

Abstract
Autophagy is a conserved cellular process required for the removal of defective organelles, protein aggregates, and intracellular pathogens. We used a network analysis strategy to identify novel human autophagy components based upon the yeast interactome centered on the core yeast autophagy proteins. This revealed the potential involvement of 14 novel mammalian genes in autophagy, several of which have known or predicted roles in membrane organization or dynamics. We selected one of these membrane interactors, FNBP1L (formin binding protein 1-like), an F-BAR-containing protein (also termed Toca-1), for further study based upon a predicted interaction with ATG3. We confirmed the FNBP1L/ATG3 interaction biochemically and mapped the FNBP1L domains responsible. Using a functional RNA interference approach, we determined that FNBP1L is essential for autophagy of the intracellular pathogen Salmonella enterica serovar Typhimurium and show that the autophagy process serves to restrict the growth of intracellular bacteria. However, FNBP1L appears dispensable for other forms of autophagy induced by serum starvation or rapamycin. We present a model where FNBP1L is essential for autophagy of intracellular pathogens and identify FNBP1L as a differentially used molecule in specific autophagic contexts. By using network biology to derive functional biological information, we demonstrate the utility of integrated genomics to novel molecule discovery in autophagy.
Autophagy is a conserved cellular process essential for the clearance of defective organelles, protein aggregates, and intracellular bacterial pathogens (1–3). Recent data have also implicated autophagy in innate and acquired immune responses and in susceptibility to Crohn's disease (4–9).
However, less is known about the exact mechanics of autophagy; the source of the membrane remains unknown, as do the necessary ancillary and accessory factors of autophagosome assembly and trafficking (10, 11). It is thought that there are subtypes of autophagy with differing cargo specificities and regulation, processes that are relatively well characterized in yeast (12, 13). In mammalian cells the process may be more sophisticated than for yeast and includes a role in the control of intracellular bacterial pathogen replication (2, 3, 14).
With this in mind, we set out to explore the autophagy apparatus to identify ancillary molecules involved in the autophagic process and their roles in different autophagy subtypes. We were particularly interested in discovering proteins involved in the membrane dynamics of autophagosome assembly and links between autophagy and the host cell membrane-handling machinery.
To this end we took a bioinformatics-based approach that involves interolog analysis (15). The mapping of interologs, protein-protein interactions conserved across species, is a powerful method for predicting and uncovering previously unidentified functional interactions by using data from high-throughput interactome screens such as two-hybrid assays and protein complementation analysis performed in model organisms (16 –18). In this article we report the construction of a hybrid “yeast-human” core autophagy network that uses interologs mapped from the yeast protein interaction network to extend the human network built around components of the core autophagy machinery. Our analysis identified 33 autophagy-associated proteins, 14 of which had no previously identified roles in mammalian autophagy. We selected a single candidate for a complete functional validation and were able to identify FNBP1L (formin binding protein 1-like) as such a molecule: essential for the genesis of anti-Salmonella autophagosomes but nonessential for serum starvation- or rapamycin-induced autophagy.
FNBP1L (also termed Toca-1) (19) is the human ortholog of yeast BZZ1 (20) and was not previously implicated in autophagy. FNBP1L has three functional domains: 1) the N-terminal FCH-BAR (FER/CIP4 homology-BIN1/amphiphysin/RVS167) domain, designated throughout as the F-BAR domain, which is thought to mediate membrane and lipid interactions; 2) the HR1 (protein kinase C-related kinase homology region 1) domain has been characterized as the site of Cdc42 interaction; and 3) an SH3 (Src homology 3) domain that serves to bind neural Wiskott-Aldrich syndrome protein during the initiation of actin polymerization. FNBP1L has previously been characterized as both essential for Cdc42-dependent actin nucleation (19) and as a vesicle trafficking regulator important for neuronal membrane morphology (21). In general it is believed that F-BAR proteins form dimers, taking on a curved shape that is able to sense and generate membrane curvature, perhaps linking membranes and organelles to the cellular trafficking machinery. In vitro F-BAR proteins such as FNBP1L are able to tubulate lipid vesicles and form wider tubules than the related BAR-domain proteins, indicating specificity toward larger membrane structures (22).
In this article we show that the HR1 domain of FNBP1L is essential for its interaction with the human ATG3 protein. Using a functional small interfering RNA (siRNA)4 approach, we identify an essential role for FNBP1L in autophagy of the intracellular pathogen Salmonella enterica serovar Typhimurium (S. Typhimurium), yet FNBP1L is dispensable for other forms of autophagy induced by serum starvation or rapamycin. In addition, we demonstrate that FNBP1L and ATG16L1 are both required for autophagy to restrict intracellular S. Typhimurium replication and, thus, we refer to the capture of Salmonella within LC3-positive vacuoles as anti-Salmonella autophagy. Full rescue of anti-Salmonella autophagy using RNA interference (RNAi)-resistant FNBP1L constructs further confirms the essential role played by FNBP1L in anti-Salmonella autophagosome assembly. We also determine that FNBP1L function during autophagy requires both the membrane-binding and the ATG3-interacting portions of FNBP1L because fragments lacking the F-BAR or HR1 domains act as dominant negatives, restricting the anti-Salmonella autophagy response. We also observe that FNBP1L and ATG3 colocate within cells, including around a subpopulation of internalized bacteria, and demonstrate that ATG3 localization to this bacterial population is dependent upon FNBP1L. Thus, we propose a model whereby FNBP1L acts as a scaffold, tethering the autophagic apparatus to the Salmonella-containing vacuole (SCV) via its interactions with membrane lipids and ATG3. By using computational analysis to derive functional biological information, we demonstrate the utility of an integrated genomic approach to novel molecule discovery in autophagy.
Materials and Methods
Integrative interolog network and literature co-citation analysis
The network was constructed by iteratively connecting interacting proteins with data obtained from the genome-wide interactome efforts of Ito et al. (23), Uetz et al. (24), and from the Human Protein Reference Database (HPRD) (25), the Molecular INTeraction (MINT) database (26), and the Protein, Signaling, Transcriptional Interactions and Inflammation Networks Gateway (pSTIING) knowledgebase (27). The network uses graph theory, which represents components (gene products) as nodes and interactions between components as edges. Graph layout descriptions were written in the Dot language (28) that implements a multidimensional scaling heuristic and uses an iterative solver (Newton-Raphson algorithm) that searches for low-energy configurations and creates a virtual physical model (Spring model) (29) to optimize the graph layout for visualization. The mapping of interacting yeast proteins onto putative human orthologs was done by identifying the reciprocal best human-yeast sequence by protein-protein basic local alignment search tool (BLASTP) analysis (reciprocal best hit or RBH approach) (30) (31). Literature co-citation analysis of autophagy proteins and their first-order interaction partners were performed using the Microarray Literature-Based Annotation (MILANO) program (32). The program identifies the number of times a protein or gene (including known aliases) was cited in articles in the PubMed database containing a specified set of terms. Vectors capturing the co-citation profiles for each of these proteins were generated for a set of terms representing cellular compartments and biological processes and log normalized against the total number of citations obtained for each protein. These were then analyzed by hierarchical clustering using Pearson's correlation coefficient as the similarity metric. The results were displayed as a heat map using Java TreeView (33).
Cell lines and bacterial strains
HEK293T and HeLa (both from American Type Culture Collection) cells were grown in DMEM (Invitrogen) with 10% FCS (HyClone) at 37°C and 5% CO2. The HeLa cell line stably expressing LC3-GFP (HeLa LC3-GFP) was generated by lentiviral transduction as previously described (7); the LC3-GFP lentiviral vector was a gift from Dr. C. Münz (The Rockefeller University, New York, NY). ATG3-deficient and control mouse embryonic fibroblasts (MEFs) were a gift from M. Komatsu (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) and have been previously described (34). The S. Typhimurium strain SL1344 was used throughout, both with and without a DsRed2 expression plasmid as has been previously described (6, 35).
Plasmids and antibodies
The human ATG3 gene (GenBank accession no. BC002830) was obtained from Open Biosystems and was subcloned into the pCMV-GFP-N (ATG3-GFP) or pCMV-3×Flag (ATG3-3×Flag) vector (pCMV-GFP-N, pCMV-3×Flag, pCMV-3×Myc, and pCMV-3×HA vectors were generated by modifying ClonTech pCMV-Myc vector with the appropriate epitope and multiple cloning site region modifications). The human FNBP1L gene (also named Toca-1) was provided by Dr. H. H. Ho and Dr. M. W. Kirschner (Harvard Medical School, Boston, MA). Myc- and GFP-tagged wild-type and mutant FNBP1Ls were constructed by subcloning the corresponding coding sequences of FNBP1L into pCMV-3 ×Myc or pCMV-GFP vectors. Dominant negative dynamin K44A was a kind gift from Dr. V. Yajnik (Massachusetts General Hospital, Boston, MA). Sequences of all cloned cDNAs were confirmed by DNA sequencing.
The following Abs were used: mouse monoclonal anti-Myc Ab (Covance); mouse monoclonal anti-GFP Ab (Covance); anti-Flag M2 mAb (Sigma-Aldrich); anti-ubiquitin mouse monoclonal (clone P4D1; Cell Signaling Technology); mouse monoclonal anti-β-tubulin (clone D10; Santa Cruz Biotechnology); rabbit polyclonal anti-Toca1(CT) (ProSci); and rabbit polyclonal anti-FNBP1L (Toca-1), a gift from Dr. M. W. Kirschner (Harvard Medical School). Secondary Abs for quantitative near-infrared detection were IRDye 680CW goat anti-rabbit and IRDye 800CW donkey anti-mouse (LI-COR Biosciences), and analysis was performed using an Odyssey imaging workstation.
Expression and immunoprecipitation of FNBP1L and ATG3
One day before transfection, 1.5 × 106 HEK293T cells in 2 ml of DMEM were plated per well in a 6-well plate. The cells were transiently transfected with the appropriate amount of plasmids by using TransFectin (Bio-Rad) according to the manufacturer's instructions. The amount of transfected plasmids was equalized among different samples by using corresponding empty vector(s). Twenty-four hours later cells were rinsed in ice-cold PBS and lysed in standard lysis buffer (50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% Triton-X-100, 5 mM EDTA, 10 mM sodium fluoride, 1 mM sodium vanadate, 0.4 mM PMSF, and a protease cocktail from Roche (one tablet for 10 ml of buffer) with rotation for 30 min at 4°C. Insoluble materials were removed by centrifugation at 14,000 rpm for 15 min. The protein concentration of the cleared lysate was determined by the DC protein assay (Bio-Rad).
For immunoprecipitation, cell lysates were incubated with the appropriate specific Abs for 3 h at 4°C and subsequently mixed with Ab affinity gel (goat affinity-purified Ab to mouse IgG, ICN Pharmaceuticals) for an additional 90 min at 4°C, or cell lysates were incubated with anti-Flag M2-agarose (Sigma-Aldrich) for 90 min at 4°C. The immunoprecipitates were washed three times with standard lysis buffer. The immunoprecipitated proteins and total cell lysates were resolved by SDS-PAGE, transferred to Immobilon-P transfer membranes (Millipore) and, immunoblotted with the indicated Abs. An HRP-conjugated anti-mouse Ab (DakoCytomation) was used as the secondary reagent. Detection was performed by ECL with the Western Lightning chemiluminescence reagent (PerkinElmer).
RNA interference of FNBP1L, ATG16L1, and Cdc42
HeLa cells (parental strain or stably transduced with LC3-GFP) were plated in 12-well plates containing 18-mm glass coverslips at a density of 1 × 105 cells per well. After 24 h, 20 pmol of modified RNA oligoduplexes (Stealth RNAi; Invitrogen) were transfected into each well using Lipo-fectamine 2000 (Invitrogen) according to the manufacturer's instructions. For FNBP1L the silencing sequences used were as follows: FNBP1L si1, 5′-GCAGUGACAUAAAUCAUCUUGUAAC-3′ and 5′-GUUACAAGAUGAUUUAUGUCACUGC-3′; FNBP1L si2, 5′-GGACACUGCAAAGCUAUCUACCCUU-3′ and 5′-AAGGGUAGAUAGCUUUGCAGUGUCC-3′; FNBP1L si3, 5′-CAAAGGUGACGGAUGGACAAGAGCU-3′ and 5′-AGCUCUUGUCCAUCCGUCACCUUUG-3′ (where si represents an siRNA). The ATG16L1 duplex sequences used were 5′-AUGUCUGCUUGAUAGCAUUUGUUGC-3′ and 5′-GCAACAAAUGCUAUCAAGCAGACAU-3′ and have been previously validated (6). Autophagy assays were begun 48 h post-transfection and 24 h later the cells were fixed and stained as described below. Knockdown was confirmed by both quantitative RT-PCR and Western blotting. For Western blots the cells were transfected in a 6-well format, lysed after 48 h in radioimmune precipitation assay buffer with protease inhibitors (Complete Mini; Roche Applied Science) and resolved by SDS-PAGE. Following transfer to polyvinylidene difluoride membranes and blocking (5% skim milk), FNBP1L was detected using rabbit anti-FNBP1L Ab, a gift from Dr. M. W. Kirschner (Harvard Medical School). Following chemiluminescent detection, membranes were stripped (Restore Western blot stripping buffer; Thermo Scientific) and reprobed with mouse anti-tubulin Abs to ensure comparable loading.
Rescue of FNBP1L was performed using myc-tagged FNBP1L constructs into which synonymous coding mutations had been introduced into the relevant siRNA target sequences. Three such single base changes were made for each siRNA target site using site-directed mutagenesis (QuikChange; Stratagene), ensuring that the siRNA targeting would be disrupted, without changing the amino acid sequence of the resulting protein. These two rescue constructs were termed FNBP1L R1 and R2. Transfections were performed as previously described, with 20 pmol of RNAi duplex and 300 ng of wild-type or rescue constructs. After 48 h the cells were lysed and the lysates were resolved by SDS PAGE and transferred to Immobilon-FL membranes (Millipore). Following blocking in 5% skim milk, membranes were incubated with both rabbit anti-FNBP1L (ProSci) and mouse anti-β-tubulin (Santa Cruz Biotechnology) Abs. After washing, membranes were probed with two near-infrared fluorescently conjugated secondary Abs (anti-mouse 800 and anti-rabbit 680), and analyzed on an Odyssey Quantitative imaging station (LI-COR Biosciences).
Cdc42 RNAi was accomplished using the same protocols, but RNAi duplexes were obtained from Qiagen. Cdc42 si1 was the validated sequence SI02757328 and Cdc42 si2 was 5′-CCUAAUUCUUGUAGAUGCA-3′ and 5′-UGCAUCUACAAGAAUUAGG-3′ (where si represents an siRNA).
Confirmation of FNBP1L and Cdc42 knockdown was performed by real-time RT-PCR. RNA from specimen wells was extracted using an RNeasy mini kit (Qiagen) and reverse transcription was performed using the iScript cDNA synthesis kit (Bio-Rad). Real-time PCR with specific primers was performed using iQ SYBR Green Supermix (Bio-Rad) on an iQ5 thermocycler (Bio-Rad). Primer sequences were as follows: FNBP1L, 5′-GGATCAGTTCGACAGCTTAGAC-3′ (forward) and 5′-CACGAGGTAAACCGTGGCT-3′ (reverse); and Cdc42, 5′-CTTGCTTGTTGGGACTCAAATTG-3′ (forward) and 5′-GGCTCTTCTTCGGTTCTGGAG-3′ (reverse). RT-PCR data were normalized to GAPDH control reactions and statistical analysis was performed using the two-tailed, unequal variance Student's t test.
Induction of autophagy by classical stimuli
Autophagy was induced in HeLa LC3-GFP cells using serum starvation or rapamycin. Cells were transfected as described above, and after 48 h the medium was changed to 1% serum or 10% serum plus 200 nM rapamycin for a further 24 h. Ammonium chloride treatment was performed with a final concentration of 50 mM for 2 h in 1% serum medium. Chase experiments to assay recovery kinetics of autophagic vesicles were performed by treating cells for 2 h with ammonium chloride as described above and then replacing it with complete medium (10% serum) for 2, 3, or 4 h.
Western blotting to demonstrate LC3-GFP cleavage was performed after cell lysis, equalization of protein amounts, and SDS-PAGE electrophoresis on a 7.5% polyacrylamide gel (Bio-Rad) as described above. Following transfer to Immobilon-P membranes (Millipore), detection was performed using anti-GFP primary (Covance) and HRP-conjugated secondary Abs as previously described.
Infection and transferrin endocytosis assays
S. Typhimurium infections were performed as previously described, with slight modifications (36). Briefly, S. Typhimurium SL1344 carrying a DsRed2 expression plasmid was grown overnight in Luria-Bertani broth containing 100 μg ml−1 ampicillin at 37°C with aeration and subcultured at a dilution of 1/33 for a further 3 h in Luria-Bertani broth. This culture was further diluted in DMEM 10% serum without antibiotics to yield a multiplicity of infection (MOI) of 100 and added to HeLa (for LAMP1 staining) or HeLa LC3-GFP (for all other experiments) cells grown on coverslips in 12-well plates. Infections were allowed to proceed for 20 min and the cells were washed once in complete medium containing 100 μg ml−1 gentamicin sulfate and then incubated in fresh high gentamicin medium for 1, 2, or 4 h; for incubations longer than 1 h the medium was exchanged for low gentamicin DMEM (containing 20 μg ml−1 gentamicin sulfate). Cells were washed twice in PBS before being fixed, permeabilized, stained, and mounted as described below. For HeLa cells transfected with FNBP1L-GFP or GFP-only constructs, infections were performed with a wild-type SL1344 strain (without DsRed expression) and all other steps were performed as described above.
For bacterial survival assays, 12-well plates of HeLa cells were infected as described above and incubated for 1, 3, or 5 h postinfection. Cells were then lysed in 1% Triton X-100 in PBS for 2 min, followed by repeated pipetting to ensure the release of all cell fragments from the plate. The resulting lysate was vigorously vortexed and serially diluted in PBS before being plated on agar plates containing 25 μg ml−1 streptomycin sulfate. Following incubation at 37°C overnight, colonies were counted to yield viable counts of internalized bacteria.
Transferrin endocytosis was performed as described previously (37) with slight modifications. HeLa cells were serum-starved for 1 h and then washed twice in ice-cold PBS. Cells were then incubated at 37°C in serum-free medium containing 25 μg ml−1 Alexa Fluor 555-conjugated transferrin (Invitrogen). At 10, 20, 30, and 40 min following transferrin addition the cells were washed twice with ice-cold PBS, acid was stripped for 5 min in ice-cold stripping buffer (150 mM NaCl, 2 mM CaCl2, and 25 mM CH3COONa (pH 4.5)), washed twice more in PBS and collected on ice. Subsequent FACS analysis was performed using a BD FACSCalibur (BD Biosciences) flow cytometer and mean fluorescence of 10,000 cells per time point was collected from triplicate samples.
Immunofluorescent staining
Following completion of experimental treatments, all cells were washed twice in PBS before being fixed in 4% formalin for 15 min. For all experiments, except LAMP1 staining, cells were permeabilized by incubation in PBS with 0.1% Triton X-100 and 1% BSA for 2 min and stained with Alexa Fluor 633 phalloidin (Invitrogen) to reveal actin filaments. For FNBP1L-GFP overexpression experiments, Hoechst 33342 (Invitrogen) was added to the phalloidin staining step to stain bacterial DNA, because bacteria used for these experiments lacked DsRed. Coverslips were then washed in PBS and mounted in Poly-Mount (PolySciences).
LAMP1 staining was performed following saponin permeabilization (0.5% saponin and 1% BSA in PBS) for 20 min and subsequent Ab steps were performed in PBS with 0.1% saponin. Rabbit anti-LAMP1 Abs (Sigma-Aldrich) were incubated >1 h; coverslips were then washed repeatedly in saponin/PBS and a secondary Alexa Fluor 488-conjugated goat anti-rabbit Ab was added. Phalloidin and DNA staining and mounting were as previously described.
Microscopy and quantification of fluorescent imaging
Slides were viewed for counting under wide-field fluorescence illumination with a ×100 lens (Zeiss Axioplan; Carl Zeiss MicroImaging). The total number of bacteria per cell and the number of LC3-GFP- or LAMP1-positive bacteria were assessed in randomly chosen fields with at least 50 cells counted for each condition. The numbers of LC3-GFP- or LAMP1-positive bacteria were then calculated as a percentage of total bacteria. Bacteria were scored as within autophagosomes only when a complete and closely conforming LC3-GFP “capsule” was visible. Significance was assessed using the two-tailed, unequal variance Student's t test. Images were obtained using laser-scanning confocal microscopy (Leica SP5; Leica Microsystems) as high-resolution z-stacks, which were subsequently projected onto single images using NIH ImageJ software. For quantification of vesicle sizes and numbers, 15 fields of cells (an average of 20 cells per field) for each condition were captured by wide-field microscopy with a ×100 lens. These images were then combined into stacks using ImageJ and identically processed for automated vesicle counting. Images of the FITC channel were processed using an automated background subtraction algorithm (Enhance plug-in) to maximize contrast without clipping and thresholded to remove any remaining background noise (representative images from each stack were examined by an operator to ensure that all autophagosome vesicles were captured by this method). The particle analysis plug-in was then used to automatically find, label, count, and measure the area of each autophagosome in each image (settings ensured that only objects greater than five pixels in area were counted, eliminating any random noise). Again, representative sections were examined manually to ensure that autophagosomes were correctly identified and counted. For this purpose, autophagosomes were defined as LC3-GFP puncta of close to spherical shape within the cell cytoplasm. Several large, irregular aggregates were manually excluded for the final data analysis. A preliminary analysis including large aggregates did not show any statistical differences in the number or size of such rare LC3-GFP aggregates between control or FNBP1L-deficient cells. These data were then pooled for each condition and average numbers of autophagosomes per cell and average autophagosome areas were calculated.
Results
A comparative bioinformatics strategy reveals links from the core autophagy apparatus to multiple genes involved in membrane dynamics
To elucidate novel components of the autophagic apparatus in mammalian cells, we used a two-step approach; first building a human protein interaction network of the core autophagy components central to the initiation and elongation of the ATG12 and LC3 conjugation systems (Fig. 1A) and then extending this using yeast protein interaction data from two-hybrid screens. Because the core autophagy “machinery” has been better characterized in yeast, we constructed protein network extensions of this core from two-hybrid screen data to identify novel ancillary factors that might potentially be involved in or recruited for autophagy. We then performed interolog mapping whereby interologs (orthologous interactions of yeast autophagy proteins and their interaction partners mapped to human orthologs) were projected onto the human network (Fig. 1B and supplemental Fig. S1A).5 The resulting “hybrid” network was used to identify novel mammalian components likely to be conserved from the yeast apparatus (Fig. 1C). Using literature (PubMed) co-citation analysis, a number of these components can be broadly annotated as having an involvement with membrane or vesicle structures (Fig. 1D). These proteins include PTK2 (a focal adhesion kinase), PLSCR1 (a phospholipid scramblase), and STX1A (syntaxin), which are known to function at the cytoskeleton-membrane interface, participating in membrane remodeling and vesicle fusion.
FIGURE 1.

Integrated network approach to uncover novel human autophagy effectors. A, Construction of protein interaction network centered on human proteins implicated in the core autophagy “machinery” (red squares) comprising the initiation factor (BECN1) and elongation components of the ATG12 conjugation system (ATG12, ATG7, ATG10, ATG5, and ATG16L1) and the LC3 conjugation cascade (MAP1LC3, ATG4, ATG7, and ATG3). Blues circles denote human proteins interacting with the core autophagy proteins. B, Yeast interolog projection. The human network is extended by projecting the yeast network centered on yeast autophagy proteins mapped to human orthologs. A more extensive figure of the yeast autophagy network is shown in supplemental Fig. S1A. C, The resulting hybrid yeast-human interaction map with orthologous interactions (or interologs) from the yeast network superimposed onto the human network. D, Combined with literature (PubMed) co-citation analysis, a number of components interacting with core ATG proteins derived from the hybrid map can be broadly annotated as having an involvement with membrane or vesicle structures. The intensity of the “heat map” denotes the extent to which each specific term is co-cited with each protein in the PubMed database.
Extending this approach to all 33 proteins (of which 14 have not been implicated in human autophagy) that were identified as potential ancillary factors of autophagy (AFA) (Fig. 1), we bioinformatically annotated these proteins and classified them in terms of cellular compartments and in the context of their functional associations (summarized diagrammatically in Fig. 2 with details in supplemental Table I). Strikingly, in addition to observing AFA having broad membrane associations as previously noted from co-citation analysis, many were found to also have associations with mitochondria (e.g., MRPS5, OGDH, PMPCA, BCL2, and SLC25A20); mitochondria are known to be degraded via autophagy and also play key roles in innate immunity and apoptosis (38–40). Interestingly, a number of AFA are involved in cell cycle regulation (e.g., GFI1B, RAD1, PSMD2, and BCL2), immune response (e.g., SQSTM1, PLSCR1, and OPRK1), or have transport roles such as vesicle-mediated transport (e.g., GOPC and STX1A), protein trafficking (GABARAP), mRNA export (e.g., UPF2), nuclear import, or export processes (e.g., XPO1). Many AFA have not been previously implicated in human autophagy, and their identification using a bioinformatics approach presents opportunities for future investigations into their contributions to as yet undiscovered multifaceted roles of autophagy in human physiology, defense, and disease.
FIGURE 2.

Cellular localization and functional associations of core and potential AFA. Core human autophagy components (red ovals) and potential AFA (blue ovals) identified from yeast-human interolog mapping and protein network extensions from two-hybrid screen data are placed in cellular compartments and in the context of functional associations (black dashed lines). This map also incorporates protein-protein interactions (gray solid lines) from the yeast-human interolog network featured in Fig. 1C. A more detailed annotation of each component is provided in supplemental Table I.
In addition to the candidates described above, we predicted an interaction between FNBP1L, an F-BAR-containing protein (and the human ortholog of the yeast BZZ1 protein) and the autophagy protein ATG3. The known involvement of F-BAR proteins in membrane tubulation and assembly suggested that FNBP1L might play a role in connecting and organizing the autophagic membrane/autophagy apparatus interface. Therefore, we used FNBP1L as a test case to experimentally validate our predicted interactions between human proteins identified as ancillary factors involved in autophagy and the core autophagic machinery.
Human FNBP1L binds ATG3 via the FNBP1L HR1 domain
We confirmed the binding of human ATG3 with FNBP1L by coexpressing ATG3 and full-length FNBP1L as well as serial truncations of FNBP1L in HEK293 cells and performing coimmunoprecipitation experiments (Fig. 3A). Full-length Myc-tagged FNBP1L was able to pull-down GFP-tagged ATG3 (Fig. 3B), and binding was confirmed by reciprocal immunoprecipitation using Flag-tagged FNBP1L and Myc-ATG3 (supplemental Fig. S2A). It has recently been reported that ATG12 can bind ATG3 (41), and therefore we tested the possibility that the ATG3/FNBP1L binding might be indirect, via ATG12. We concluded that the FNBP1L/ATG3 interaction was not mediated by ATG12 because attempts to pull-down FNBP1L with ATG5, ATG12, or ATG16L1 constructs were unsuccessful (supplemental Fig. S2B), yet ATG3 was consistently able to immunoprecipitate FNBP1L. Unfortunately, Abs suitable for use in immunoprecipitations of endogenous FNBP1L and ATG3 proteins are not currently available. To further characterize the interaction, we made serially truncated FNBP1L constructs, each lacking one or more potential binding domains, and performed immunoprecipitation experiments (Fig. 3, A and B). These results implicated the protein kinase C-related kinase HR1 (homology region 1) domain of FNBP1L as the key mediator of ATG3 binding, because truncations containing solely the FCH (FER/CIP4 homology) domain and coiled-coil (FNBP1L-1-293) or only the SH3 (Src homology 3; FNBP1L-483–547) domains were unable to bind ATG3, but all those containing the HR1 domain bound reliably to ATG3, even in the absence of the FCH and coiled-coil domains (Fig. 3B). We therefore concluded that FNBP1L binds ATG3 via the HR1 domain in a fashion similar to that of the characterized FNBP1L ligand Cdc42.
FIGURE 3.
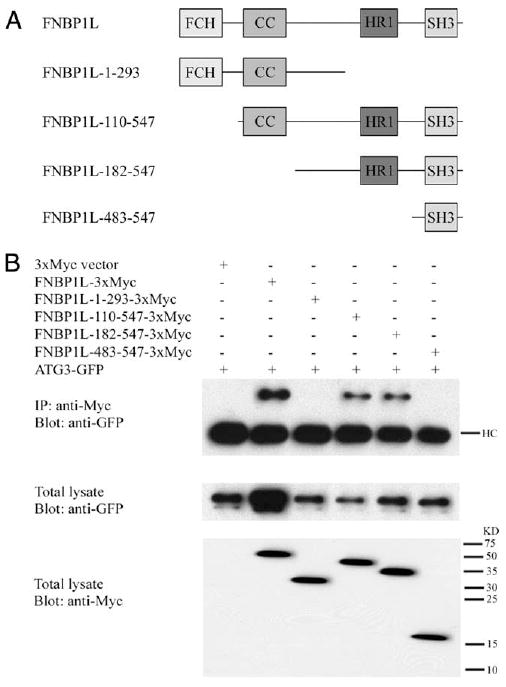
FNBP1L interacts with ATG3 via the FNBP1L HR1 domain and locates to internalized bacteria. A, Schematic representation of FNBP1L constructs used in this study. FCH, FER/CIP4 homology (FCH) domain; CC, coiled-coil domain; HR1, protein kinase C-related kinase homology region 1; SH3, Src homology 3 domain. B, Full-length FNBP1L or FNBP1L mutants and ATG3 were expressed in HEK293T cells either together or individually as indicated. Twenty-four hours after transfection the expressed proteins were immunoprecipitated (IP) and probed with the indicated Abs. FNBP1L/ATG3 binding was observed to be HR1 domain dependent. HC, Heavy chain; KD, kDa.
Having characterized the ATG3-FNBP1L interaction, we set out to study the functional role of FNBP1L in both classical and antibacterial autophagy using an siRNA strategy.
RNA silencing of FNBP1L has no observed effects upon classically induced autophagy, as quantified by accumulation of LC3-GFP+ vesicles
To determine the role of FNBP1L in autophagy, we obtained modified RNA duplexes (Stealth siRNA; Invitrogen) to perform siRNA silencing of the FNBP1L transcript. We transfected these duplexes into a HeLa cell line stably expressing LC3-GFP (see Materials and Methods) according to the manufacturer's instructions alongside a control duplex (nontargeting, GC-content matched). Assays were performed 48 h later and knockdown was confirmed by quantitative real-time RT-PCR upon RNA extracted from specimen wells. We determined that all three duplexes were capable of robust knockdown in HeLa cells (between 71 and 78% reduction in mRNA level; Fig. 4A) and performed experiments using duplexes 1 and 2 (see Materials and Methods for sequences). Compared with control duplex-treated cells, FNBP1L knockdown cells showed no apparent morphological differences. Careful examination of actin cytoskeletal organization, as revealed by phalloidin staining, showed little effect of loss of FNBP1L upon actin structures (Fig. 4, B and C).
FIGURE 4.
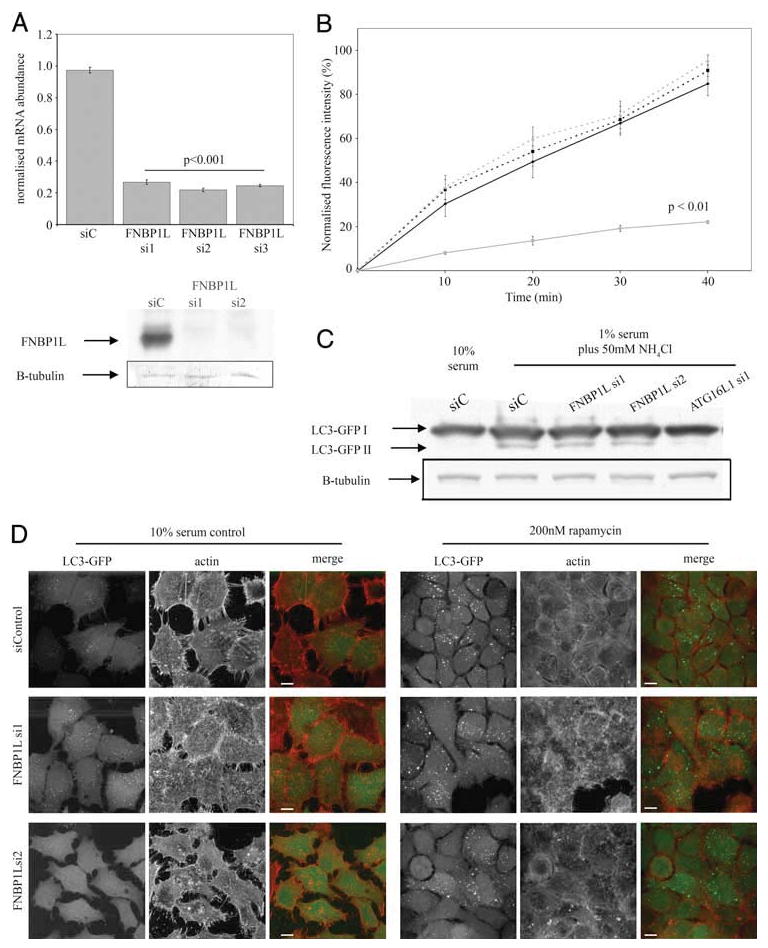
siRNA-mediated knockdown of FNBP1L has no effect upon transferrin endocytosis or autophagy induced by classical stimuli. A, Effective knockdown of FNBP1L mRNA in HeLa cells is achieved by all three siRNA duplexes (si1, si2, and si3) directed against FNBP1L, compared with a control (nontargeting) duplex. Mean FNBP1L mRNA abundance values, normalized to GAPDH control reactions, are shown (±SEM). The p values were calculated using a one-way ANOVA followed by a post hoc Student's t test using the Bonferroni correction compared to siControl (siC) values. Data were calculated from three separate samples for each condition, with PCR performed in duplicate. The lower panels show endogenous FNBP1L protein, detected using FNBP1L-specific Abs, in cells transfected with control and the FNBP1L si1 and si2 RNAi duplexes. The same blots, reprobed for β-tubulin (B-tubulin) are shown to confirm equal loading. B, Transferrin endocytosis is unaffected by FNBP1L knockdown. HeLa cells were transfected with control or FNBP1L-directed siRNAs or a dominant negative dynamin (K44A) construct (as a control for endocytosis inhibition) and 48 h later were serum-starved for 1 h and exposed to fluorescently labeled transferrin for up to 40 min. Following washing, cells were placed on ice and analyzed by FACS. Normalized average fluorescent intensities from triplicate samples are shown (±SEM). Statistical comparison by two-way ANOVA, followed by post hoc t tests (Bonferroni corrected) showed only dominant negative dynamin-expressing samples to be significantly different (p < 0.01 at all time points). C, FNBP1L knockdown does not inhibit classical autophagy induced by serum starvation and ammonium chloride treatment. HeLa LC3-GFP cells were transfected with control and FNBP1L-directed or ATG16L1-directed siRNAs and autophagy was induced after 48 h by incubation in 1% serum medium with 50 mM ammonium chloride for 4 h. Cells were then lysed, equalized for protein amounts, and blotted with anti-GFP Abs following SDS-PAGE. Autophagy was quantified by the generation of the lipidated LC3-GFP product (LC3-GFPII); note the absence of this product in 10% serum control-treated cells and the loss of product in ATG16L1-deficient cells (as previously published in Ref. 6). No differences in the amount of LC3-GFP II produced were observed between similarly treated control and FNBP1L-deficient cells. D, FNBP1L knockdown does not inhibit classical autophagy induced by rapamycin. HeLa LC3-GFP cells transfected with control or FNBP1L-directed siRNAs were either grown in 10% serum (left panels) or treated with 200 nM rapamycin for 24 h (right panels). No differences were observed in either case, with robust LC3-GFP (green in merge columns) vesicle and aggregate formation seen in all cases upon rapamycin treatment. The actin cytoskeleton of cells (red in merge columns) was also unaffected by siRNA treatments. Images are projections of confocal z-stacks; scale bars represent 10 μm.
We considered the possibility that any autophagic defect observed in FNBP1L knockdown cells might be due to a broader alteration in major vesicle trafficking and handling processes and not a specific autophagic process. The yeast homologue of FNBP1L, BZZ1, and the related human protein FNBP1 have both been previously implicated in the internalization phase of receptor-mediated endocytosis (22, 42, 43), and therefore we performed transferrin-mediated endocytosis assays upon siRNA-treated cells and controls. We were unable to observe an effect of FNBP1L knockdown upon the rate or amount of transferrin endocytosis as quantified by FACS analysis of cells treated with fluorescently labeled transferrin (Fig. 4B). However, we were able to observe significant endocytosis inhibition following transfection of a dominant-negative dynamin, validating the sensitivity of the assay. We also performed staining of lysosomes by using anti-LAMP1 Abs. In both FNBP1L knockdown and control cells the LAMP1-positive compartment appeared to be identical (data not shown). These results suggest that FNBP1L-deficient cells do not exhibit loss of endocytic activity, nor does FNBP1L deficiency result in profound alterations to the morphology of the lysosomal compartment. Although we cannot rule out more subtle effects of FNBP1L depletion upon these and other cellular processes, these data, along with the lack of global actin morphological alterations, led us to conclude that transient FNBP1L knockdown was well tolerated by HeLa cells without gross effects upon endocytosis or lysosomes. Therefore, we tested the effects of FNBP1L ablation upon classical autophagic induction.
Induction of autophagy by serum starvation or rapamycin treatment was equally effective in both FNBP1L-silenced and control cells, as determined by accumulation of the cleaved LC3-GFP II product (detected by anti-GFP Western blotting) and LC3-GFP positive vesicles within cells. Western blotting revealed equal levels of lipid-conjugated LC3-GFP II in FNBP1L-deficient cells compared with control siRNA-treated cells when incubated in 1% serum and 50 mM ammonium chloride for 4 h (Fig. 4C). We have found in our prior studies that blockade of lysosomal fusion using ammonium chloride yields more robust accumulation of LC3-GFP-positive vesicles compared with starvation alone because autophagosomes accumulate without the loss of GFP fluorescence exhibited upon acidification of the autolysosomal compartment. In contrast to the lack of an FNBP1L-deficient phenotype, the use of siRNA directed against ATG16L1, a vital component for all forms of autophagy (6), resulted in a loss of LC3-GFP II under the same autophagy-inducing conditions. Treatment with either rapamycin for 24 h or ammonium chloride in the presence of 1% serum for 2 h was sufficient to generate robust levels of LC3-GFP vesicles in both control and specific siRNA-treated cells (Fig. 4D and supplemental Fig. S2C). Under both rapamycin and ammonium chloride conditions almost 100% of cells showed accumulation of LC3-GFP vesicles regardless of siRNA treatment. The size and number of these vesicles was determined using an automated image processing method with ImageJ software as described in Materials and MethodS. Vesicle size was unchanged between FNBP1L-deficient and control cells under both rapamycin and ammonium chloride treatments. There was a trend toward FNBP1L-deficient cells possessing more LC3-GFP-positive vesicles and hence having a greater overall vesicle area than control cells, but these differences failed to reach statistical significance (data not shown).
Despite the apparently normal induction of autophagic vesicles, we did not know whether subsequent lysosomal fusion and processing events might be affected by the absence of FNBP1L. To eliminate the possibility of altered autophagosome clearance kinetics, we performed “chase” experiments where cells were treated for 2 h with 1% serum and 50 mM ammonium chloride and then washed and incubated in complete DMEM with 10% serum. Examination of vesicle loss in these cells revealed no significant differences, with both FNBP1L knockdown and control cells eliminating the majority of LC3-positive vesicles within 4 h of the addition of complete medium (data not shown), a similar time scale to that previously described (44). Similar chase experiments using rapamycin treatment gave the same results, with removal of most GFP-bright vesicles within 3–4 h of exposure to fresh serum-replete medium.
Previous studies have shown that autophagy can be induced by intracellular bacterial pathogens such as S. Typhimurium and Shigella flexneri (3, 14, 45). To investigate the role of FNBP1L in this process, we used a well-characterized model system, S. Typhimurium infection of HeLa cells.
FNBP1L is essential for efficient autophagy of intracellular S. Typhimurium
We and others have previously used HeLa cells and S. Typhimurium to study the role of the autophagic process in antibacterial responses (3, 6). In these experiments, we infected stably transfected LC3-GFP HeLa cells with a DsRed2-expressing SL1344 strain under short-incubation, high MOI conditions, thus ensuring synchronous infection. Cells were analyzed after 1, 2 or 4 h of infection; under control conditions 1 h is known to be the peak of bacterial capture within autophagosomes, yet we wanted to eliminate the possibility that FNBP1L deficiency resulted in delay of autophagy. In control siRNA-treated LC3-GFP cells we observed a mean of 22 ± 3% of intracellular bacteria in LC3-GFP-positive vacuoles following 1 h of infection (Fig. 5A). HeLa LC3-GFP cells treated with either of two siRNAs directed against FNBP1L showed a marked reduction in this percentage to a mean of just 5 or 6 ± 2%. In control cells after 2 and 4 h this percentage dropped markedly to 9 and 6 ± 2%, respectively. FNBP1L deficient cells yielded just 6 or 7 ± 2% encapsulation after 2 h and 3 or 4 ± 2% bacterial autophagy after 4 h. These data showed a significant loss of anti-Salmonella autophagy in FNBP1L-deficient cells after 1 h, with no recovery of autophagy at later time points, indicating that FNBP1L ablation results in anti-Salmonella autophagy failure, not merely delay. The percentage of LC3 membrane engulfment of SL1344 in control cells is within the range reported by ourselves and others for HeLa cells transiently transfected with LC3-GFP constructs (∼15–25% at 1 h, falling to 5% after 2 h) (3, 6). For the experiments described in this article, MOI was maintained at 1:100 and a comparison of S. Typhimurium infection of cells receiving siRNA directed against FNBP1L with control-treated cells showed no significant alteration in the numbers of invading bacteria nor in the kinetics of invasion (data not shown). Similar numbers of bacteria were found attached to or internalized within (at 20 min and 1 or 2 h postinfection, respectively) both siFNBP1L- and siControl-treated cells.
FIGURE 5.

FNBP1L is essential for autophagy of internalized S. typhimurium. A, FNBP1L-directed siRNA significantly reduces autophagy of S. Typhimurium. HeLa cells stably transduced to express LC3-GFP were subjected to siRNA knockdown and 48 h later were infected for 1, 2, or 4 h (filled, gray, and open bars, respectively) with S. Typhimurium. Following fixation and mounting, the percentage of internalized SL1344 encapsulated within LC3-GFP positive membranes were calculated. A statistically significant difference between control and FNBP1L deficient cells was observed at 1 h postinfection when compared with siControl at 1 h; *, p < 0.005. At least 50 cells per condition were used to count total internalized SL1344 and LC3-GFP+ SL1344 in each experiment. Data shown are means of three independent experiments ± SEM; significance was assessed using a two-way ANOVA followed by post hoc Student's t tests with Bonferroni's correction. B, The effects of FNBP1L-siRNA duplexes upon anti-Salmonella autophagy can be specifically rescued using appropriately modified FNBP1L sequences. LC3-GFP HeLa cells were cotransfected with siRNA (si) alone (filled bars), siRNA plus wild-type (WT) FNBP1L (dark gray bars), siRNA plus the FNBP1L R1 rescue construct (light gray bars), or siRNA plus the FNBP1L R2 rescue construct (open bars). Forty-eight hours later the cells were infected for 1 h with S. Typhimurium and the autophagy rate was assayed as described, or the cells were lysed for Western blotting (lower panels). Rescue of both the autophagy phenotype induced by siRNA against FNBP1L and FNBP1L expression was observed only in the presence of the appropriate rescue construct, indicating that the loss of FNBP1L was fully responsible for the siRNA phenotype observed. Significant differences between siControl samples and matched siFNBP1L groups were assessed by one-way ANOVA, followed by post hoc t tests with Bonferroni correction; *, p < 0.005. Expression of the appropriate rescue construct in FNBP1L-deficient cells returned the autophagy rate to a level comparable to that of the siControl samples (as indicated by the loss of statistically significant differences compared with control cells). Autophagy rate data is shown as means of two independent experiments ± SEM. C, Autophagy of S. Typhimurium requires FNBP1L. Representative images of the experiments summarized in A. S. Typhimurium (red in merge column) is frequently targeted for autophagic encapsulation by LC3-GFP-positive membranes (green in merge column) in control cells; this encapsulation is largely absent following FNBP1L knockdown, although small cytoplasmic LC3-GFP puncta are still visible. Actin is shown as blue in the merged image. Images are confocal projections of z-stacks and scale bars represent 10 μm. D, FNBP1L-dependent autophagy serves to control S. Typhimurium replication within epithelial cells. HeLa cells were transfected with control or ATG16L1- or FNBP1L-directed siRNA duplexes (si1 and si2) and infected with S. Typhimurium 48 h later. Following 1, 3, or 5 h of gentamicin protection, cells were lysed and serially diluted and the lysates were plated to determine viable bacterial numbers. We observed significant elevations in bacterial numbers in both ATG16L1- and FNBP1L-deficient cells compared with controls after 5 h. Data shown as means of three independent experiments, each performed in triplicate ± SEM. Significance was assessed using two-way ANOVA followed by post hoc t tests (Bonferroni correction) comparing ATG16L1- or FNBP1L-deficient cells to controls.
Because we had identified FNBP1L as interacting with ATG3, we also performed experiments to determine whether ATG3 was essential to anti-Salmonella autophagy by using ATG3-replete or -deficient MEFs. MEFs were seeded into 12-well plates as described above, transfected with 0.5 μg of LC3-GFP plasmid, and subsequently infected with SL1344-DsRed2 after 48 h. In ATG3-deficient cells the percentage of bacteria observed within LC3-GFP-positive membranes was markedly reduced at all time points studied compared with control cells in a similar fashion to that seen in FNBP1L knockdown. In ATG3-replete cells 19 ± 3% of bacteria were within LC3-GFP-compartments after 1 h of infection, falling to 5 and 4 ± 2% after 2 and 4 h, respectively. In contrast, ATG3-deficient MEFs failed to encapsulate more than 4 ± 2% of internalized Salmonella within LC3-positive membranes at any time point. These results (pooled from two independent experiments) showed significantly less anti-Salmonella autophagy in ATG-deficient cells than ATG3-replete MEFs at 1 h postinfection (p > 0.005); however, the difference was lost by 2 h after infection. We thus concluded, as has been observed for classical autophagy (34), that ATG3 was essential for the autophagy of S. Typhimurium.
Previous studies have shown that FNBP1L associates with the active form of Cdc42 (GTP bound) and is essential for activation of the neural Wiskott-Aldrich syndrome protein, both in vitro and in vivo (19, 46). Therefore, we explored whether Cdc42 plays an active role in anti-Salmonella autophagy and whether the Cdc42-FNBP1L interaction is required for this process. Using siRNAs directed to Cdc42, we established robust knockdown of Cdc42 mRNA message in HeLa-LC3-GFP cells (supplemental Fig. S3A). Infection of these cells with S. Typhimurium resulted in levels of bacterial internalization similar to those in control-treated cells, as has been previously established (47). In addition, we observed no overt effect of Cdc42 knockdown upon cytoskeletal morphology at the time points studied (48 h post-transfection), similar to previous studies (47). Cdc42 knockdown did not result in a significant reduction of bacteria within autophagosomes (supplemental Fig. S3B). Autophagosomes in Cdc42-deficient cells formed around bacteria within the same time scale as in control cells, and no overt differences in autophagosome cellular location or shape were observed (supplemental Fig. S3C).
The FNBP1L-deficient phenotype can be fully and specifically rescued by restoration of FNBP1L expression
To ensure that the observed loss of anti-Salmonella autophagic ability was due to FNBP1L depletion, we undertook rescue experiments using FNBP1L constructs with synonymous mRNA mutations within the siRNA target sequences. Cotransfection of HeLa LC3-GFP cells with control siRNA and wild type or either rescue sequence resulted in no significant alteration in the anti-Salmonella autophagic response to Salmonella infection (Fig. 5B, upper graph). FNBP1Lsi1-treated cells showed a marked loss of autophagic activity with siRNA alone and were fully rescued only with FNBP1L R1 rescue construct but not the wild-type or the R2 rescue construct sequence. Likewise, rescue of FNBP1Lsi2-transfected cells was only accomplished with FNBP1L R2 and not with the wild-type or R1 construct. This result was confirmed by Western blotting, with the expression of FNBP1L observed only under the appropriate rescue conditions (Fig. 5B, lower panels). These data confirm that FNBP1L is required for anti-Salmonella autophagy.
Detailed microscopic examination showed that cells in which FNBP1L was silenced displayed a lack of bacterial/LC3 colocalization despite having normal levels of LC3-GFP expression and often showing many small LC3-GFP vesicles throughout the cytoplasm (Fig. 5C). Autophagic capture of bacteria was almost entirely absent in FNBP1l-deficient cells, and no partial LC3-positive membrane enclosure events (e.g., autophagic cups or putative pre-autophagosomal structures) were observed, suggesting an initiation failure rather than an autophagosomal completion defect. These findings are indicative of a failure to assemble the autophagic apparatus in adequate quantity or density to generate sufficient membrane to engulf bacteria despite the ability to generate smaller autophagic vesicles in response to bacterial infection or starvation signals.
To further confirm these results and demonstrate conclusively that FNBP1L-dependent autophagy is an antibacterial mechanism in epithelial cells, we performed gentamicin protection assays in control, ATG16L1-, or FNBP1L-deficient HeLa cells followed by quantification of viable bacteria (Fig. 5D). We had previously established that ATG16L1 is essential for anti-Salmonella autophagy in HeLa cells (6) and therefore included an ATG16L1-directed siRNA as a positive control for loss of autophagy. We observed no significant differences in numbers of viable internalized bacteria at 1 or 3 h, but at 5 h postinfection the control cells harbored significantly fewer viable bacteria than ATG16L1- or FNBP1L-deficient cells. This result demonstrates that anti-Salmonella autophagy is both a potent restraint upon bacterial replication within the intracellular niche and is dependent upon FNBP1L.
FNBP1L is associated with a subpopulation of internalized bacteria, colocates with ATG3, and is required for ATG3 association with internalized S. typhimurium
We reasoned that if FNBP1L was associated with the autophagy apparatus via ATG3, it should be possible to place both FNBP1L and ATG3 around internalized bacteria during infection. Currently available Abs are not sensitive enough to permit immunofluorescent detection of endogenous FNBP1L, and therefore we overexpressed FNBP1L-GFP in HeLa cells and examined its location during infection with S. typhimurium. We were able to observe that, 40 min after infection, internalized bacteria could be found closely surrounded by rings or “halos” of FNBP1L-GFP in a manner not observed at earlier time points nor with GFP alone (Fig. 6A). The kinetics of the bacterial FNBP1L-GFP localization was shifted toward earlier time points than that of LC3-GFP, peaking at 40 min postinfection (Fig. 6B) rather than at 1 h. This apparent association of FNBP1L to the SCV was lost post-autophagic encapsulation because we did not observe colocalization of mRFP-LC3 and FNBP1L-GFP around bacteria (data not shown). Expressing the ATG3-GFP construct also resulted in localization to bacteria within a similar time scale, and at 40 min postinfection an average of 10% of internalized bacteria were surrounded by ATG3-GFP “halos.” This ATG3 localization was FNBP1L-dependent, because knockdown of FNBP1L by siRNA resulted in a significant loss of ATG3 halos (Fig. 6C). Using the ATG3-GFP construct along with cotransfection of FNBP1L-Myc, we observed colocalization of these proteins both around internalized bacteria and in other vesicular structures in proximity to internalized Salmonella. It is of note that none of the FNBP1L- or ATG3-associated bacteria or the costained vesicular membranes were associated with F-actin (as determined by phalloidin staining). Confocal z-reconstruction of image stacks also showed the bacteria to be within the cytoplasmic volume. Therefore, we do not believe that these observations were caused by bacterial entry artifacts, nor were they simply surface ruffles or tubules induced by FNBP1L overexpression.
FIGURE 6.

FNBP1L localizes to a subset of internalized bacteria; ATG3 localization to Salmonella is FNBP1L dependent. A, FNBP1L-GFP-transfected cells and control GFP-expressing cells were infected with S. Typhimurium for 40 min, fixed, and examined by confocal microscopy. A subset of internalized S. Typhimurium was surrounded by FNBP1L-GFP “halos” (arrows); these structures were absent in GFP-expressing cells. In merged images S. Typhimurium is shown in blue and FNBP1L-GFP/GFP is green; the scale bar represents 10 μm. Images were cropped and were taken from high-resolution confocal z-projections. B, Colocalization of FNBP1L and S. Typhimurium was observed to peak at 40 min postinfection, with a marked drop at 1 h. The peak of 21% colocalization is of a similar magnitude to that of S. Typhimurium and LC3-GFP at 1 h. Data are shown as means and SEM and were pooled from two independent experiments, each counting at least 50 cells per condition. C, Localization of ATG3-GFP to S. Typhimurium is FNBP1L dependent. HeLa cells were cotransfected with control or FNBP1L-directed siRNA and ATG3-GFP plasmid and infected 48 h later. Forty minutes postinfection the cells were fixed, stained, and examined for ATG3-GFP/bacterial association. In control cells, 10% of internalized S. Typhimurium were placed within ATG3-GFP “halos,” and this percentage dropped to 1% in the absence of FNBP1L. Data are presented as means with SEM; data were pooled from two independent experiments, each counting at least 50 cells per condition. Significance was assessed using one-way ANOVA, followed by post hoc t tests using the Bonferroni correction, comparing control siRNA to FNBP1L-directed siRNA samples. D, ATG3-GFP and FNBP1L-Myc colocate to a subset of bacteria within infected cells. HeLa cells cotransfected with ATG3-GFP and FNBP1L-Myc were infected with S. Typhimurium for 40 min and fixed and stained. Upper row shows overview of a confocal z-stack projection with the lower row showing high magnification crops of the area marked in the merge column. ATG3-GFP is shown as green, FNBP1L-Myc as red, and DNA as blue in the merged images. Bacteria surrounded by both FNBP1L and ATG3 are marked by arrows in the lower panelS. Scale bar represents 10 μm.
Salmonella-containing vesicle handling and bacterial ubiquitination is unaffected by FNBP1L knockdown
To control for the possibility that FNBP1L knockdown blocked SCV maturation or maintenance, we performed control experiments in which we examined SCV maturation. SCV maturation is a process by which the nascent SCV acquires a number of cellular markers, including LAMP1, through Rab7-dependent interactions with the endosomal recycling system (48). It is worth noting that SCVs exclude most lysosomal enzymes and avoid direct lysosomal fusion; thus, acquisition of a subset of lysosomal glycoproteins does not signify lysosome-mediated killing of internalized bacteria. LAMP1 was chosen as a marker because it is acquired by the SCV with similar kinetics that of to anti-Salmonella autophagy, with maximal LAMP1/SCV-colocalization 1 h postinfection (49). SCV maturation was assessed by the staining of infected cells with anti-LAMP1 Abs 1 h postinfection followed by the counting of LAMP1-bacteria colocalization. In both control and FNBP1L-knockdown HeLa cells, the proportion of SCVs acquiring LAMP1 after 1 h remained constant at between 48 and 53% (Fig. 7A). This figure is similar to that observed by others in this model system (49) and indicates that SCV maturation kinetics are not affected by FNBP1L ablation.
FIGURE 7.

FNBP1L is not required for the maturation of the Salmonella-containing vacuole, and FNBP1L truncations act as dominant negatives. A, FNBP1L knockdown does not affect the maturation of the SCV. HeLa cells were transfected with control or FNBP1L siRNAs and infected with S. Typhimurium 48 h later. One hour postinfection the cells were fixed and stained for LAMP1. The percentage of LAMP1-positive SCVs observed for each siRNA is shown. No significant differences in LAMP1 accumulation, and hence SCV maturation, were observed. Data are shown as the means of two independent experiments ± SEM, counting at least 100 cells for each condition with p values calculated by Student's t test, compared to siControl values. The accompanying micrographs show representative high-magnification crops of LAMP1 (green in merge) and DNA to mark bacteria (blue in merge). B, FNBP1L knockdown does not affect the ubiquitination (Ubq) of internalized Salmonella. HeLa cells treated with control or anti-FNBP1L siRNAs were infected with S. Typhimurium as above; following fixation, cells were stained with anti-ubiquitin Abs and examined for bacterial ubiquitination. The number of bacteria colocalizing with ubiquitin staining were quantified and no significant differences were observed between control and FNBP1L-deficient cells. Data are shown as the means of two independent experiments ± SEM, counting at least 100 cells for each condition with p values calculated by Student's t test, compared to siControl values. The accompanying micrographs show representative high magnification crops of staining for ubiquitinated proteins (green in merge) and DNA to mark bacteria (blue in merge). C, FNBP1L truncations act as dominant negatives, reducing autophagic efficiency against S. Typhimurium. LC3-GFP HeLa cells were transfected with 500 ng of Myc vector, full-length FNBP1L, or one of two truncations corresponding to amino acids 1–293 or 110–547 of the full-length protein. Twenty-four hours later the cells were infected with S. Typhimurium and fixed after 1 h. The number of bacteria captured within autophagic vacuoles was quantified as previously detailed. Significant loss of autophagy was observed with both truncated constructs when compared with full-length FNBP1L expression. Data are shown as means from at least 120 cells per condition ± SEM, with p values calculated by Student's t test, compared to full-length FNBP1L values.
Autophagosome formation around intracellular S. Typhimurium has been linked to permeabilization of the SCV membrane by the SPI1 secretion apparatus (3). This results in bacterial ubiquitination, and this or another uncharacterized signal from the cytoplasm appears essential for anti-Salmonella autophagy. We considered that the loss of FNBP1L might reduce or eliminate ubiquitination of SCV-permeabilizing bacteria or otherwise alter SCV membrane dynamics, and we therefore performed staining for ubiquitination of bacteria in FNBP1L-deficient and control HeLa cells. In both control and FNBP1L-deficient cells the ubiquitination of bacteria was maintained at ∼25%, and no significant differences were observed between conditions (Fig. 7B).
FNBP1L constructs lacking either the F-BAR or HR1 domains act as dominant negatives
We reasoned that the role of FNBP1L in autophagy would require both membrane and ATG3 interactions, and therefore truncations lacking either one of these features would act as dominant negatives. This was indeed the case, with both FNBP1L-1–293 and 110–547 significantly reducing the proportion of Salmonella within autophagic compartments at 1 h postinfection (Fig. 7C). Compared with vector control, overexpression of FNBP1L did not result in significant alteration in autophagy rates; however, FNBP1L-1–293 markedly reduced autophagy compared with full-length FNBP1L. This reduction from 22 ± 2%) to just 7.0 ± 2%) was not as dramatic in the case of the FNBP1L-100–547 construct, where S. Typhimurium captured by autophagy only fell to 12 ± 2%); however, both results were statistically significant.
Discussion
In this study we demonstrate a strategy for discovering novel human autophagy components by building upon the yeast interaction network. The mapping of yeast autophagy components onto human orthologs identified some 14 novel putative autophagy-related components. In parallel, we also extended the yeast interolog network to encompass both first and second order interactions, which revealed a number of interesting, functionally related clusters (supplemental Fig. S1B). ATG5 and ATG12 exhibited extensive second order connectivities with a cluster of proteosomal components via PSMD2, itself a regulatory subunit of the 26S proteasome. It has become clear that under some conditions autophagy and the proteasome share substrates and that homeostatic protein turnover is accomplished through a regulated balance of their activities (50). This interaction of a regulatory proteasome subunit with the autophagy apparatus highlights a potentially important interplay between autophagy and the proteasome machinery.
In our initial “hybrid” mapping we found that ATG12 interacts with the phospholipid scramblase PLSCR1, a protein with a known role in movement of phospholipids, response to growth factors, acute phase responses, and IFN-mediated antiviral activities and whose expression level is highly responsive to TLR and dectin receptor ligands (51–53). In addition, PLSCR1 is thought to exhibit antileukemic activity via its interactions with Bcl-2 and c-Myc (54, 55), which is especially intriguing because Beclin-1 also is known to interact with Bcl-2 (56). Almost all of these functions have plausible links to autophagy regulation, especially in phagocytes. It is also known that during programmed cell death, dying cells expose specific phospholipids (e.g., phosphatidylserine) on their cell surfaces as a signal for their rapid phagocytosis and disposal and that both phosphatidylserine display and cell corpse disposal are autophagy dependent (57). It is tempting to speculate that PLSCR1 may in part be responsible for such activities, either in generating the cell membrane signal during cell death or by performing a similar role upon membrane-bound organelles within the cell, e.g., mitochondria, marking them as autophagic cargo.
Also intriguing is the recent identification of two focal adhesion-associated proteins that play key roles within autophagy, namely paxillin and FIP200 (58, 59). In mammalian cells, paxillin, vinculin, and FIP200 can be localized to classical autophagosomes following starvation. However, at least in the case of FIP200, this appears to occur in a focal adhesion kinase (PTK2)-independent fashion via interaction with ULK/ATG1, suggesting that the entire focal adhesion complex is not required for autophagy (58). In our yeast interactome we identified PTK2 as an interactor with ATG12 and we note that ULK overexpression also yields changes in cell morphology, suggesting that additional interplay between focal adhesion and autophagy proteins remains to be discovered. There is likely to be a role for specific membrane trafficking and fusion events within the autophagy process, and Rab5 is known to be essential for the assembly of autophagosomes from smaller membrane compartments (60). Previously, the only known role of Rab5 was in endocytosis, also a process with which FNBP1L has been associated (20, 43). These recent results suggest that components of the cytoskeleton/membrane interface apparatus are key mediators of autophagosome formation.
It is known that FNBP1L is involved in the generation of membrane structures and dynamic membrane folding events (19, 21, 61) and that BZZ1 performs a partially redundant role in yeast endocytosis, linking the cytoskeleton to sites of clathrin accumulation (20, 43). We were able to demonstrate via siRNA that FNBP1L is essential for anti-Salmonella autophagosome formation and that the siRNA-mediated phenotype can be rescued by the restoration of FNBP1L expression. Our data also show that in epithelial cells FNBP1L is essential for the restriction of S. Typhimurium replication within the intracellular niche. This control of replication was established postinvasion, because similar numbers of bacteria were internalized by FNBP1L-deficient and control cells, and differences were only observed during the replicative phase of infection (3–5 h postinvasion). The ability of cells to generate and process classical autophagosomes and the presence of small LC3 puncta in Salmonella-infected, FNBP1L-deficient cells indicated that FNBP1L did indeed play a crucial role solely in the assembly of anti-Salmonella autophagosomes. We were able to place FNBP1L-GFP surrounding internalized bacteria, an association with the SCV membrane that suggests FNBP1L may act as a scaffold between the cargo and the growing autophagosome. The FNBP1L-bacteria localization was largely matched in both frequency and location by ATG3, with both proteins coassociating with a subset of internalized bacteria at 40 min postinfection. We demonstrated that ATG3 was required for successful anti-Salmonella autophagy and showed that ATG3 recruitment to the SCV was dependent upon FNBP1L, further validating the functional significance of the ATG3/FNBP1L interaction. Based upon these findings, we postulate that FNBP1L is required for the autophagic encapsulation of intracellular bacteria. It is thought that the antibacterial autophagic process proceeds via the assembly of large autophagosomes that conform to the shape of their cargo. This is in contrast to other forms of autophagy, where spherical vesicles do not conform to their contents (62). It has been suggested that the BAR domain-containing protein Bif-1 is essential for classical autophagy (63), acting as a scaffold to localize class III PI3K activity to the autophagic membrane. In antibacterial autophagy following the formation of an autophagosomal cup, the double membrane must be extended and fused to form a complete autophagosome, enveloping the bacterium (3). This process is likely to require significantly enhanced membrane generation and extension than that required for smaller classical autophagosomes. It has been shown that the amount of available ATG8 (LC3) serves to control the size of autophagic vesicles (64) in yeast. Thus, we postulate that FNBP1L serves as a scaffold to tether the ATG3/7 complex close to the autophagic cargo, localizing LC3-PE generation to the nascent autophagosome, driving formation of large autophagic membranes.
Although the exact nature and stimuli behind the process of anti-Salmonella autophagosome initiation remain largely unresolved, in this study we present FNBP1L as a molecule with a specific role in anti-Salmonella autophagy as distinct from classical autophagy. The identification of FNBP1L using a network method built upon the yeast interactome demonstrates both the large level of evolutionary conservation of autophagy components from yeast to mammals, as well as the evolution of mammalian autophagy to a more diverse and sophisticated system. We postulate that it is likely that many as yet unknown mammalian autophagy components will also be conserved from yeast, and we believe that our approach provides an insight into these previously unidentified mammalian autophagy components.
Supplementary Material
Acknowledgments
We sincerely thank Drs. M. W. Kirschner, V. Yajnik, and C. Munz for gifts of reagents. Aimee Landry and Chun Li provided invaluable technical assistance. Additional thanks go to Dr. N. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) for critical discussions of the manuscript.
Footnotes
This work was supported National Institutes of Health Grants AI062773, DK83756, and DK043351 (to R.J.X., M.J.D., and A.H.) and DK060049 and DK043351 (to D.K.P.). A.N. is supported by a Crohn's and Colitis Foundation of America fellowship.
Abbreviations used in this paper: siRNA, small interfering RNA; AFA, ancillaryfactors of autophagy; MEF, mouse embryonic fibroblast; MOI, multiplicity of infection;RNAi, RNA interference; SCV, Salmonella-containing vacuole; S. Typhimurium, Salmonella enterica serovar Typhimurium.
The online version of this article contains supplemental material.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neu-rodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 2.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 3.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–11383. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 4.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 5.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, Drummond H, Lees CW, Khawaja SA, Bagnall R, Burke DA, Todhunter CE, Ahmad T, Onnie CM, McArdle W, Strachan D, Bethel G, Bryan C, Lewis CM, Deloukas P, Forbes A, Sanderson J, Jewell DP, Satsangi J, Mansfield JC, Cardon L, Mathew CG. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 9.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 11.Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281:36303–36316. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- 12.Scott SV, Hefner-Gravink A, Morano KA, Noda T, Ohsumi Y, Klionsky DJ. Cytoplasm-to-vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc Natl Acad Sci USA. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutchins MU, Veenhuis M, Klionsky DJ. Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J Cell Sci. 1999;112:4079–4087. doi: 10.1242/jcs.112.22.4079. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, Garrels J, Vincent S, Vidal M. Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs”. Genome Res. 2001;11:2120–2126. doi: 10.1101/gr.205301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavithra SR, Kumar R, Tatu U. Systems analysis of chaperone networks in the malarial parasite Plasmodium falciparum. PLoS Comput Biol. 2007;3:1701–1715. doi: 10.1371/journal.pcbi.0030168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JD, Bertin N, Chung S, Vidal M, Gerstein M. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14:1107–1118. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarassov K, Messier V, Landry CR, Radinovic S, Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–1470. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- 19.Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 20.Soulard A, Lechler T, Spiridonov V, Shevchenko A, Shevchenko A, Li R, Winsor B. Saccharomyces cerevisiae Bzz1p is implicated with type I myosins in actin patch polarization and is able to recruit actin-polymerizing machinery in vitro. Mol Cell Biol. 2002;22:7889–7906. doi: 10.1128/MCB.22.22.7889-7906.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kakimoto T, Katoh H, Negishi M. Regulation of neuronal morphology by Toca-1, an F-BAR/EFC protein that induces plasma membrane invagination. J Biol Chem. 2006;281:29042–29053. doi: 10.1074/jbc.M604025200. [DOI] [PubMed] [Google Scholar]
- 22.Tsujita K, Suetsugu S, Sasaki N, Furutani M, Oikawa T, Takenawa T. Coordination between the actin cytoskeleton and membrane deformation by a novel membrane tubulation domain of PCH proteins is involved in endocytosis. J Cell Biol. 2006;172:269–279. doi: 10.1083/jcb.200508091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito T, Tashiro K, Muta S, Ozawa R, Chiba T, Nishizawa M, Yamamoto K, Kuhara S, Sakaki Y. Toward a protein-protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 25.Mishra GR, Suresh M, Kumaran K, Kannabiran N, Suresh S, Bala P, Shivakumar K, Anuradha N, Reddy R, Raghavan TM, et al. Human protein reference database—2006 update. Nucleic Acids Res. 2006;34:D411–D414. doi: 10.1093/nar/gkj141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–D574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng A, Bursteinas B, Gao Q, Mollison E, Zvelebil M. pSTIING: a ‘systems’ approach towards integrating signalling pathways, interaction and transcriptional regulatory networks in inflammation and cancer. Nucleic Acids Res. 2006;34:D527–D534. doi: 10.1093/nar/gkj044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gansner ER, North SC. An open graph visualization system and its applications to software engineering. Softw Pract Exper. 2000;30:1203–1233. [Google Scholar]
- 29.Kamada T, Kawai S. An algorithm for drawing general undirected graphs. Inform Proc Lett. 1989;31:7–15. [Google Scholar]
- 30.Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, Harris MA, Dolinski K, Mohr S, Smith T, et al. Comparison of the complete protein sets of worm and yeast: orthology and divergence. Science. 1998;282:2022–2028. doi: 10.1126/science.282.5396.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mushegian AR, Garey JR, Martin J, Liu LX. Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 1998;8:590–598. doi: 10.1101/gr.8.6.590. [DOI] [PubMed] [Google Scholar]
- 32.Rubinstein R, Simon I. MILANO–custom annotation of microarray results using automatic literature searches. BMC Bioinformatics. 2005;6:12. doi: 10.1186/1471-2105-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saldanha AJ. Java Treeview– extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 34.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–4775. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 36.Beuzon CR, Meresse S, Unsworth KE, Ruiz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 2000;19:3235–3249. doi: 10.1093/emboj/19.13.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engqvist-Goldstein AE, Zhang CX, Carreno S, Barroso C, Heuser JE, Drubin DG. RNAi-mediated Hip1R silencing results in stable association between the endocytic machinery and the actin assembly machinery. Mol Biol Cell. 2004;15:1666–1679. doi: 10.1091/mbc.E03-09-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 40.Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;2006:pe44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- 41.Fujita N, Itoh T, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamioka Y, Fukuhara S, Sawa H, Nagashima K, Masuda M, Matsuda M, Mochizuki N. A novel dynamin-associating molecule, formin-binding protein 17, induces tubular membrane invaginations and participates in endocytosis. J Biol Chem. 2004;279:40091–40099. doi: 10.1074/jbc.M404899200. [DOI] [PubMed] [Google Scholar]
- 43.Soulard A, Friant S, Fitterer C, Orange C, Kaneva G, Mirey G, Winsor B. The WASP/Las17p-interacting protein Bzz1p functions with Myo5p in an early stage of endocytosis. Protoplasma. 2005;226:89–101. doi: 10.1007/s00709-005-0108-4. [DOI] [PubMed] [Google Scholar]
- 44.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 46.Leung Y, Ally S, Goldberg MB. Bacterial actin assembly requires toca-1 to relieve N-WASP autoinhibition. Cell Host Microbe. 2008;3:39–47. doi: 10.1016/j.chom.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel JC, Galan JE. Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol. 2006;175:453–463. doi: 10.1083/jcb.200605144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meresse S, Steele-Mortimer O, Finlay BB, Gorvel JP. The rab7 GTPase controls the maturation of Salmonella typhimurium-containing vacuoles in HeLa cells. EMBO J. 1999;18:4394–4403. doi: 10.1093/emboj/18.16.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith AC, Cirulis JT, Casanova JE, Scidmore MA, Brumell JH. Interaction of the Salmonella-containing vacuole with the endocytic recycling system. J Biol Chem. 2005;280:24634–24641. doi: 10.1074/jbc.M500358200. [DOI] [PubMed] [Google Scholar]
- 50.Scott CM, Kruse KB, Schmidt BZ, Perlmutter DH, McCracken AA, Brodsky JL. ADD66, a gene involved in the endoplasmic reticulum-associated degradation of α-1-antitrypsin-Z in yeast, facilitates proteasome activity and assembly. Mol Biol Cell. 2007;18:3776–3787. doi: 10.1091/mbc.E07-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong B, Zhou Q, Zhao J, Zhou A, Harty RN, Bose S, Banerjee A, Slee R, Guenther J, Williams BR, et al. Phospholipid scramblase 1 potentiates the antiviral activity of interferon. J Virol. 2004;78:8983–8993. doi: 10.1128/JVI.78.17.8983-8993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu B, Sims PJ, Wiedmer T, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. Expression of the phospholipid scramblase (PLSCR) gene family during the acute phase response. Biochim Biophys Acta. 2007;1771:1177–1185. doi: 10.1016/j.bbalip.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 53.Zhou Q, Zhao J, Wiedmer T, Sims PJ. Normal hemostasis but defective hematopoietic response to growth factors in mice deficient in phospholipid scramblase 1. Blood. 2002;99:4030–4038. doi: 10.1182/blood-2001-12-0271. [DOI] [PubMed] [Google Scholar]
- 54.Huang Y, Zhao Q, Zhou CX, Gu ZM, Li D, Xu HZ, Wiedmer T, Sims PJ, Zhao KW, Chen GQ. Antileukemic roles of human phospholipid scramblase 1 gene, evidence from inducible PLSCR1-expressing leukemic cells. Oncogene. 2006;25:6618–6627. doi: 10.1038/sj.onc.1209677. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Rogulski K, Zhou Q, Sims PJ, Prochownik EV. The negative c-Myc target onzin affects proliferation and apoptosis via its obligate interaction with phospholipid scramblase 1. Mol Cell Biol. 2006;26:3401–3413. doi: 10.1128/MCB.26.9.3401-3413.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 57.Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 58.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen GC, Lee JY, Tang HW, Debnath J, Thomas SM, Settleman J. Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy. 2008;4:37–45. doi: 10.4161/auto.5141. [DOI] [PubMed] [Google Scholar]
- 60.Ravikumar B, Imarisio S, Sarkar S, O'Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci. 2008;121:1649–1660. doi: 10.1242/jcs.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 62.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 63.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–3298. doi: 10.1091/mbc.E07-12-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.