

Abstract
Non-small cell lung cancer (NSCLC)-associated epidermal growth factor receptor (EGFR) mutants are constitutively active and induce ligand-independent transformation in nonmalignant cell lines. We investigated the possibility that the ability of mutant EGFRs to transform cells reflects a constitutive cooperativity with Src using a system in which the overexpression of mutant but not wild-type EGFR induced anchorage-independent cell growth. Src was constitutively activated and showed enhanced interaction with mutant EGFRs, suggesting that constitutive EGFR-Src cooperativity may contribute to mutant EGFR-mediated oncogenesis. Indeed, the mutant EGFR-mediated cell transformation was inhibited by Src- as well as EGFR-directed inhibitors. Importantly, a tyrosine to phenylalanine mutation of the major Src phosphorylation site on EGFR, Y845, reduced the constitutive phosphorylation of NSCLC EGFR mutants as well as of STAT3, Akt, Erk and Src, and reduced the mutant EGFR-Src association as well as proliferation, migration, and anchorage-independent growth. Reduced anchorage-independent growth and migration were also observed when DN-Src was expressed in mutant EGFR-expressing cells. Overall, our findings demonstrate that mutant EGFR-Src interaction and cooperativity play critical roles in constitutive engagement of the downstream signaling pathways that allow NSCLC-associated EGFR mutants to mediate oncogenesis, and support the rationale to target Src-dependent signaling pathways in mutant EGFR-mediated malignancies.
Keywords: mutant EGFR, Src, transformation
Introduction
Epidermal growth factor receptor (EGFR) is a member of the ErbB family (ErbB1-4) of receptor tyrosine kinases that plays important physiological functions by controlling critical processes including proliferation, survival, migration, and differentiation (Herbst, 2004; Wiley and Burke, 2001). Aberrant EGFR signaling through overexpression and/or mutation occurs in many cancers including breast cancer, head-and-neck cancer, NSCLC, renal cancer, ovarian cancer and colon cancer (Herbst, 2004). Notably, specific subtypes of NSCLC patients exhibit somatic EGFR mutations that impart a higher sensitivity to EGFR-directed tyrosine kinase inhibitors (TKIs) such as gefitinib (Iressa) or erlotinib (Tarceva) (Gschwind et al., 2004; Lynch et al., 2004). Consistent with in vivo findings, NSCLC cell lines with mutant EGFRs are hypersensitive to growth inhibition upon gefitinib treatment (Sordella et al., 2004).
Gefitinib-sensitizing NSCLC-associated EGFR mutations are located in the catalytic domain of EGFR near the ATP-binding pocket, which is also the binding site for TKIs (Paez et al., 2004; Sordella et al., 2004). The most common NSCLC-associated mutations are in-frame deletions of codons 746-750 (EGFR Δ746-750) and a leucine to arginine point mutation at codon 858 (EGFR L858R) of the full-length EGFR (Mitsudomi et al., 2005).
Previous studies of cells expressing mutant EGFRs indicate that mutant receptors are constitutively active (Lynch et al., 2004; Yun et al., 2007; Zhang et al., 2006) and transform nonmalignant cell lines in a ligand-independent manner (Greulich et al., 2005). Furthermore, cells harboring mutant EGFRs undergo “oncogene addiction” and require the mutant receptor activity for survival (Sharma et al., 2007). In transgenic mouse models, reduced expression of mutant EGFR or inhibition of kinase activity caused rapid tumor regression, demonstrating that mutant EGFR is required for tumor maintenance (Politi et al., 2006). These analyses support the overall rationale provided by clinical observations for EGFR-directed targeted therapy of mutant EGFR-expressing cancers. However, resistance to these drugs can develop rapidly in part due to secondary mutations that prevent TKI binding (Pao et al., 2005). Therefore, understanding mechanisms that allow mutant EGFRs to engage in constitutive oncogenic signaling are likely to point to alternate strategies for treatment of mutant EGFR-driven cancers.
In contrast to mutant EGFRs, overexpression of wild-type EGFR (wtEGFR) in primary cells is not oncogenic. High levels of exogenous ligands have been reported to promote oncogenesis in certain cell systems (Di Fiore et al., 1987). Several studies have revealed cooperative signaling between EGFR and Src to be an important determinant of EGFR-mediated oncogenesis (Bromann et al., 2004). Overexpression of both EGFR and Src in a mouse fibroblast model system led to synergistic increase in EGF-induced DNA synthesis, soft agar colony formation, and tumor formation in nude mice (Bromann et al., 2004). This cooperativity has also been demonstrated in the context of epithelial cell transformation: loss of polarity in three-dimensional cultures of nonmalignant human mammary epithelial cells as well as their anchorage-independent growth were only seen when both EGFR and Src were co-overexpressed (Dimri et al., 2007). Consistent with these initial studies, EGFR and Src are often overexpressed in human cancers (Silva, 2004). In NSCLC, Src expression and its activity are elevated in tumor compared to normal tissue (Masaki et al., 2003).
The mechanism of EGFR and Src synergy involves reciprocal trans-activation. Src becomes activated upon EGF stimulation and mediates phosphorylation of EGFR, including that on tyrosine 845 (Y845, Y869 in the full-length EGFR) (Biscardi et al., 1999). Mutational analysis has shown that phosphorylation of EGFR Y845 is critical for EGF-induced DNA synthesis (Biscardi et al., 1999).
Altogether, these findings suggest that one likely mechanism of oncogenic signaling by mutant EGFRs may be a constitutive engagement of the Src pathway. Consistent with this idea, Src was hyperphosphorylated and associated with EGFR in NSCLC cell lines harboring mutant EGFRs, and these cells are more sensitive to Src inhibitors for cell death than cells expressing wtEGFR (Song et al., 2006; Yang et al., 2008; Zhang et al., 2007). To systematically define the role of Src in mutant EGFR-mediated oncogenesis, we used an NIH 3T3 cell overexpression system in which the mutant but not the wild-type EGFR induces oncogenic transformation. Our findings suggest that a constitutive cooperativity with Src is critical for mutant EGFR-mediated oncogenic transformation, supporting the rationale for concurrent or alternative targeting of Src-dependent signaling for targeted therapy of mutant EGFR-driven cancers.
Results
Constitutive activity of mutant EGF receptors and cellular transformation upon stable expression in NIH 3T3 cells
We first established retroviral transductants of NIH 3T3 cells stably expressing the vector (V), wtEGFR (WT), EGFR L858R (LR) or EGFR Δ746-750 (DEL) to confirm the mutant EGFR-induced transformation. Previous studies have established that introduction of NSCLC-associated EGFR mutants into NIH 3T3 cells, which expresses very low levels of endogenous EGFR, leads to transformation (Greulich et al., 2005). We used this model system to examine the role of EGFR-Src interaction in oncogenesis. Consistent with prior studies (Sordella et al., 2004), mutant EGFRs showed constitutive phosphorylation (Supplementary Figure S1A), while wtEGFR showed phosphorylation only upon EGF stimulation.
We next used the soft agar colony assay to assess the transforming ability of mutant EGFRs (Supplementary Figure S1B). Cells expressing mutant EGFRs but not the vector control or wtEGFR-expressing cells showed colony formation in soft agar. Interestingly, colony growth in low serum medium (0.1 % FBS) was comparable to that in complete growth medium (10 % FBS) (Supplementary Figure S1C). Thus, consistent with previously published data, mutant EGFRs expressed in NIH 3T3 cells are constitutively active and induce cellular transformation, providing a validated set of cellular reagents to test the role of mutant EGFR cooperation with Src.
Constitutive Src signaling downstream of mutant EGF receptors
In an effort to identify molecular pathways through which mutant EGFRs trigger cell transformation, we examined the activation and expression levels of STAT3, Akt and Erk, major downstream targets of EGFR that control a wide array of cellular processes. Similar to EGFR phosphorylation (Supplementary Figure S1A), the phosphorylation levels of STAT3, Akt and Erk upon EGF stimulation did not differ greatly between wtEGFR- and mutant EGFR-expressing cells (Figure 1A). In contrast, cells expressing mutant EGFRs showed enhanced phosphorylation of each of the analyzed signaling proteins in the absence of EGF stimulation when compared to the vector control or wtEGFR cells.
Figure 1. Constitutive activation of EGFR downstream targets and Src by mutant EGFRs.

NIH 3T3 cells stably expressing the vector (V), wtEGFR (WT), EGFR L858R (LR) or EGFR Δ746-750 (DEL) were serum-deprived for 3 hr and then either left unstimulated (−) or stimulated (+) with 10 ng/ml EGF for 10 min. (A) 50 μg aliquots of whole cell lysates were used for immunoblotting with antibodies against the indicated proteins. (B) 50 μg aliquots of whole cell lysates were used for immunoblotting with antibodies against phospho-Src (pSrc, Y416) and total Src (SRC 2).
Since STAT activation mediates EGFR and Src synergism (Kloth et al., 2003) and Akt and Erk can also be activated by Src (Bromann et al., 2004), these findings suggest that mutant EGFRs induce a constitutive activation of the Src signaling pathway. Consistent with this suggestion, Src is hyperphosphorylated in NSCLC cell lines expressing mutant receptors, and these cell lines are sensitive to Src inhibition (Song et al., 2006; Zhang et al., 2007). Indeed, levels of active Src were elevated in cells expressing mutant EGFRs (Figure 1B). While Src phosphorylation was dependent upon EGF stimulation in wtEGFR-expressing cells, Src was hyperphosphorylated and its phosphorylation was constitutive in mutant EGFR-expressing cells.
Next, we examined the complex formation between EGFR and Src. Previous studies have shown that, unlike ErbB2, interaction of EGFR with Src is not stable (Kim et al., 2005; Muthuswamy and Muller, 1995). The differential association of Src with ErbB2 versus EGFR is mediated through differences in the kinase domain (Kim et al., 2005). As mutant EGFRs also share structural differences with wtEGFR in the kinase domain (Zhang et al., 2006), we surmised that mutant EGFRs may exhibit more stable association with Src. Therefore, we carried out co-immunoprecipitation experiments in unstimulated as well as EGF-stimulated cells to evaluate EGFR-Src association (Figure 2A). Consistent with a previous report (Yang et al., 2008), when EGFR was immunoprecipitated and probed for Src, mutant EGFR-expressing cells showed increased levels of co-immunoprecipitated Src in the presence and absence of EGF stimulation compared to wtEGFR. Co-IP experiments on NSCLC cell lines expressing wtEGFR (H1666) versus mutant EGFR (HCC4006) confirmed these findings (Figure 2B). Thus, mutant EGFRs interact more stably with Src compared to wtEGFR. Preincubating cells with erlotinib abrogated mutant EGFR phosphorylation and association with Src, indicating that mutant EGFR kinase activity is critical for the enhanced association with Src (Figure 2C).
Figure 2. Constitutive interaction of mutant EGFRs with Src.
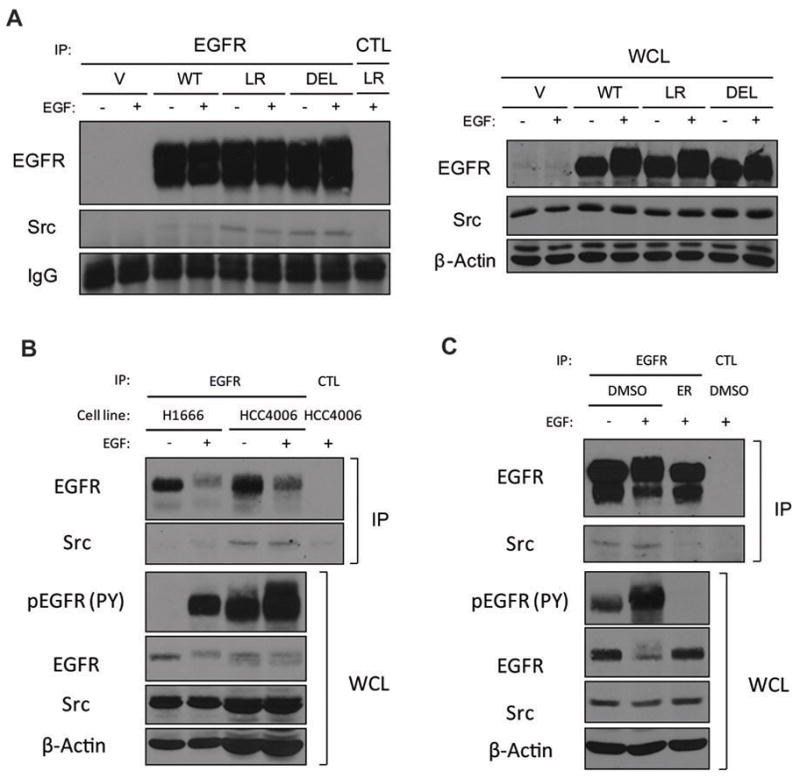
(A) NIH 3T3 cells were treated as described in Figure 1. 1 mg aliquots of cell lysates immunoprecipitated (IP) with anti-EGFR (528) or control (CTL) antibody or 50 μg aliquots of whole cell lysates (WCL) were resolved directly followed by immunoblotting with antibodies against the indicated proteins. (B) H1666 or HCC4006 cells were serum-deprived for 48 hr and then either left unstimulated (−) or stimulated (+) with 10 ng/ml EGF for 30 min. 667 μg (H1666) or 2 mg (HCC4006) aliquots of cell lysates immunoprecipitated with anti-EGFR (528) or control (CTL) antibody or 66.7 (H1666) or 200 (HCC4006) μg aliquots of whole cell lysates (WCL) were resolved directly followed by immunoblotting with antibodies against the indicated proteins. Different amounts of lysates were used to compensate for the difference in EGFR and Src expression levels between two cell lines. (C) NIH 3T3 LR cells were serum-deprived for 3 hr, preincubated in DMSO or 1 μM erlotinib (ER) for 1 hr, and either left unstimulated (−) or stimulated (+) with 10 ng/ml EGF for 30 min. 1 mg aliquots of cell lysates immunoprecipitated with anti-EGFR (528) or control (CTL) antibody or 50 μg aliquots of whole cell lysates (WCL) were resolved directly followed by immunoblotting with antibodies against the indicated proteins.
Src is required for mutant EGFR-mediated cell transformation
We then asked whether mutant EGFR-driven oncogenic transformation required the Src signaling pathway. We assessed the anchorage-independent growth of mutant EGFR-expressing cells in the presence or absence of the Src inhibitor PP2 (Figure 3A). Similar to Supplementary Figure S1B, only cells expressing mutant EGFRs showed evidence of colony growth in soft agar. As expected based on mutant EGFR-driven transformation, EGFR inhibition with erlotinib abolished the colony formation in mutant EGFR-expressing cells. Interestingly, increasing concentrations of PP2 also resulted in dose-related decreases in colony formation (Figure 3B). Treating mutant EGFR-expressing cells with erlotinib or PP2 also decreased phosphorylation of downstream targets (Supplementary Figure S2). These results support the integral role of Src activity in mutant EGFR-driven oncogenic transformation.
Figure 3. Src activity is required for mutant EGFR-mediated transformation.
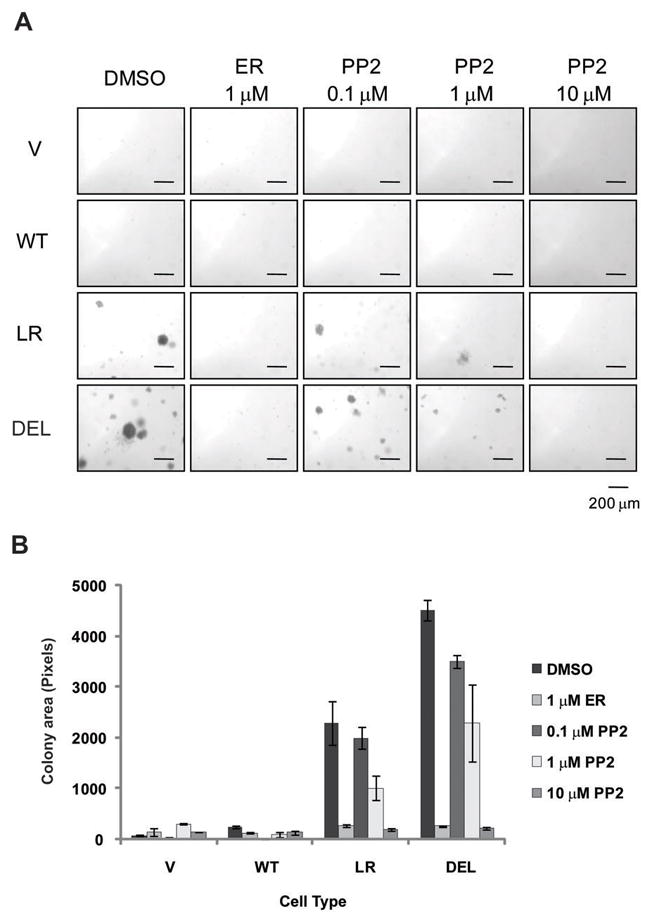
(A) Cells expressing the indicated EGFRs were grown on soft agar with media containing DMSO, 1 μM erlotinib (ER), or indicated concentration of PP2. After three weeks, pictures were taken with a phase-contrast microscope. (B) Images from (A) were quantified using the ImageJ software. Each condition was prepared in triplicate with four high power fields per replicate. Data points represent combined colony areas of four high power fields. Data were expressed as colony area (mean ± S.D.) of cells expressing indicated EGFRs. Analysis of Variance (ANOVA) performed on the colony area yielded a statistically significant difference between conditions.
Previous studies have shown that a key mechanism of EGFR-Src cooperative signaling in cell transformation is the Src-dependent phosphorylation on EGFR Y845 (Biscardi et al., 1999). Therefore, to genetically define the involvement of Src in the mutant EGFR-mediated transformation, we examined the importance of EGFR Y845 phosphorylation. For this purpose, we engineered Y845F mutations in wtEGFR, EGFR L858R, and EGFR Δ746-750 and expressed these mutants in NIH 3T3 cells; the corresponding cell lines are designated as YF (EGFR Y845F), YF/LR (EGFR Y845F/L858R) and YF/DEL (EGFR Y845F/Δ746-750).
Each cell line had a comparable level of introduced EGFRs (Figure 4A). Interestingly, while overall levels of EGF-induced tyrosine phosphorylation of EGFRs bearing Y845F mutations were comparable, we observed that there was a notable decrease in levels of ligand-independent phosphorylation of YF/LR compared to LR and to lesser degree of YF/DEL versus DEL (Figure 4A). Surprisingly, the use of phosphotyrosine 845-specific commercial antibody (pY845) for immunoblotting showed very little difference, if any, in EGF-mediated phosphorylation of Y845F mutants (data not shown). This was not due to reversion of Y845F mutation as sequencing of genomic PCR products from stable NIH 3T3 transductants confirmed the presence of appropriate mutants in each cell line (data not shown). The lack of significant decreases in the pY845 level among Y845F mutants may be due to a relative lack of specificity of the antibody at higher EGFR levels, as was previously noted (Song et al., 2006).
Figure 4. The Src phosphorylation site on EGFR, Y845, is important for constitutive activation of mutant EGF receptors.
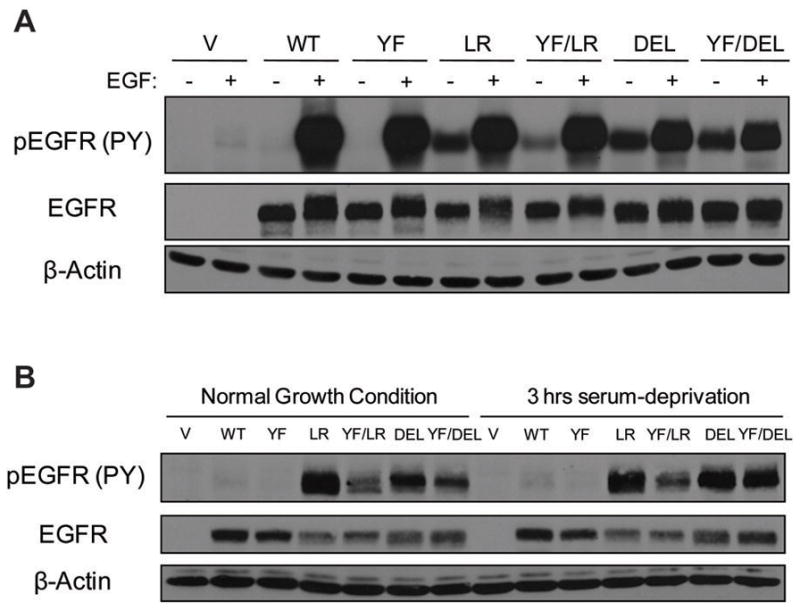
(A) Cells expressing the indicated EGFRs were serum-deprived for 3 hr and then either left unstimulated (−) or stimulated (+) with 10 ng/ml EGF for 10 min., or (B) Cells were placed in media with 10 % FBS (Normal Growth Condition) or 0.1 % FBS for 3 hr (3 hr serum-deprivation). 50 μg aliquots of whole cell lysates were used for immunoblotting with antibodies against the indicated proteins.
In contrast to relatively similar levels of EGF-induced phosphorylation, mutant EGFRs with Y845F mutation showed a substantial reduction in the basal phosphorylation. Cells were cultured in normal growth (10 % FBS) or in serum-starvation (0.1 % FBS) medium for 3 hr, harvested and analyzed for EGFR phosphorylation (Figure 4B). In both cases, YF/LR and YF/DEL showed lower levels of phosphorylation when compared to LR and DEL, respectively. YF/LR and YF/DEL still showed high levels of phosphorylation over wtEGFR. These data suggest that while Y845F mutation does not significantly alter the overall phosphorylation of the receptor in the presence of EGF stimulation, it reduces the constitutive phosphorylation of mutant EGFRs.
Src phosphorylation on Y845 is critical for the mutant EGFR mediated cell transformation, proliferation, and migration
To assess the biological impact of Y845F mutation on mutant EGFR-driven oncogenic transformation, we compared the anchorage-independent growth of cell lines expressing NSCLC-associated EGFR mutants versus their Y845F mutants (Figure 5A). Cells expressing NSCLC mutants showed colony formation, while vector, wtEGFR-, or EGFR Y845F-expressing cells did not. However, Y845F mutations decreased the abilities of EGFR mutants to promote colony growth. While the decrease is more profound with YF/LR than with YF/DEL (Figure 5B), reduction in both cases was statistically significant.
Figure 5. Src and its phosphorylation on EGFR Y845 is critical for mutant EGFR-mediated cell transformation and proliferation.
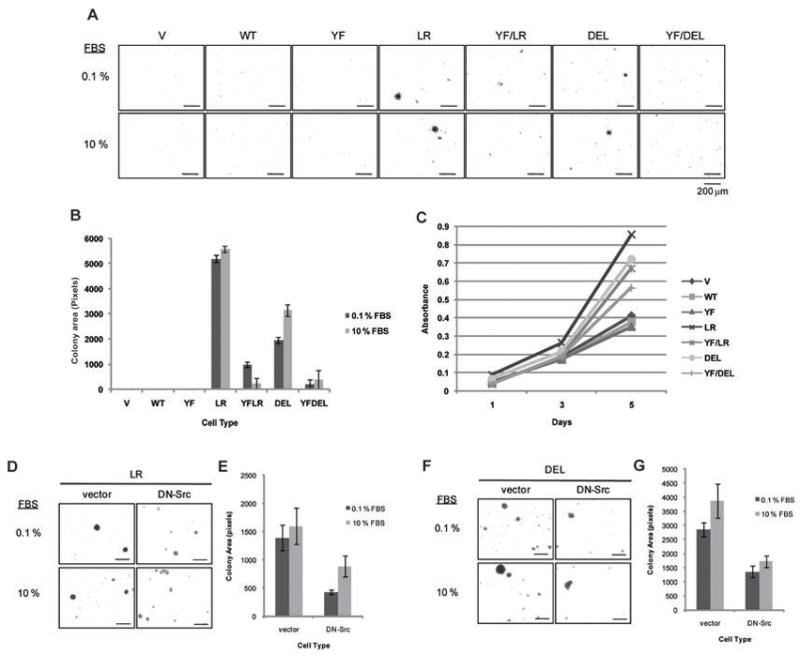
(A) Cells expressing the indicated EGFRs were grown in soft agar with media containing 0.1 % or 10 % FBS. After 3 weeks, pictures were taken with a phase-contrast microscope. (B) Images from (A) were quantified using the ImageJ software. Each condition was prepared in triplicate with four high power fields per replicate. Data points represent combined colony areas of four high power fields. Data were expressed as colony area (mean ± S.D.) of cells expressing indicated EGFRs. Analysis of Variance (ANOVA) performed on the colony area yielded a statistically significant difference between cell types. (C) Cells were plated at day 0 and grown in media with 10 % FBS. MTT dye incorporation was measured as an index of cell proliferation at day 1, 3, and 5. For each data point, 6 replicates were performed. Data were expressed as absorbance at 570 nm (mean ± S.D.) on days 1, 3 and 5 of cells expressing indicated EGFRs. LR (D) or DEL (F) cells expressing vector control or dominant-negative Src (DN-Src) were grown as described in (A) and quantified and analyzed as described above in (E) and (G), respectively. Analysis of Variance (ANOVA) performed on the colony area yielded a statistically significant difference between cell types.
We also examined the effect of Y845F mutations of mutant EGFRs on the rate of proliferation of transduced cells. As expected, similar rates of cell growth were observed between vector, wtEGFR- and EGFR Y845F-expressing cells (Figure 5C). As anticipated, cells expressing NSCLC mutants showed a dramatic increase in cell proliferation over those expressing vector, wtEGFR and EGFR Y845F. However, the advantage in cell growth was partially reduced by Y845F mutation of mutant EGFRs (Figure 5C).
To further confirm the requirement of Src and its activity in mutant EGFR-mediated transformation, vector or dominant-negative Src (DN-Src) was stably expressed in mutant EGFR-expressing cells (Supplementary Figure S3), and soft agar colony formation was assessed (Figures 5D–H). Anchorage-independent growth of both LR (Figure 5D) and DEL (Figure 5G) cells was reduced by DN-Src expression as compared to vector controls; the reduction was statistically significant (Figures 5E and H, respectively).
We further examined the impact of Y845F mutation on the ability of NSCLC mutants to drive increased cell migration, another critical oncogenesis-related biological outcome (Figure 6A). Unstimulated vector control or wtEGFR-expressing cells showed minimal migration, while the migration was substantially enhanced in the presence of EGF with a higher level of migration seen in cells overexpressing wtEGFR (Figure 6A). Notably, EGFR Y845F-expressing cells showed a level of EGF-induced cell migration that was comparable to the vector control. Importantly, the basal migration of cells expressing NSCLC mutants was substantially higher compared to those of wtEGFR and higher levels of migration were seen upon EGF stimulation. Both the basal and EGF-stimulated cell migration was reduced in cells expressing NSCLC mutants with the Y845F mutation. Furthermore, the expression of DN-Src reduced the EGF-mediated cell migration of mutant EGFR-expressing cells (Figure 6C). Use of EGFR, Src and Akt inhibitors also confirmed the requirement of these proteins in the mutant EGFR-mediated cell migration (Supplementary Figure S4). Collectively, the analyses of cells expressing Y845F mutations or DN-Src provide strong support for the conclusion that constitutive activation of mutant EGFRs is transduced into oncogenic signaling through cooperative signaling with Src and involves obligatory role of the major Src phosphorylation site on EGFR.
Figure 6. Src and its phosphorylation on EGFR Y845 is critical for mutant EGFR-mediated cell migration.
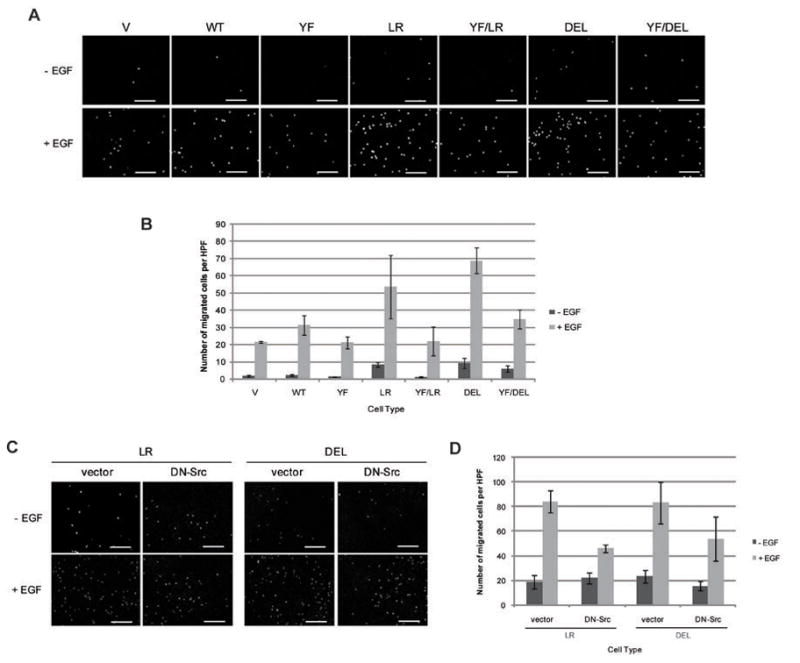
Transwell migration was performed on cells expressing the indicated EGFRs (A) or indicated Src (C) with or without 10ng/ml EGF on the bottom chamber. After 16 hr, cells were fixed, their nuclei stained with propidium iodide and pictures were taken with a fluorescence microscope. Each condition was prepared in triplicate with four high power fields per replicate. (B) The number of cells migrated from (A) or (D) number of cells migrated from (C) were quantified using the ImageJ software. Data points represent the average number of migrated cells in four high power fields (HPF). Data were expressed as number of migrated cells per HPF (mean ± S.D.). Analysis of Variance (ANOVA) performed on the number of migrated cells yielded a statistically significant difference between cell types.
Y845 phosphorylation is important in constitutive downstream signaling by EGFR mutants
Given the substantial effects of the Y845F mutation and DN-Src on the biological behavior of NSCLC-associated EGFR mutants, it was of considerable interest to investigate if the phosphorylation of Y845 was important to link mutant EGFRs to downstream signaling pathways that are constitutively activated by these mutants (Figure 1A & B). In view of the apparent lack of requirement for exogenous ligand for oncogenicity of mutant EGFRs, we focused on constitutive phosphorylation of the downstream effectors. For this purpose, we compared the phosphorylated and total protein levels of STAT3, Akt and Erk in NIH 3T3 cells expressing wtEGFR, EGFR L858R or Δ746-750 versus their Y845F mutants under normal growth conditions (Supplementary Figure S5) or after 3 hr serum-deprivation (Figure 7A). While cells grown under normal growth conditions or those stimulated with EGF (Supplementary Figure S6) showed little to modest difference in signaling pathways, difference in signaling became evident when cells were serum-deprived. Similar to Figure 1B, STAT3, Akt and Erk phosphorylation in EGFR L858R-and, more significantly, Δ746-750-expressing cells remained relatively high when compared to vector and wtEGFR-expressing cells upon serum-deprivation (Figure 7A). Notably, cells expressing Y845F mutants showed reduced phosphorylation levels of STAT3, Akt and Erk compared to cells expressing mutant EGFRs without Y845F mutations (quantified in Supplementary Table S2). Src phosphorylation between normally-grown and serum-deprived cells harboring particular EGFRs was comparable (Supplementary Figure S5A and Figure 7B). However, Src phosphorylation levels decreased substantially as a result of Y845F mutation under both growth conditions.
Figure 7. Y845 phosphorylation is key to constitutive downstream signaling by EGFR mutants.

NIH 3T3 cells were placed in media with 0.1 % FBS for 3 hr. (A) 50 μg of whole cell lysates were used for immunoblotting with antibodies against the indicated proteins; and (B) 1 mg aliquots of cell lysates were immunoprecipitated with anti-Src (B-12) antibody followed by immunoblotting with antibodies against the indicated proteins. The levels of phosphorylated and total proteins were quantified by densitometry using the ImageJ software. Mean phosphorylated protein levels to the total levels of cells with Y845F mutation relative to that of corresponding cells without Y845F mutation from at least three experiments are noted.
As mutant EGFRs showed enhanced interactions with Src compared to wtEGFR (Figure 1C), the impact of Y845F mutations on EGFR-Src association was also examined. For this purpose, anti-EGFR immunoprecipitation from cells grown in complete growth medium followed by anti-Src immunoblotting was performed (Figure 8). In both wtEGFR and mutant EGFRs, Y845F mutation led to a weaker co-immunoprecipitation by Src (quantified in Supplementary Table S2). Similar analysis performed on serum-deprived cell lysates yielded identical results (data not shown). Thus, Y845F mutation decreases the interaction of mutant EGFRs with Src.
Figure 8. Y845F mutation reduces Src interaction with NSCLC-associated EGFR mutants.
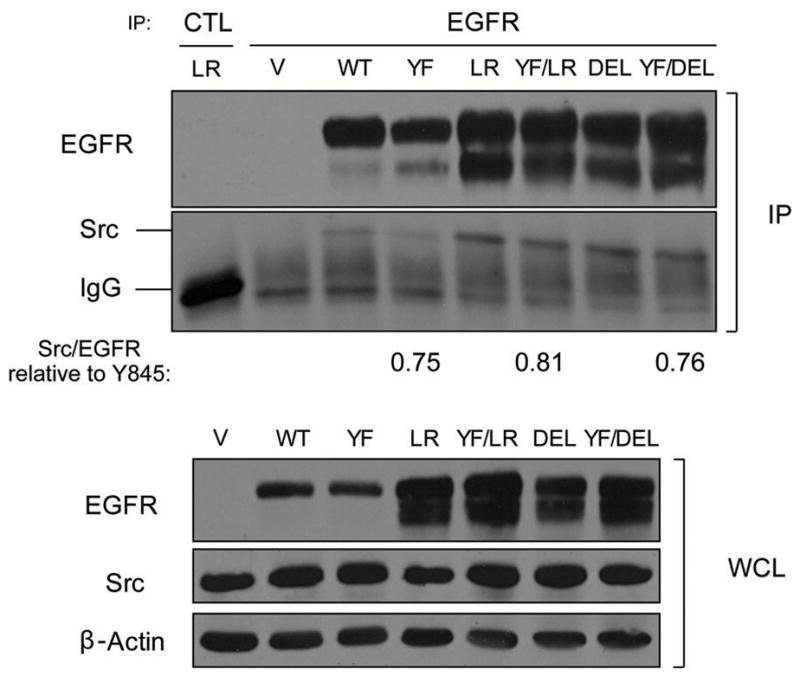
NIH 3T3 cells expressing the indicated EGFRs were grown in complete growth medium. 1 mg aliquots of cell lysates were immunoprecipitated (IP) with anti-EGFR (528) antibody or control (CTL) antibody or 50 μg of whole cell lysates (WCL) were resolved directly followed by immunoblotting with antibodies against the indicated proteins. Co-immunoprecipitated levels of Src relative to immunoprecipitated EGFRs (Src/EGFR) were determined by densitometry using the ImageJ software. Mean Src/EGFRs of cells with Y845F mutation relative to that of corresponding cells without Y845F mutation from at least three experiments are noted.
Discussion
Somatic mutations in the kinase domain of EGFR impart increased binding to tyrosine kinase inhibitors. Coupled with evidence that cells expressing mutant EGFRs undergo “oncogene addiction”, these findings have led to selective targeting of NSCLC patients with EGFR mutations with EGFR TKIs (Sharma et al., 2007). However, modulation of TKI sensitivity by acquired secondary mutations in EGFR or due to other genetic or epigenetic alteration in cancer cells necessitates the development of alternate strategies against mutant EGFR-driven NSCLC.
Recent studies have revealed that, in the process of oncogene addiction, cells become dependent not only on the oncogene itself, but also on the activity of signaling pathways that mediate the oncogenic activity (Sharma et al., 2007). A better understanding of how downstream signaling pathways are coupled to mutant EGFRs is therefore likely to identify critical therapeutic targets in NSCLC. Here, we used a NIH 3T3 cell transformation system to underscore the critical involvement of signaling through Src, orchestrated by the Src phosphorylation site on EGFR (Y845), which appears crucial in linking mutant EGFRs to oncogenesis and constitutive activation of specific signaling pathways.
Src dependence of EGFR Y845 phosphorylation is well established (Biscardi et al., 1999; Bromann et al., 2004; Ishizawar and Parsons, 2004). However, it has been suggested that phosphorylation of Y845 on one of NSCLC-associated EGFR mutants, L861Q, represents an autophosphorylation site and is Src activity-independent based on the use of EGFR and Src inhibitors (Yang et al., 2008). However, Y845 phosphorylation in EGFR L858R-expressing cells was still sensitive to Src inhibitors (Yang et al., 2008). While it is reasonable that activated mutant EGFRs might acquire the ability to autophosphorylate Y845, constitutive activity of Src and the enhanced EGFR-Src interaction in mutant EGFR-expressing cells suggest that phosphorylation of Y845 is quite likely to be predominantly Src-dependent. Different EGFR mutations may utilize these mechanisms (autophosphorylation vs. Src-dependent phosphorylation) to different extents. It is also important to note that in addition to questions about available pY845 antibody, as mentioned above, the specificity of Src inhibitors has been questioned, as they appear to also inhibit other kinases, such as Abl (Golas et al., 2003; Lombardo et al., 2004; Tatton et al., 2003) and TGF-beta receptors (Maeda et al., 2006). Therefore, our studies of the potential role of Src in the oncogenic transforming ability of NSCLC-associated EGFR mutants used genetic ablation of this potential interaction rather than the use of Src-inhibitors alone.
Previous studies have established the cooperativity between EGFR and Src signaling pathways using overexpression of both tyrosine kinases in fibroblasts (Maa et al., 1995) and more recently in epithelial cells (Dimri et al., 2007). However, a specific role of Src in mediating oncogenic transformation by NSCLC-associated EGFR mutants has not been clearly established nor have the specific signaling pathways linked to mutant EGFR-Src interaction been delineated. In this regard, our studies provide new insights that clarify the importance of Src interaction with mutant EGFR in oncogenic transformation.
Our studies demonstrate that not only does the chemical inhibition of Src kinase activity inhibit oncogenic transformation driven by mutant EGFRs (Figure 3), but that specific mutation of a Src tyrosine kinase phosphorylation site on mutant EGFRs or the expression of DN-Src substantially reduced their transforming ability (Figure 5). Concomitant with the overall decrease in transformation, the Y845F mutation also reduced the ability of mutant EGFRs to drive cell proliferation and migration, important determinants of a transformed phenotype. Consistent with these findings, a recent study showed that an intact Y845 is important to maintain the overall phosphotyrosine level of mutant EGFRs (Fu et al., 2008).
The model system in our studies provides important insights into altered EGFR-Src interactions as a result of NSCLC-associated EGFR kinase domain mutations and the role of a Src phosphorylation site in EGFR as a critical determinant of mutant EGFR-Src interactions. Src was constitutively activated (Figure 1B), and constitutively-enhanced yet EGFR kinase activity-dependent EGFR-Src interactions (Figure 2) were observed in mutant EGFR- but not wtEGFR-expressing cells. While the precise mechanism of the enhanced interaction is not clear, several factors could account for this. As noted above, Src associates more stably with ErbB2 than with EGFR. It has been suggested that mutant EGFRs may dimerize with ErbB2 (Shtiegman et al., 2007). Mutant EGFRs also appear to acquire ErbB2-like properties, such as an association with molecular chaperone Hsp90 and sensitivity to Hsp90 inhibitors (Kim et al., 2005; Shimamura et al., 2005; Yang et al., 2006); this may account for a higher ErbB2-like affinity for Src.
Use of mutant EGFRs with Y845F mutation for parallel biological analyses and examination of downstream signaling targets also allowed us to determine the importance of the EGFR-Src interaction in activating key pathways important in cellular transformation and oncogene addiction. NSCLC-associated mutants enhance the activation of Erk, Akt and STAT3, and Akt and STAT pathways are critical for cell survival (Alvarez et al., 2006; Haura et al., 2005; Lynch et al., 2004; Sordella et al., 2004). Our results demonstrate that similar to mutant EGFR-expressing NSCLC cell lines, NIH 3T3 cells expressing mutant EGFRs exhibit a constitutive activation of major downstream targets of EGFR including STAT3, Akt and Erk (Figure 1A).
Together with effects of Y845F mutations in mutant EGFRs and EGFR and Src inhibitors on STAT3, Akt and Erk activity (Figure 7A and Supplementary Figure S2), it is reasonable to suggest that increased EGFR-Src interaction contributes to oncogenic signaling by linking mutant EGFRs to downstream signaling pathways. Consistent with this suggestion, activation of STATs has been reported as a key mediator of wtEGFR and Src synergism, and STATs are required for oncogenic transformation by Src (Kazansky and Rosen, 2001; Kloth et al., 2003; Turkson et al., 1998). EGFR-Src interaction has also been linked to enhanced Akt activation (Dimri et al., 2007).
Also rather unexpectedly, we observed that Y845F mutation of NSCLC-associated EGFR mutants decreased the pool of activated Src (Figure 7B) and reduced the level of EGFR-Src association (Figure 8). Unlike the requirement of Y845 in EGFR signaling and mitogenesis (Bromann et al., 2004), the role of this phosphorylation site in EGFR-Src association is not known. It is likely that reduced kinase activity of Y845F mutants is in part responsible for reduced Src phosphorylation and EGFR-Src association. It is likely that the cooperative activation of these two tyrosine kinases involves a complex mechanism that requires further investigation.
Overall, our data suggests that Src interaction and phosphorylation of mutant EGFRs at Y845 play a significant role in mutant EGFR-driven and Src-dependent activation of downstream signaling pathways and in mutant EGFR-driven oncogenesis. Further studies to elucidate the mechanism of interplay between mutant EGFRs and Src should help design more effective ways to treat mutant EGFR-dependent NSCLC.
Materials and Methods
Retroviral constructs
The retroviral expression vectors pMSCV-puro-EGFR and pMSCV-hygro-Src K297R have been described previously (Dimri et al., 2007). EGFR mutants and dominant-negative Src (K297R/Y529F) were generated using the QuikChange II XL Site-Directed Mutagenesis Kit (Strategene, La Jolla, CA) following the manufacturer’s instructions; the primers used are shown in Supplementary Table S1. All constructs were verified by DNA sequencing.
Cell culture and retroviral infections
NIH 3T3 cells (ATCC, Manassas, VA) were grown in Alpha Modified Eagle Medium (Invitrogen, Carlsbad, CA) containing 10 % fetal bovine serum (FBS, Hyclone Inc., Logan, UT), 20 mM HEPES, pH 7.35, 1 mM sodium pyruvate, 1 mM each of nonessential amino acids, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM L-Glutamine and 55 μM 2-Mercaptoethanol (all supplements were from Invitrogen) at 37 °C in 5 % CO2. Retroviral supernatants were generated and infected as previously described (Reddi et al., 2007).
Antibodies and other reagents
The following antibodies were obtained from commercial sources: rabbit polyclonal (pAb) anti-EGFR (1005), pAb anti-Src (SRC 2), pAb anti-phosphorylated AKT (pAKT1/2/3) (Ser 473), pAb anti-pErk 1/2 (Thr 202/Tyr 204), pAb anti-Erk1 (K-23), mouse monoclonal (mAb) anti-AKT (B-1), mAb anti-Syk (4D10) and mAb anti-Src (B-12) were from Santa Cruz Biotechnology (Santa Cruz, CA); pAb anti-phospho-EGFR (Tyr1068), pAb anti-phospho-EGFR (Tyr845), pAb anti-phospho-Src (Tyr416), pAb anti-STAT3, and Rabbit monoclonal anti-phospho-STAT3 (Tyr705) were from Cell Signaling Technology (Danvers, MA); mAb anti-β actin (Clone AC-15) was from Sigma-Aldrich (St Louis, MO); mAb anti-EGFR (clone 528; ATCC) was Protein G purified from hybridoma supernatants. Purified anti-phosphotyrosine (PY) mAb 4G10 was provided by Dr. Brian Druker (Oregon Health Science University, Portland, OR). Purified mouse EGF was from Sigma-Aldrich. EGFR inhibitor, erlotinib (Tarceva), was obtained from the Hospital Pharmacy. Src inhibitor PP2 was from Calbiochem (San Diego, CA).
Preparation of cell lysates, SDS-PAGE and immunoblotting
Cells at 80–90 % confluence were incubated in the 10 or 0.1 % FBS-containing medium for 3 hr. For EGF stimulation, cells previously incubated for 3 hr in 0.1 % FBS-containing medium were either left unstimulated or stimulated with EGF (10 ng/ml) for 10 min. Cell lysates were prepared in cold lysis buffer as previously described (Dimri et al., 2007). SDS-PAGE and immunoblotting was performed as previously described (Reddi et al., 2007).
Immunoprecipitation
Cells were grown, EGF stimulation performed, and cell lysates prepared as above with exceptions that the lysis buffer contained 0.25% NP-40 (instead of Triton X-100), 50 mM Tris (pH 8.0), and 100 mM sodium chloride. Cell lysate aliquots were incubated with anti-EGFR (528) or anti-Src (B-12) antibody and anti-Syk antibody as a control, and immune complexes captured using the Protein A-Sepharose beads (GE Healthcare, Piscataway, NJ). Subsequent SDS-PAGE and immunoblotting were performed as above.
Anchorage-independent growth in soft agar
Soft agar colony growth assay was performed as previously described (Dimri et al., 2007). Phase-contrast images were processed using the ImageJ software (http://rsb.info.nih.gov/ij) to quantify colony areas. All conditions were performed in triplicates and four high power fields were imaged per replicate.
MTT Assay
1000 cells were plated per well of 96-well plate at Day 0. Absorbance at 570 nm after adding the MTT dye was measured at Day 1, 3, and 5 using the Vybrant MTT Cell Proliferation Assay Kit (Molecular Probes, Eugene, OR).
Transwell cell migration assay
The Transwell chambers with 8 μm pores (VWR, West Chester, PA) were coated overnight at 4°C with 10 μg/ml fibronectin (Sigma-Aldrich) followed by blocking with 0.1 % FBS-containing starvation medium for 1 hr at 4°C. The cells (2.5 × 104) were serum-deprived for 4 hr in starvation medium, and plated in the top chamber for 3 hr to allow attachment. Starvation medium, with or without 10 ng/ml EGF, was then added in the bottom chamber. After 16 hr, cells were washed, fixed in methanol at −20°C and stained with propidium iodide. Cotton swab was used to remove cells from the top chamber. Fluorescence images of migrated cells were processed using the ImageJ software to quantify the number of cells migrated. All conditions were performed in triplicates and four high power fields were imaged per replicate.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank members of the Band laboratories for helpful suggestions and discussion. This work was supported by: the NIH grants CA87986, CA105489, CA99900, CA116552 and CA99163 to HB, and CA94143, CA96844 and CA81076 to VB; Department of Defense Breast Cancer Research Grants W81XVVH-08-1-0617 (HB) and DAMD17-02-1-0508 (VB); the NCI Cancer Center of Nanotechnology Excellence Grant NCI 1U54 CA119341-01 (HB and VB); the NCI Breast Cancer SPORE and AVON Breast Cancer Fund at Robert H. Lurie Cancer Center, Northwestern University; the H Foundation, Chicago, IL; the Jean Ruggles-Romoser Chair of Cancer Research (HB) and the Duckworth Family Chair of Breast Cancer Research (VB); and the Malkin Scholarship (BMC). UNMC-Eppley Cancer Center is supported by an NCI Cancer Center Core Grant.
Footnotes
The work presented here was initiated while the principal investigators were at: Evanston Northwestern Healthcare Research Institute, Department of Medicine, Feinberg School of Medicine, Department of Biochemistry, Molecular Biology and Cell Biology, Weinberg College of Arts and Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Evanston, IL.
Supplementary information is available at the Oncogene website
References
- Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- Bromann PA, Korkaya H, Courtneidge SA. The interplay between src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, Fleming TP, Hazan R, Ullrich A, King CR, et al. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 1987;51:1063–1070. doi: 10.1016/0092-8674(87)90592-7. [DOI] [PubMed] [Google Scholar]
- Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, et al. Modeling breast cancer-associated c-src and EGFR overexpression in human MECs: C-src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007;67:4164–4172. doi: 10.1158/0008-5472.CAN-06-2580. [DOI] [PubMed] [Google Scholar]
- Fu YN, Yeh CL, Cheng HH, Yang CH, Tsai SF, Huang SF, et al. EGFR mutants found in non-small cell lung cancer show different levels of sensitivity to suppression of src: Implications in targeting therapy. Oncogene. 2008;27:957–965. doi: 10.1038/sj.onc.1210684. [DOI] [PubMed] [Google Scholar]
- Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, et al. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of src and abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63:375–381. [PubMed] [Google Scholar]
- Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Haura EB, Zheng Z, Song L, Cantor A, Bepler G. Activated epidermal growth factor receptor-stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin Cancer Res. 2005;11:8288–8294. doi: 10.1158/1078-0432.CCR-05-0827. [DOI] [PubMed] [Google Scholar]
- Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59:21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- Ishizawar R, Parsons SJ. c-src and cooperating partners in human cancer. Cancer Cell. 2004;6:209–214. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Kazansky AV, Rosen JM. Signal transducers and activators of transcription 5B potentiates v-src-mediated transformation of NIH-3T3 cells. Cell Growth Differ. 2001;12:1–7. [PubMed] [Google Scholar]
- Kim H, Chan R, Dankort DL, Zuo D, Najoukas M, Park M, et al. The c-src tyrosine kinase associates with the catalytic domain of ErbB-2: Implications for ErbB-2 mediated signaling and transformation. Oncogene. 2005;24:7599–7607. doi: 10.1038/sj.onc.1208898. [DOI] [PubMed] [Google Scholar]
- Kloth MT, Laughlin KK, Biscardi JS, Boerner JL, Parsons SJ, Silva CM. STAT5b, a mediator of synergism between c-src and the epidermal growth factor receptor. J Biol Chem. 2003;278:1671–1679. doi: 10.1074/jbc.M207289200. [DOI] [PubMed] [Google Scholar]
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-src: Implications for the etiology of multiple human cancers. Proc Natl Acad Sci U S A. 1995;92:6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M, Shintani Y, Wheelock MJ, Johnson KR. Src activation is not necessary for transforming growth factor (TGF)-beta-mediated epithelial to mesenchymal transitions (EMT) in mammary epithelial cells. PP1 directly inhibits TGF-beta receptors I and II. J Biol Chem. 2006;281:59–68. doi: 10.1074/jbc.M503304200. [DOI] [PubMed] [Google Scholar]
- Masaki T, Igarashi K, Tokuda M, Yukimasa S, Han F, Jin YJ, et al. Pp60c-src activation in lung adenocarcinoma. Eur J Cancer. 2003;39:1447–1455. doi: 10.1016/s0959-8049(03)00276-4. [DOI] [PubMed] [Google Scholar]
- Mitsudomi T, Kosaka T, Endoh H, Horio Y, Hida T, Mori S, et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol. 2005;23:2513–2520. doi: 10.1200/JCO.2005.00.992. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Muller WJ. Direct and specific interaction of c-src with neu is involved in signaling by the epidermal growth factor receptor. Oncogene. 1995;11:271–279. [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi AL, Ying G, Duan L, Chen G, Dimri M, Douillard P, et al. Binding of cbl to a phospholipase Cgamma1-docking site on platelet-derived growth factor receptor beta provides a dual mechanism of negative regulation. J Biol Chem. 2007;282:29336–29347. doi: 10.1074/jbc.M701797200. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65:6401–6408. doi: 10.1158/0008-5472.CAN-05-0933. [DOI] [PubMed] [Google Scholar]
- Shtiegman K, Kochupurakkal BS, Zwang Y, Pines G, Starr A, Vexler A, et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene. 2007;26:6968–6978. doi: 10.1038/sj.onc.1210503. [DOI] [PubMed] [Google Scholar]
- Silva CM. Role of STATs as downstream signal transducers in src family kinase-mediated tumorigenesis. Oncogene. 2004;23:8017–8023. doi: 10.1038/sj.onc.1208159. [DOI] [PubMed] [Google Scholar]
- Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006;66:5542–5548. doi: 10.1158/0008-5472.CAN-05-4620. [DOI] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Tatton L, Morley GM, Chopra R, Khwaja A. The src-selective kinase inhibitor PP1 also inhibits kit and bcr-abl tyrosine kinases. J Biol Chem. 2003;278:4847–4853. doi: 10.1074/jbc.M209321200. [DOI] [PubMed] [Google Scholar]
- Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998;18:2545–2552. doi: 10.1128/mcb.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley HS, Burke PM. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2:12–18. doi: 10.1034/j.1600-0854.2001.020103.x. [DOI] [PubMed] [Google Scholar]
- Yang S, Park K, Turkson J, Arteaga CL. Ligand-independent phosphorylation of Y869 (Y845) links mutant EGFR signaling to stat-mediated gene expression. Exp Cell Res. 2008;314:413–419. doi: 10.1016/j.yexcr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Qu S, Perez-Tores M, Sawai A, Rosen N, Solit DB, et al. Association with HSP90 inhibits cbl-mediated down-regulation of mutant epidermal growth factor receptors. Cancer Res. 2006;66:6990–6997. doi: 10.1158/0008-5472.CAN-06-1042. [DOI] [PubMed] [Google Scholar]
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kalyankrishna S, Wislez M, Thilaganathan N, Saigal B, Wei W, et al. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol. 2007;170:366–376. doi: 10.2353/ajpath.2007.060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.