μM) |
| 9m | H | NH2CH2CH2CH2OH | 0.031 |
| 9n | H | NH2CH2CH2CH2CH2OH | 0.035 |
| 9o | H | NH2CH2CH2OMe | 0.046 |
| 9p | H | NH2(CH2CH2OH)2 | 0.047 |
| 9q | H | 0.082 | |
| 9r | H | 0.459 | |
| 9s | H | 0.285 | |
| 9t | F | NH2CH2CH2CH2OH | 0.056 |
| 9u | F | NH(CH2CH2OH)2 | 0.127 |
In the second series, when the hydroxyethyl chain was increased by one or two additional carbon atoms (9m, 9n), or a second hydroxyethyl side chain was added (9p), these changes did not affect the potency of the initial hit 9a (Tables 1 and 2). The most potent compound in this series was the 2,3,4-trichlorophenyl analogue 9v (Table 3). This compound showed exceptional activity with an in vitro IC50 of 5 nM (Table 3). Furthermore, compound 9v was inactive (IC50 > 10 μM) against both the related placental alkaline phosphatase (PLAP) isozyme and the house keeping enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), indicating a selectivity for TNAP of at least 2000-fold.
Table 3.
Summary of in vitro data for trichloro- and trifluorophenyl derivatives.
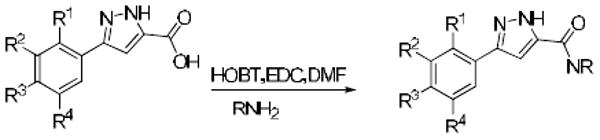 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | RNH2 | IC50 (μM) |
| 9v | CI | CI | CI | H | NH2CH2CH2OH | 0.005 |
| 9w | F | H | F | F | NH2CH2CH2OH | 0.134 |
| 9x | F | H | F | H | NH2CH2CH2OH | 0.035 |
We next performed a series of experiments to elucidate the mechanism of action (MOA) of the novel TNAP inhibitors. The catalytic mechanism by which TNAP degrades PPi consists of rapid phosphorylation of the active site in the presence of the phosphor-donor substrate and a rate-limiting dephosphorylation by the phospho-acceptor substrate, either water or amino-containing alcohols (Figure 2). All the TNAP inhibitors known to date are uncompetitive with respect to phospho-donors7,8 and are likely to be non- or uncompetitive with diethanolamine (DEA).
Figure 2.

The catalytic mechanism of the alkaline phosphatase reaction.12 The initial alkaline phosphatase (E) catalyzed reaction consists of a substrate (DO-Pi) binding step, phosphate-moiety transfer to Ser-93 (in the TNAP sequence of its active site) and product alcohol (DOH) release. In the second step of the reaction, phosphate is released through hydrolysis of the covalent intermediate (E-Pi) and the non-covalent complex (E·Pi) of the inorganic phosphate in the active site. In the presence of nitrogen-containing alcohol molecules (AOH), such as the buffer diethanolamine (DEA), phosphate is also released via a transphosphorylation reaction.
The latter conclusion is based on the fact that the majority of the alkaline phosphatase assays are performed in the presence of saturating concentrations of DEA or other phosphor-acceptors.9
To characterize the mode of action (MOA) of the novel TNAP inhibitor series we selected compound 9v for additional studies. By performing detailed kinetic studies,13 we demonstrated that 9v is competitive with respect to both substrates, the water-soluble 1,2-dioxetane reagent disodium 2-chloro-5-(5′-chloro-4-methoxyspiro[1,2-dioxetane-3,2′-tricyclo[3.3.1.13,7]decan]-4-yl)-phenol-1-(dihydrogen phosphate) (CDP-star) and DEA (Figure 3). This is the first time that a competitive MOA has been established for an inhibitor of TNAP.
Figure 3.

Mechanism of action studies for compound 9v: Lineweaver-Burke plots. TNAP kinetic data were obtained in the presence of (a) 6.25 mM DEA or (b) 18.8 μM CDP-star. The concentration of the second substrate was varied in the presence of different concentrations of 9v. Closed circle– 1000 nM, closed square – 500 nM, open square – 250 nM, closed triangle - 125 nM, open triangle – 62.5 nM, closed upside-down triangle – 31 nM, open upside-down triangle – 15.6 nM.
To gain some structural insight into how the inhibitors interact with TNAP protein, we performed in silico docking studies to investigate the potential binding mode of 9a in the active site of TNAP using the Schrodinger Glide program.14
Since the TNAP 3-dimensional crystal structure is not available, a homology model, built using the placental alkaline phosphatase (PLAP) crystal structure (PDB code: 1EW2) as the template, was used for docking15. The sequence identity of TNAP and PLAP is 56% according to the sequence alignment algorithm blastp provided by NCBI BLAST (http://blast.ncbi.nlm.nih.gov/), while the homology is 74%. The binding mode suggested by Glide is shown in Figure 4. In this model, the carbonyl of the amide chain coordinates to the Zn2+ ion and the 2,4-dichloro phenyl ring of compound 9a contributes to the binding through a stacking interaction with the side chain of Tyr371.
Figure 4.
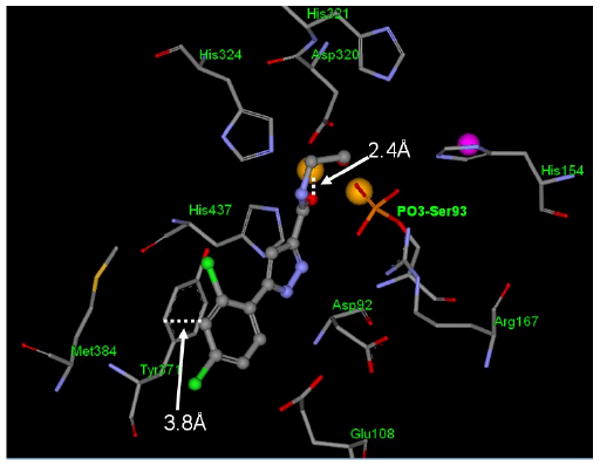
Proposed binding mode of 9a in the catalytic site of the enzyme. Compound 9a is displayed in ball and stick mode and the protein residues within 4Å of the ligand are displayed in stick mode. Dashed lines highlight the potential key interactions between protein and 9a. The zinc ions are shown in gold, magnesium ion in magenta.
In agreement with the importance of the stacking interaction, replacement of the phenyl group by an acyl group results in complete loss of activity (data not shown). Compound 9a was docked into the catalytic site either with serine 93 phosphorylated or with free serine 93. Interestingly, the binding modes are the same, suggesting that compound 9a functions as a competitive inhibitor. This predicted inhibition mechanism is consistent with the kinetic/mechanistic studies.
In summary, we have described the design, synthesis and chemical optimization of a series of pyrazole amide derivatives that are potent inhibitors of TNAP. The hit-to-lead optimization of the screening hit 1 led to compound 9v, which is approximately 200 times more potent against TNAP, and shows a high degree of selectivity against the related PLAP isozyme. Mechanistic studies demonstrated a novel MOA for 9v, and these data were supported by in silico docking studies. Compound 9v should prove to be an extremely useful small molecule tool to facilitate investigations into the biochemistry and pharmacology of TNAP.
Acknowledgments
This work was supported by NIH grants U54HG003916 and DE12889.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Millán JL. Mammalian alkaline phosphatases. From biology to applications in medicine and biotechnology. Wiley-VCH Verlag GmbH & Co; Weinheim, Germany: 2006. pp. 1–322. [Google Scholar]
- 2.Shioi A, Nishizawa Y, Jono S, Koyama H, Hosoi M, Morii H. Arterioscler Thromb Vasc Biol. 1995;15:2003. doi: 10.1161/01.atv.15.11.2003. [DOI] [PubMed] [Google Scholar]
- 3.Shioi A, Katagi M, Okuno Y, Mori K, Jono S, Koyama H, Nishizawa Y. Circ Res. 2002;91:9. doi: 10.1161/01.res.0000026421.61398.f2. [DOI] [PubMed] [Google Scholar]
- 4.Lomashvili KA, Garg P, Narisawa S, Millan JL, O'Neill WC. Kidney Int. 2008;73:1024. doi: 10.1038/ki.2008.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Addison WN, Azari F, Sorensen ES, Kaartinen MT, McKee MD. J Biol Chem. 2007;282:15872. doi: 10.1074/jbc.M701116200. [DOI] [PubMed] [Google Scholar]
- 6.Kaunitz JD, Yamaguchi DT. J Cell Biochem. 2008;105:655. doi: 10.1002/jcb.21885. [DOI] [PubMed] [Google Scholar]
- 7.Kozlenkov A, Hoylaerts MF, Ny T, Le Du MH, Millán JL. J Bone Miner Res. 2004;19:1862. doi: 10.1359/JBMR.040608. [DOI] [PubMed] [Google Scholar]
- 8.Narisawa S, Harmey D, Yadav MC, O'Neill WC, Hoylaerts MF, Millán JL. J Bone Miner Res. 2007;22(11):1700. doi: 10.1359/jbmr.070714. [DOI] [PubMed] [Google Scholar]
- 9.Sergienko EA, Millán JL. unpublished results. [Google Scholar]
- 10.TNAP activity was measured on an EnVision plate reader in the presence of 100 mM diethanolamine-HCl, pH 9.8, 1 mM MgCl2, 20 μM ZnCl2, and 50 μM CDP-star.
- 11.Representative synthesis of 9v: A mixture of 2,3,4-trichloroacetophenone (4.46 g, 0.02 mol) and dimethyl oxalate (2.36 g, 0.02 mol) was dissolved in Et2O (100 mL) and stirred at room temperature. To this solution was added a freshly prepared solution of NaOMe, prepared from sodium metal (0.55 g, 0.0024 mol) dissolved in MeOH (10 mL). A yellow precipitate immediately formed and the reaction was stirred for 1 h at room temperature. The precipitate was filtered and dissolved in water (200 mL) and the solution was adjusted to pH 3 with HOAc. The resulting precipitate was filtered and dried to give 5.20 g (84%) of methyl 2,4-dioxo-4-(2,3,4-trichlorophenyl)butanoate (6). A mixture 6 (5.20 g, 0.018 mol) and HOAc (100 mL) was stirred at room temperature. To this solution was added hydrazine (7.5 mL, 0.24 mol) and the reaction mixture was stirred overnight. The precipitate was filtered, washed with hexane and dried to give 5.19 g (95%) of methyl 3-(2,3,4-trichlorophenyl)-1H-pyrazole-5-carboxylate (7). A mixture of 7 (5.19 g, 0.017 mol), MeOH (100 mL), THF (100 mL) and LiOH solution (2M, 10 mL) was heated at reflux overnight. The reaction was cooled and the solvents removed under reduced pressure. The residue was dissolved in water (100 mL) and EtOAc (100 mL) and the pH of the solution was adjusted to neutral with 1 M HCl. The organic layer was separated, dried and the solvents were removed under reduced pressure to give 5.01 g (96%) of 3-(2,3,4-trichlorophenyl)-1H-pyrazole-5-carboxylic acid (8). A mixture of 8 (0.29 g, 0.001 mol), EDC (0.19 g, 0.001 mol), HOBt (0.14, 0.001 mol) and DMF (10 mL), was stirred for 1 h. To this solution was added ethanolamine (1 mL, 0.017 mol). The reaction was stirred overnight and the solvents were removed under reduced pressure. The residue was dissolved in EtOAc (25 mL) and water (25 mL). The organic layer was separated, and washed with HCl (1 M) followed by sat. sodium bicarbonate solution and the organic layer was dried over magnesium sulfate and evaporated. The residue was purified using automated medium pressure silica gel chromatography (ISCO) eluting with DCM:EtOAc 100:0 to 70:30 gradient to yield 0.27 g (80%) of 9v. 1H NMR (300 MHz, DMSO-d6,TMS) δ 8.48 (s, 1H), 7.71 (s, 2H), 7.34 (s, 1H), 4.82 (s, 1H), 3.54-3.35 (m, 4H); 13C NMR (75 MHz, DMSO-d6,TMS) δ 160.3, 145.1, 142.0, 133.2, 132.4, 131.9, 131.7, 130.0, 129.7, 106.9, 60.4, 42.2. HRMS calcd. for C12H10Cl3N3O2: 333.9911; found: 333.9919.
- 12.Holtz KM, Stec B, Kantrowitz ER. J Biol Chem. 1999;274:8351. doi: 10.1074/jbc.274.13.8351. [DOI] [PubMed] [Google Scholar]
- 13.TNAP MOA studies: the activity of TNAP was measured in the presence of varied concentrations of substrate at different concentrations of test compound. Parallel studies were performed for DEA and CDP-star substrates. The concentration of the second substrate was maintained constant (see Figure 2 legend for details).
- 14.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 15.Le Du MH, Millán JL. J Biol Chem. 2002;277:49808. doi: 10.1074/jbc.M207394200. [DOI] [PubMed] [Google Scholar]