

Abstract
In myotonic dystrophy type 1 (DM1) the muscle fibers express RNA containing an expanded CUG repeat (CUGexp). The CUGexp RNA is retained in the nucleus, forming ribonuclear foci. Splicing factors in the muscleblind (MBNL) family are sequestered in ribonuclear foci, resulting in abnormal regulation of alternative splicing. In extrajunctional nuclei, these effects on splicing regulation lead to reduced chloride conductance and altered insulin receptor signaling. Here we show that CUGexp RNA is also expressed in subsynaptic nuclei of muscle fibers and in motor neurons in DM1, causing sequestration of MBNL1 protein in both locations. In a transgenic mouse model, expression of CUGexp RNA at high levels in extrajunctional nuclei replicates many features of DM1, but the toxic RNA is poorly expressed in subsynaptic nuclei and the mice fail to develop denervation-like features of DM1 myopathology. Our findings indicate that subsynaptic nuclei and motor neurons are at risk for DM1-induced spliceopathy, which may affect function or stability of the neuromuscular junction.
Keywords: myotonic dystrophy, DMPK, dystrophia myotonica kinase, MBNL1, muscleblind, neuromuscular junction, subsynaptic nuclei, RNA disease
1. Introduction
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy in adults. The clinical features include myotonia, progressive myopathy, defects of cardiac conduction, and cataracts.
DM1 is caused by expansion of a CTG repeat in the gene encoding dystrophia myotonica-protein kinase (DMPK) [1]. A multi-step model for DM1 pathogenesis has been proposed [reviewed in reference 2]: (1) the mutant gene is transcribed, giving rise to transcripts that contain an expanded CUG repeat (CUGexp) [3]; (2) the CUGexp transcripts accumulate in RNA nuclear (ribonuclear) foci [4]; (3) RNA binding proteins, including muscleblind 1 (MBNL1), are sequestered in the ribonuclear foci [5, 6]; (4) altered activity of splicing factors, such as MBNL1 and CUG binding protein 1, leads to abnormal alternative splicing for a sub-group of pre-mRNAs [7, 8]; and (5) expression of inappropriate splice products leads to symptoms of DM1. For example, CUGexp RNA triggers abnormal alternative splicing of the ClC-1 chloride channel, and the predominant ClC-1 splice products expressed in DM1 muscle are devoid of ion channel activity [9-11]. Deficiency of ClC-1 channels contributes to myotonia in DM1, and can be reversed in a transgenic mouse model by overexpressing MBNL1 to levels that exceed the capacity of CUGexp RNA to sequester proteins [12]. Thus, effects of DM1 in a particular nucleus depend, at least in part, on the level of CUGexp RNA in relation to supplies of MBNL proteins.
The muscle endplate is specialized for post-synaptic functions of the neuromuscular junction (NMJ). These specializations result from increased transcription of synapse-specific genes in the junctional nuclei [13-15], coupled with posttranscriptional regulatory mechanisms in the subsynaptic domain [14]. Initial studies of DMPK protein showed immunolocalization at the endplate, suggesting a role for this kinase in the function or maintenance of the NMJ [16, 17]. However, neuromuscular transmission was normal in DMPK knockout mice [18], and endplate staining was not observed using monospecific anti-DMPK monoclonal antibodies [19], raising questions about whether DMPK is expressed significantly at the NMJ. To address this question and determine whether subsynaptic nuclei and motor neurons are at risk for spliceopathy in DM1, we examined the expression of CUGexp RNA, DMPK protein, and MBNL1 protein at the NMJ in DM1 patients and in transgenic mouse models.
2. Materials and Methods
2.1. Tissue samples
Neuromuscular junctions were identified by Alexa 488-tagged α-bungarotoxin (α–BTX, Invitrogen) binding (see below). By examining 35 DM1 muscle biopsies, we identified NMJs in 3 samples. We also identified NMJs in 3 disease controls. Spinal cord tissue was obtained at autopsy and stored at -70°C. HSALR transgenic mice that express CUGexp RNA in skeletal muscle were described previously [20]. CUGexp expression in these transgenic mice is controlled by regulatory elements from the human skeletal actin gene, which results in ribonuclear foci, myotonia, myopathy, and a spliceopathy that is similar to human DM1 [6]. Transgenic mice that express human DMPK (hDMPK) and mice with a targeted disruption of the DMPK gene were described previously [21].
2.2. Fluorescence in situ hybridization (FISH) and immunofluorescence (IF)
FISH and IF were performed on frozen sections as described previously [22]. The probe for FISH was a 2-O-methyl RNA 20mer oligonucleotide, consisting of CAG repeats and labeled at the 5’ end with Texas red. Rabbit anti-hDMPK polyclonal antibody 5244 was generated against an E. coli-expressed 25 kD fusion protein. This fusion protein contains a polyhistidine peptide (MRGSHHHHHGMAS) fused to the coiled coil region of hDMPK (residues 367-581 in accession no. AAC14449). This region excludes the kinase homology domains of DMPK. IF was performed with primary antibodies against the following proteins: DMPK (antibody 5244, 1:2000), developmental myosin heavy chain (MHC) (1:60; Novacastra), MBNL1 (antibody A2764, 1:10,000 [6]), or choline acetyl transferase (Chemicon, 1:100). Secondary antibodies were labeled with Alexa 488, 568, or 594 (Invitrogen, 1:400). Where indicated, Alexa488-α–BTX (1:500) was incubated concurrently with secondary antibody.
2.3. Immunoblot
COS-7 transformed monkey kidney cells were grown in DMEM with 10% fetal bovine serum. Cells were transiently transfected with a plasmid encoding full-length hDMPK using SuperFect Reagent (Qiagen) according to the manufacturer’s recommendations. Lysates of COS-7 cells or tibialis anterior muscle from wild-type or hDMPK transgenic mice were prepared by homogenization in buffer (1% NP-40, 150 mM NaCl, 20 mM Tris, pH 7.5, 0.1% SDS, 0.5% Na deoxycholate, and 5 mM EDTA). Lysates were resolved on 10% polyacrylamide gels, transferred to nitrocellulose membranes, blocked in 5% milk, and incubated at 4°C overnight in primary antibody (anti-DMPK at 1:2000, anti-GAPDH at 1:10,000, Biogenesis). After washing and incubation with secondary antibody (goat anti-rabbit IRDye 800, Rockland, goat anti-mouse Alexa 688, Invitrogen; 1:10,000), membranes were imaged using an infrared laser scanner (LiCor).
3. Results
3.1. Ribonuclear foci in subsynaptic nuclei
In each of 3 DM1 patients examined, FISH combined with α–BTX staining showed ribonuclear foci in both subsynaptic and extrajunctional nuclei (Fig. 1a). The most prominent ribonuclear foci in sections of skeletal muscle tended to occur in subsynaptic nuclei. In addition, co-staining with α–BTX and MBNL1 antibodies showed that MBNL1 was expressed and highly recruited into nuclear foci in subsynaptic nuclei in DM1 (Fig 1b). By contrast, in HSALR transgenic mice, ribonuclear foci were abundant in extrajunctional myonuclei but sparse or entirely absent in subsynaptic nuclei (Fig 1c).
Figure 1.
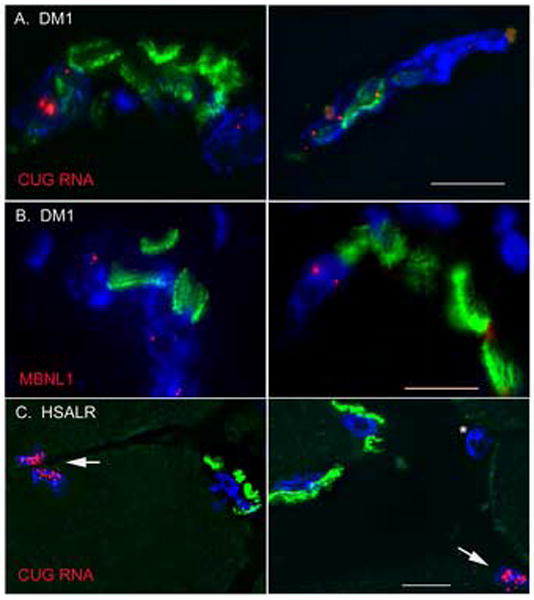
Ribonuclear foci in subsynaptic nuclei of muscle fibers. (a) In sections of DM1 skeletal muscle, FISH demonstrates foci of CUGexp RNA (red) in subsynaptic nuclei (DAPI, blue). Endplates are shown by staining with α–BTX (green). (b) Immunofluorescence for MBNL1 (red) reveals MBNL1 foci in subsynaptic myonuclei (DAPI, blue; α–BTX, green), suggesting sequestration of MBNL1 by CUGexp RNA. (c) In HSALR mice, foci of CUGexp RNA (red) are abundant in extrajunctional myonuclei (arrows), but sparse or absent in myonuclei (DAPI, blue) subadjacent to the endplate (α–BTX, green). (*) identifies an interstitial nucleus. Bar = 10 μM.
3.2. Ribonuclear foci in motor neurons
In each of 3 DM1 spinal cords examined, FISH combined with IF for choline acetyl transferase (ChAT) demonstrated ribonuclear foci in motor neurons (Fig 2a,b). The most prominent nuclear foci in sections of lumbosacral and cervical cord tended to occur in large, ChAT-positive cells of the anterior horn. Ribonuclear foci were not observed in spinal cord from controls without neurologic disease, and in these samples the distribution of MBNL1 in the cytoplasm and nucleus of motor neurons was diffuse (Fig 2c). In DM1 motor neurons, however, MBNL1 was highly localized to ribonuclear foci (Fig 2d-f).
Figure 2.
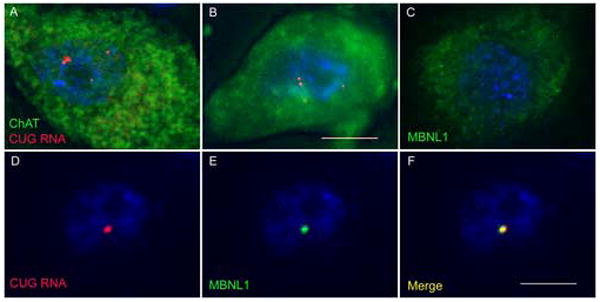
Ribonuclear foci in motor neurons. (a,b) In frozen sections of DM1 spinal cord, FISH combined with immunofluorescence demonstrates foci of CUGexp RNA (red) in large, choline acetyltransferase-positive neurons (green) in the anterior horn. (c) In non-DM1 motor neurons, MBNL1 protein (green) is localized throughout the cytoplasm and nucleus. (d-f) In DM1 motor neurons, MBNL1 (green) is sequestered in nuclear foci of CUGexp RNA (red, FISH). Bar = 10 μM.
3.3. DMPK protein at the NMJ
Because of questions raised [19] about antibody specificity in previous immunolocalization studies [16, 17], we generated polyclonal antibodies against the coiled coil domain of hDMPK. This region of DMPK was selected because sequence similarity with other proteins was low. Ab5244 recognized bands of expected molecular weight by immunoblot of COS-7 cells transfected with an hDMPK expression construct (Fig 3a) and in muscle lysates prepared from hDMPK transgenic mice (Fig 3b). Although the immunogen had 79% sequence identity with mouse DMPK, Ab5244 recognition of murine DMPK by immunoblot was weak. As further evidence for specifity, IF with Ab5244 showed diffuse cytoplasmic staining in muscle fibers from hDMPK transgenic mice, whereas staining was absent in DMPK knockout mice (Fig 3c). In human muscle from disease controls, DMPK staining was increased in regenerating fibers, identified by co-expression of developmental MHC (Fig 3d). In muscle from DM1 patients and disease controls, DMPK staining was markedly increased at the endplate, but did not coincide precisely with staining for acetylcholine receptors. Instead, DMPK localized to the subsynaptic region directly beneath the layer of α-BTX binding (Fig 3e,f).
Figure 3.
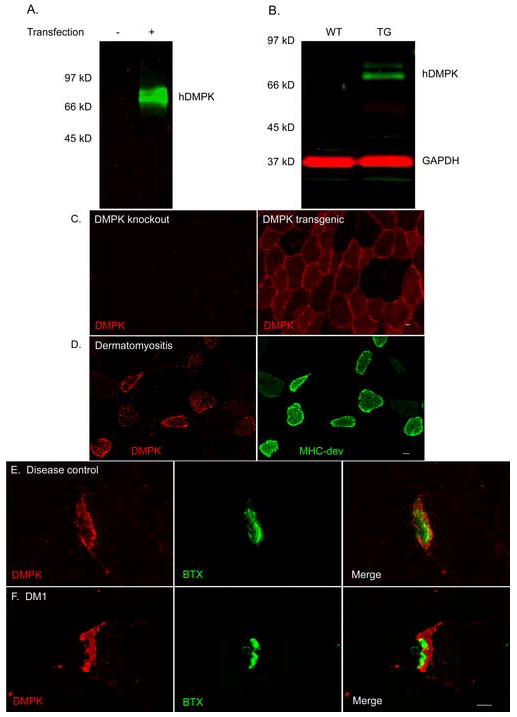
DMPK protein localization in muscle tissue. (a) Antibody 5244 recognized hDMPK by immunoblot of COS-7 cells transfected with hDMPK expression construct. (b) This antibody also recognized hDMPK by immunoblot in lysates prepared from hDMPK transgenic mouse muscle, but not in wild type muscle. (c) Immunofluorescence reveals cytoplasmic hDMPK in muscle sections from hDMPK transgenic mouse that is absent in DMPK knockout mouse muscle. (d) DMPK (red) is upregulated in regenerating fibers labeled with developmental myosin (green), shown here in dermatomyositis. (e,f) DMPK protein (red) accumulates at the NMJ in DM1 and disease control muscle, localizing to a subsynaptic domain immediately beneath the acetylcholine receptors labeled by α-BTX (green). Bar = 10 μM.
4. Discussion
Our results indicate that CUGexp RNA is expressed and MBNL1 protein is sequestered in presynaptic and postsynaptic nuclei of the NMJ in DM1. Muscleblind proteins are nuclear factors that participate in the developmental regulation of alternative splicing [6, 23], and they also may regulate the transport and decay of mRNA [24]. However, except for microtubule-associated protein tau, it is not known which transcripts expressed in subsynaptic nuclei or motor neurons are subject to CUGexp-induced spliceopathy.
Alternative splicing of tau pre-mRNA is abnormally regulated in DM1. The pattern of tau splice products expressed in adult DM1 brain is similar to that normally expressed in fetal brain [22, 25], which may contribute to the development of neurofibrillary tangles and hyperphosphorylated tau in DM1 [26]. It is noteworthy that forced expression of fetal tau in motor neurons of transgenic mice led to motor axonopathy and muscle wasting [27]. However, morphometric analysis of the deep peroneal nerve showed no evidence for motor axon retraction in 4 patients with DM1 [28].
We confirmed previous observations that expression of DMPK protein is increased at the endplate [16, 17], and we also found increased expression in regenerating fibers. If DMPK expression in these locations is upregulated at the level of transcription, we could expect that increased synthesis of CUGexp RNA would intensify the RNA-mediated disease process in subsynaptic nuclei and regenerating fibers. The latter possibility would point to an auto-accelerating disease process in which CUGexp RNA induces muscle injury and regeneration, which leads in turn to increased transcription of DMPK and higher levels of RNA toxicity.
Subsynaptic nuclei of HSALR transgenic mice display very few foci of CUGexp RNA. In this transgenic mouse model, the expanded repeat is inserted in a genomic fragment containing the entire human skeletal actin gene, including the 5’ regulatory sequences, and the pattern of transgene expression is similar to endogenous skeletal actin [20, 29]. To our knowledge, the paucity of ribonuclear foci at the NMJ in HSALR transgenic mice is the first evidence that genes encoding myofibrillar proteins are downregulated in subsynaptic nuclei. These findings also indicate that features of DM1 reproduced in HSALR mice, such as, myotonia, deficiency of ClC-1 chloride channels, internal nuclei, ring fibers, and sarcoplasmic masses, do not require expression of CUGexp RNA in subsynaptic nuclei.
Our findings raise the possibility that expression of CUGexp RNA may influence the function or maintenance of the NMJ in DM1. Indeed, two of the most striking features of the myopathology in DM1, angular atrophic fibers and pyknotic nuclear clumps, are similar to the histologic changes in denervated muscle. It is noteworthy that HSALR transgenic mice reproduce several features of myopathology in DM1, but they do not display pyknotic nuclear clumps or angular atrophic fibers, raising the question of whether expression of CUGexp RNA in subsynaptic nuclei, motor neurons, or both is required to develop these features. Previous studies of the NMJ in DM1 showed several abnormalities, including irregular and expanded terminal arborizations and profuse axonal proliferation, multiple endplates on the same muscle fiber, and abnormally large endplate size [30-33], suggesting that this disorder may affect development or maintenance of the NMJ. However, a careful ultrastructural study showed no consistent abnormalities of the NMJ in DM1 (n=4 patients, 69 endplates examined), although there was a slight decrease in density of the pre-synaptic vesicles [34]. Also, the expression of extrajunctional acetylcholine receptors (AChRs), a sensitive marker for muscle denervation, was not increased in atrophic DM1 muscle fibers [35]. These observations, however, do not eliminate the possibility of NMJ impairment or functional denervation in DM1. For example, in a transgenic mouse model of Huntington’s disease that displayed polyglutamine inclusions in subsynaptic myonuclei, Ribchester and colleagues observed progressive muscle atrophy, denervation-like properties in muscle fibers, and ultimately failure of neuromuscular transmission at some endplates, yet the ultrastructural changes at the NMJ were slight [36]. These findings were attributed to dissociation of trophic signaling between motor neurons and subsynaptic nuclei. Furthermore, previous studies have shown that upregulation of extrajunctional AChRs does not occur following denervation of ClC-1 deficient muscle fibers [37, 38], presumably because myotonic discharges may substitute for nerve activity in suppressing the expression of extrajunctional AChRs. This indicates that effects of DM1 on ClC-1 expression may suppress certain biochemical markers of muscle denervation. Ultimately it may be necessary to express CUGexp RNA conditionally in subsynaptic nuclei or motor neurons to determine the effects of RNA toxicity on the NMJ.
Acknowledgments
This work comes from the University of Rochester Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (NIH/NS48843) with support from NIH/NIAMS (AR49077, AR46806), the Muscular Dystrophy Association, and the Saunders Family Neuromuscular Research Fund. The authors thank Drs. B. Wieringa, C. Perez-Terzic, and H. Epstein for the kind gift of muscle tissue from hDMPK transgenic mice and lines of DMPK knockout mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 2.Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15(Suppl 2):R162–9. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- 3.Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A. 1997;94:7388–93. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller JW, Urbinati CR, Teng-Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J. 2000;19:4439–48. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–97. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 7.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–41. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 8.Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–80. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 9.Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 10.Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 11.Berg J, Jiang H, Thornton CA, Cannon SC. Truncated ClC-1 mRNA in myotonic dystrophy exerts a dominant-negative effect on the Cl current. Neurology. 2004;63:2371–5. doi: 10.1212/01.wnl.0000148482.40683.88. [DOI] [PubMed] [Google Scholar]
- 12.Kanadia RN, Shin J, Yuan Y, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci U S A. 2006;103:11748–53. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaeffer L, de Kerchove d’Exaerde A, Changeux JP. Targeting transcription to the neuromuscular synapse. Neuron. 2001;31:15–22. doi: 10.1016/s0896-6273(01)00353-1. [DOI] [PubMed] [Google Scholar]
- 14.Chakkalakal JV, Jasmin BJ. Localizing synaptic mRNAs at the neuromuscular junction: it takes more than transcription. Bioessays. 2003;25:25–31. doi: 10.1002/bies.10205. [DOI] [PubMed] [Google Scholar]
- 15.Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 16.van der Ven PF, Jansen G, van Kuppevelt TH, et al. Myotonic dystrophy kinase is a component of neuromuscular junctions. Hum Mol Genet. 1993;2:1889–94. doi: 10.1093/hmg/2.11.1889. [DOI] [PubMed] [Google Scholar]
- 17.Whiting EJ, Waring JD, Tamai K, et al. Characterization of myotonic dystrophy kinase (DMK) protein in human and rodent muscle and central nervous tissue. Hum Mol Genet. 1995;4:1063–72. doi: 10.1093/hmg/4.6.1063. [DOI] [PubMed] [Google Scholar]
- 18.Reddy S, Smith DB, Rich MM, et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet. 1996;13:325–35. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- 19.Pham YC, Man N, Lam LT, Morris GE. Localization of myotonic dystrophy protein kinase in human and rabbit tissues using a new panel of monoclonal antibodies. Hum Mol Genet. 1998;7:1957–65. doi: 10.1093/hmg/7.12.1957. [DOI] [PubMed] [Google Scholar]
- 20.Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–73. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 21.Jansen G, Groenen PJ, Bachner D, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet. 1996;13:316–24. doi: 10.1038/ng0796-316. [DOI] [PubMed] [Google Scholar]
- 22.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–88. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 23.Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. Embo J. 2004;23:3103–12. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adereth Y, Dammai V, Kose N, Li R, Hsu T. RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat Cell Biol. 2005;7:1240–7. doi: 10.1038/ncb1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sergeant N, Sablonniere B, Schraen-Maschke S, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. 2001;10:2143–55. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- 26.Vermersch P, Sergeant N, Ruchoux MM, et al. Specific tau variants in the brains of patients with myotonic dystrophy. Neurology. 1996;47:711–7. doi: 10.1212/wnl.47.3.711. [DOI] [PubMed] [Google Scholar]
- 27.Probst A, Gotz J, Wiederhold KH, et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–81. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 28.Pollock M, Dyck PJ. Peripheral nerve morphometry in myotonic dystrophy. Arch Neurol. 1976;33:33–9. doi: 10.1001/archneur.1976.00500010035006. [DOI] [PubMed] [Google Scholar]
- 29.Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J Biol Chem. 1993;268:719–25. [PubMed] [Google Scholar]
- 30.MacDermot V. The histology of the neuromuscular junction in dystrophia myotonica. Brain. 1961;84:75–84. doi: 10.1093/brain/84.1.75. [DOI] [PubMed] [Google Scholar]
- 31.Allen DE, Johnson AG, Woolf AL. The intramuscular nerve endings in dystrophia myotonica--a biopsy study by vital staining and electron microscopy. J Anat. 1969;105(Pt 1):1–26. [PMC free article] [PubMed] [Google Scholar]
- 32.Coers C, Telerman-Toppet N, Gerard JM. Terminal innervation ratio in neuromuscular disease. II. Disorders of lower motor neuron, peripheral nerve, and muscle. Arch Neurol. 1973;29:215–22. doi: 10.1001/archneur.1973.00490280027003. [DOI] [PubMed] [Google Scholar]
- 33.Fardeau M, Tome FMS. Light and electron microscopic study of motor endplates in the adult and neo-natal forms of dystrophia myotonica. In: Taxi J, editor. Ontogenesis and Functional Mechanisms of Peripheral Synapses: Proceedings of the INSERM Symposium on Ontogenesis and Functional Mechanisms of Peripheral Synapses. Amsterdam: Elsevier/North-Holland Biomedical Press; 1980. pp. 287–298. [Google Scholar]
- 34.Engel AG, Jerusalem F, Tsujihata M, Gomez MR. The neuromuscular junction in myopathies: a quantitative ultrastructural study. In: Bradley WG, Gardner-Medwin D, Walton JN, editors. Recent Advances in Myology: Proceedings of the Third International Congress on Muscle Diseases, International Congress Series no. 360. New York: Elsevier; 1975. pp. 132–143. [Google Scholar]
- 35.Drachman DB, Fambrough DM. Are muscle fibers denervated in myotonic dystrophy? Arch Neurol. 1976;33:485–8. doi: 10.1001/archneur.1976.00500070027005. [DOI] [PubMed] [Google Scholar]
- 36.Ribchester RR, Thomson D, Wood NI, et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur J Neurosci. 2004;20:3092–114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- 37.Klocke R, Steinmeyer K, Jentsch TJ, Jockusch H. Role of innervation, excitability, and myogenic factors in the expression of the muscular chloride channel ClC-1. A study on normal and myotonic muscle. J Biol Chem. 1994;269:27635–9. [PubMed] [Google Scholar]
- 38.Wang X, Li Y, Engisch KL, et al. Activity-dependent presynaptic regulation of quantal size at the mammalian neuromuscular junction in vivo. J Neurosci. 2005;25:343–51. doi: 10.1523/JNEUROSCI.3252-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]