

Abstract
Apolipoprotein E (apoE) is the primary apolipoprotein synthesized in the brain in response to injury with known neuroprotective effects exerted through antioxidant, antiinflammatory, antiexcitotoxic, and neurotrophic mechanisms. We have previously demonstrated that COG1410, an apoE mimetic peptide, exerts neuroprotective and antiinflammatory effects in a murine model of traumatic brain injury (TBI). As in TBI, ischemia-reperfusion injury is a component of acute stroke, which displays a pharmacogenetic association with the APOE4 gene. Using an intraluminal middle cerebral occlusion (MCAO) model in rats, we found that a single intravenous injection of COG1410 at 120 min post-MCAO significantly improved vestibulomotor function, decreased poststroke locomotor asymmetry, and decreased infarct volume of the ipsilateral hemisphere. These results support further exploration of a novel apoE-mimetic peptide, COG1410, as a therapeutic treatment for stroke.
Keywords: apolipoprotein E, MCAO, neuroprotection
Apolipoprotein E (ApoE) is a 299-amino-acid protein with a growing list of functions in humans. Although ApoE was originally described as a transport and targeting molecule for lipoproteins (Mahley, 1988), critical roles for ApoE in CNS integrity, function, and repair after injury have also been demonstrated (Strittmatter and Roses, 1996; Arendt et al., 1997; Laskowitz et al., 1998). In addition, the epsilon-4 (e4) allele of the APOE gene is the main risk factor for the most common sporadic form of Alzheimer’s disease (AD; Corder et al., 1993). In contrast, inheritance of the more frequent e3 allele, or of the rarer e2 allele, lowers the likelihood of developing AD (Corder et al., 1994; Farrer et al., 1997). In the brain, apoE protein is synthesized primarily by astrocytes and microglia. Neurons express apoE at lower levels than astrocytes in response to various physiological and pathological conditions (Huang et al., 2001; Harris et al., 2003; Brecht et al., 2004; Chang et al., 2005). ApoE generated in cells surrounding small blood vessels may maintain the integrity and normal function of the blood-brain barrier (BBB; Xu et al., 2006). ApoE deficiency is associated with lack of function as observed with increased permeability of the blood vessels (Fullerton et al., 2001; Methia et al., 2001).
Neuroprotective effects of ApoE are associated with multiple mechanisms, including antioxidant, antiinflammatory, antiexcitotoxic, and neurotrophic mechanisms (Nathan et al., 1994; Miyata and Smith, 1996; Laskowitz et al., 1997, 2001; Lomnitski et al., 1999; Lynch et al., 2001, 2003, 2005; Aono et al., 2002, 2003; Colton et al., 2002). These reports suggest a role for apoE in recovery from cerebral injury. In spite of its useful properties for neuroprotection, however, the dimeric-apoE complexes do not have immediate therapeutic potential. Because of the dimer’s large size of about 68 kDa, apoE does not readily cross the BBB, which makes it less useful for pharmacological treatment of CNS disease (Linton et al., 1991).
Given the strong association between APOE polymorphisms and outcome after brain injury, we developed COG133, an apoE-mimetic peptide derived from amino acids 133-149 located in the receptor-binding region of apoE, as an antiinflammatory and neuroprotective agent (Laskowitz et al., 2001). Interestingly, COG133 does not include the polymorphic residues at positions 112 and 158 that define the E2, E3, and E4 isoforms of the apoE protein in humans. COG133 retains the neuroprotective, antioxidant, antiexcitotoxic, and antiinflammatory properties of the intact apoE holoprotein in functional tests both in vitro and in vivo (Laskowitz et al., 2001; Misra et al., 2001; Aono et al., 2003; Lynch et al., 2003, 2005; McAdoo et al., 2005). Using a murine closed-head, traumatic brain injury (TBI) model, we have demonstrated that a single intravenous administration of COG133 at 30 min following TBI resulted in significant improvements in mortality, short-term vestibulomotor function, long-term cognitive function, and survival of hippocampal neurons compared with head-injured mice that received vehicle (Lynch et al., 2005). However, a therapeutic window of 30 min is not sufficient to ensure adequate access to treatment in the clinical setting. Therefore, we developed COG1410, a shorter analog composed of apoE residues 138-149 with aminoisobutyric acid (Aib) substitutions at positions 140 and 145. COG1410 possesses many of the same properties as COG133 but is superior because of its enhanced efficacy and potency in vitro (Laskowitz et al., 2006) and in vivo (Laskowitz et al., 2007). In a previous investigation, we demonstrated that treatment with COG1410 expands the therapeutic window for the treatment of TBI by a factor of 4, to 120 min post-TBI compared with 30 min post-TBI for COG133. These data provide substantial support for the utility of this peptide in the treatment of TBI (Laskowitz et al., 2007).
Although COG1410 (like COG133 heretofore) has been tested in vivo on a TBI model, it can be very useful for therapeutic application in other cases of acute brain injury, such as stroke. Ischemic stroke is one of the main causes of human death and disability (Bonita, 1992; The World Health Report, 1999) and is a huge clinical problem that is being addressed by investigation of new neuroptotective substances. In the current study, we demonstrate the neuroprotective activity of COG1410 in the setting of a focal stroke model in CD rats.
MATERIALS AND METHODS
Animals
Sprague-Dawley rats (32 males; The Animals Breeding Center, Pushchino, Russia), weight 300-340 g at surgery were maintained on 12-hr light/dark schedule (lights on at 8 am), with food available ad libitum.
Middle Cerebral Artery Occlusion Procedure
All procedures were approved by the Institutional Animal Care and Use Committee of Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry Russian Academy of Sciences (branch). Animals were anesthetized with a ketamine-xylazine mixture (30 mg/kg ketamine and 12 mg/kg xylazine, total volume of injection -1.2 ml/kg intramuscular). The right temporal muscle was dissected, the temporal bone was dried with cotton balls, and a laser Doppler flow (LDF) probe (PeriFlux 5001; PR407-1 Small Straight Probe) was attached to the temporal bone to measure regional cerebrovascular blood flow (rCBF). A midline cervical incision was made to expose the right common carotid artery (CCA). Loose ligatures were placed on the CCA and internal carotid artery (ICA), followed by two loose ligatures on the external carotid artery (ECA) at a point 2-3 mm distal from the bifurcation of CCA. The occipital artery was ligated permanently, and the pterygopalatine artery (PPA) was ligated for the time of middle cerebral occlusion (MCAO) duration. Occluding sutures were prepared using a silicon-Teflon monofilament of 0.13 mm diameter (Stroft GTM, Germany) with a heatblunted end (0.3 mm diameter), coated with 0.5% poly-llysine, and dried for 1 hr under a heating lamp (Belayev et al., 1996). The intraluminal suture was inserted through a small puncture opening in ECA and advanced into the ICA to approximately 18 mm from the bifurcation (depending on rat weight) until slight resistance was felt. When decreased blood flow was observed, the occlusion suture was left in place for 90 min. Ischemia was confirmed by a decrease of rCBF greater than 70% from the baseline value. After 90 min of occlusion, the suture was withdrawn. Blood flow restitution to 90-120% of baseline in 10 min was recognized as a successful reperfusion. The ligatures were removed from the CCA, ICA, and PPA, and the ECA was permanently ligated at both sides above and below the opening. The LDF probe was detached, and wounds were closed with a twisted suture. Sham-operated rats were subjected to the same surgical procedure except for the MCAO. With the aid of a heating pad, the rectal temperature was maintained at 37°C until the animals recovered from anesthesia. After 30 min of reperfusion (120 min after ischemia onset), COG1410 (0.8 mg/kg, volume of injection -1 ml/kg) or vehicle (Ringer’s solution) was injected into the caudal vein. Rats were returned to their cages and allowed free access to food and water.
Behavioral Testing
Each rat was subjected to a series of behavioral tests at days 1, 2, 3, 5, 7, 14, 21, 28, and 35 after stroke. Motor deficits of integrated balance and coordination were assessed with rotarod testing. Spontaneous and drug-induced rotation asymmetry was tested. Spontaneous locomotor asymmetry evaluation was conducted in an open field (plexiglass box 50 × 50 cm). All behavioral testing was done during the animals’ light cycle.
Rotarod Test
Quantitative assessment of motor coordination and balance was performed by using an automated rotarod (Dual Species Economex Rota-Rod; Columbus Instruments). On three consecutive days during the week before the MCAO procedure, all animals were trained on the rotarod. This training consisted of three trials per day on each of the 3 days for a total of nine trials. Each trial consisted of placing the animal onto the rotarod, which was revolving at a constant speed of 5 rpm/min, and then the machine was switched to acceleration mode to accelerate at 0.6 rpm/min. Latency is defined as the length of time for which the animal remains of the rotating rod beginning at time 0 with the switch to acceleration mode and continuing until the animal falls off or holds onto the rod for two complete revolutions (720° of rotation). After these 3 days of testing, all animals were randomized into three groups, where the performance of each group was not significantly different from the performance of the other groups. The purpose of this pretesting was to identify groups of animals with similar performance so that we could more accurately determine how MCAO and MCAO plus COG1410 treatment might alter that performance.
On the first day after MCAO, the consecutive sessions of rotarod testing were started. In each daily session, testing was repeated three times, with 5-min intervals between each trial. Animals unable to grasp the rotating rod were given a latency value of 0 sec. Latency times were measured as described above.
Locomotor Asymmetric Activities
Drug-induced rotation
Drug-induced rotation asymmetry was recorded manually over 5 min following injection of ketamine (10 mg/kg i.m.; Lebedev et al., 2003) in a container with a round, smooth bottom (30 cm diameter and 40 cm wall height). A full 360° arc of turning in the same direction was scored as one rotation.
Spontaneous rotation asymmetry
Assessment of spontaneous locomotor asymmetry was performed in a plexiglass box (50 × 50 cm). Spontaneous rotations were measured manually over 3 min. An observer, blinded to group assignment, scored rotations to the right and to the left.
Infarct Analysis
After 35 days of testing, animals were euthanized with CO2 inhalation. The brains were then perfusion fixed. The heart was exposed by a median sternotomy, and the ascending aorta was catheterized via the left ventricle. The blood was removed from animals with 150 ml physiologic saline solution containing heparin (100 U/liter) perfusion at a constant perfusion rate of 30 ml/min. The perfusion medium was then switched to a mixture of 3% formaldehyde-buffered solution (pH 7.4) with 1% glutaraldehyde (150 ml). After perfusion fixation of the brain, animals were decapitated., and the brain was then removed from the skull. Brains were cryoprotected in sucrose-buffered solutions (pH 7.4) of graded concentrations (10%, 20%, 30%), after which they were immediately frozen in the cryostat (Microm HM 525; Carl Zeiss) at -65°C. The coronal cryosections of 50 lm thickness at 1-mm intervals were obtained from each brain and stained with cresyl violet. Stained sections were then scanned (Umax Astra 4450) and processed with a 3D-reconstruction program, “Reconstruct” (http://synapses.bu.edu/tools/download.htm). The volumes of both hemispheres were calculated. The Cavalieri method was used for the object volume estimation (Gundersen and Jensen, 1987). The indirect lesion area was calculated as the intact area of the ipsilateral hemisphere subtracted from the area of the contralateral (uninjured) hemisphere. Lesion volume was calculated as a percentage in which the lesion volume was compared with the contralateral hemisphere volume (Swanson et al., 1990).
Statistical Analysis
The behavioral data and infarct size were analyzed by nonparametric Mann-Whitney U-test in Statistica for Windows 7.0. All values are presented as mean ± SEM.
RESULTS
COG1410 Improves Motor Performance After MCAO in Rotarod Test
Animals treated with COG1410 showed significantly improved performance in rotarod test at days 5 and 7 after injury (P < 0.05) compared with vehicletreated animals. Animals treated with COG1410 showed performance that was not significantly different from that of sham-treated animals at days 5 and 7 postinjury. In contrast, performance in vehicle-treated animals did not significantly improve until 14 days postinjury (Fig. 1A). These results suggest an increased rate of recovery of motor function following a single administration of COG1410 when given 120 min following ischemia-reperfusion injury.
Fig. 1.
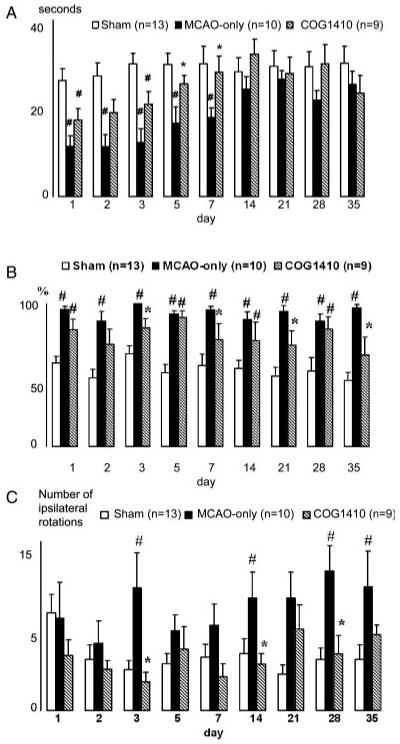
Vestibulomotor performance measured as latency of animals falling from a rotating rod (A). Data plotted as mean ± SEM in seconds. Animals treated with COG1410 showed significant improvement of vestibule-motor performance at 5 and 7 days vs. vehicletreated MCAO-only animals (*P < 0.05), whereas MCAO-only animals lagged from shams until 14 day (#P < 0.05). In the testing of ketamine-induced asymmetry (B), rotational behavior of COG1410-treated animals was significantly more symmetrical compared with MCAO-only animals (*P < 0.05), which showed asymmetrical behavior vs. shams over the whole testing period (#P < 0.05). Data are plotted as mean ± SEM percentage of ipsilateral-to-lesion rotations. Stroke-associated spontaneous asymmetry in the rotational behavior, measured in the open field (C), was not evident in COG1410-treated animals compared with shams, whereas MCAO-only animals showed increased amount of rotations ipsilateral to the lesion side vs. COG1410-treated animals (*P < 0.05) and sham animals (#P < 0.05). Data are presented as mean ± SEM number of ipsilateral rotations.
COG1410 Reduces the MCAO-Associated Asymmetry in Ketamine-Induced Rotation
To evaluate better the function of neurons in animals suffering an ischemia-reperfusion injury on one side of the brain, we measured rotational behavior in untreated and ketamine-treated animals. In this procedure, unilateral damage to one side of the brain is associated with preferential rotations to the ipsilateral side. If the unilateral damage is less following treatment with COG1410, then ketamine-induced rotational behavior should be more symmetrical and parallel the behavior seen in sham-treated animals (Lebedev et al., 2003). With this approach, COG1410-treated animals consistently showed more symmetrical rotational behavior than their vehicle-treated/MCAO-injured counterparts. Although the rotational behavior was not as symmetric as that in sham-treated animals, it was not significantly different from sham on days 2, 3, 7, 21, or 35 (Fig. 1B).
COG1410 Reduces Spontaneous Locomotor Asymmetry After MCAO
To complement the ketamine-induced rotational behavior, we also investigated spontaneous locomotion by measuring rotations in an open-field test. As shown in Figure 1C, COG1410-treated animals consistently displayed low numbers of turns to the ipsilateral side of the unilateral MCAO lesion, which were not significantly different from those of sham-treated animals. In contrast, vehicle-treated animals had dramatically more rotational behavior than either the sham or the COG1410-treated groups.
COG1410 Significantly Reduces Infarct Volume
Improvements in motor function and in rotational behaviors are typically consistent with improved protection of neuronal survival following the MCAO-induced ischemia-reperfusion injury. To assess the damage in the brains, infarcted regions were stained on serial coronal slices (Fig. 2A), followed by three-dimensional reconstruction of the damaged regions. As shown in Figure 2B, the total volume of infarcted regions was significantly greater in the MCAO-treated animals than in the sham-operated controls. Consistently with the improvements in behavioral measures, the infarct volumes in the COG1410-treated animals were significantly less than in their vehicle-treated counterparts (23% ± 4% vs. 34% ± 3%, P < 0.05).
Fig. 2.
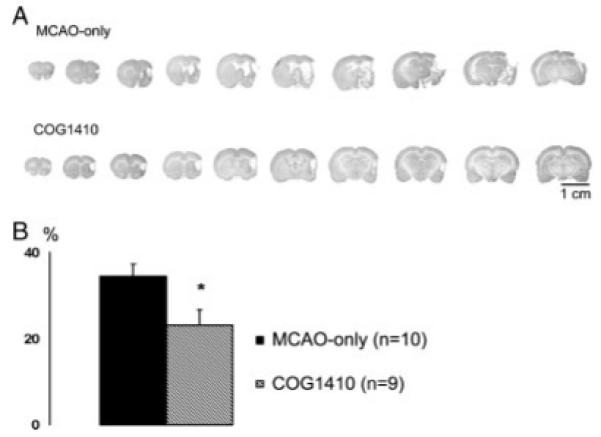
Representative brain coronal sections of a COG1410-treated MCAO animal and an MCAO-only animal (A). B: Cerebral infarct analysis was determined at 12 areas per section and displayed a rather uniform pattern of cell survival within each treatment group. Infarct volume analysis showed that treatment with COG1410 reduced infarct volume by 10% vs. vehicle-treated animals (*P < 0.05). Data are plotted as mean ± SEM percentage of infarct volume from volume of undamaged hemisphere.
DISCUSSION
Strokes result in significant ischemia-reperfusion injury that leads to significant behavioral and motor impairments in its patients. MCAO is an experimentally induced stroke that can be applied to animal models that reproduces many of the behavioral and neurodegenerative traits observed in stroke victims. With an MCAO injury paradigm, our results show that COG1410, a novel apoE-derived peptide, has significant neuroprotective properties when intravenously injected at 120 min following a 90 min, experimentally induced focal ischemic stroke. Specifically, COG1410 was effective in reducing motor impairments as measured with a rotorod assay, in reducing asymmetric ketamine-induced rotations, in reducing spontaneous asymmetric rotations in an open-field paradigm, and in reducing infarct volumes as determined following 3D reconstruction of brain lesions. Importantly, these significant improvements were observed even though administration of COG1410 was delayed by 30 min postreperfusion and a total of 120 min postocclusion. These data strongly correlate with the results of our previous study, in which COG1410 exhibited substantial neuroprotective effects when administered as a single intravenous injection at 120 min following TBI. Significant CNS penetration was shown for COG1410 in that study (Laskowitz et al., 2007) and is also apparent in naïve mice administered COG1410 via intravenous, intraperitoneal, or subcutaneous routes (Deane, Zlokovic, and Vitek, unpublished; Christensen and Vitek, unpublished). Combined with increased permeability of the vasculature at the site of infarct, COG1410 is likely to be increased locally at the site of damage following the MCAO procedure and more generally by crossing the BBB. Thus, COG1410 is in the physical location within the brain to effect its antiinflammatory and neuroprotective activities.
COG1410 significantly reduced the locomotor asymmetry that typically results from the neuronal dysfunction and degeneration that develop after focal ischemic insult in rats. The locomotor asymmetry is caused by damage of dopaminergic neurons of cortex and subcortical structures (Lopez-Martin et al., 1998) and ascending innervation disturbance within these structure that is ultimately connected with movement performance. Significant reduction of ketamine-induced rotation asymmetry and reasonably complete elimination of spontaneous locomotor asymmetry were observed in the COG1410-treated group. Although ketamine is known to have its own neuroprotective properties, the levels of ketamine that show these neuroprotective properties are tenfold greater than the dose used in our experiments (Fujikawa, 1995; Ferro, 2007). Our consistent use of ketamine in all groups should result in similar protection (if any) among all the groups, which further demonstrates that the results obtained in this test reliablely result from treatment with COG1410. Based on COG1410-treated group performance, a single injection of COG1410 at 120 min after focal stroke attenuated neuronal death in cortex and subcortical structures.
Assessment of vestibulomotor functions also demonstrated the beneficial effects of COG1410. The complete compensation of damaged coordination and balance functions in COG1410-treated animals was observed by day 5 after injury, whereas vehicle-treated MCAO-subjected animals showed such compensation only by day 14 after injury. Measurement of vestibulomotor function is a clinically relevant outcome insofar as it models everyday skills relevant to human stroke patients, e.g., balance, coordination, and walking (Scandinavian Stroke Study Group, 1985; Spilker et al., 1997; Lyden et al., 2001). Consistent with improvements in behavioral performance, histological evaluation of right-hemisphere infarct volume exhibited a reduction in COG1410-treated animals vs. MCAO-only animals.
A therapeutic window of 120 min opens the potential for COG1410 usage in ischemic stroke treatment. Undoubtedly, further study of this novel apoE-mimetic in the focal stroke model at different doses and with different administration schemes is needed to understand more completely the limits of the therapeutic window for effective treatments of stroke with COG1410.
Acknowledgments
Contract grant sponsor: NIH; Contract grant number: AG 020473 (to M.P.V.).
REFERENCES
- Aono M, Lee Y, Grant ER, Zivin RA, Pearlstein RD, Warner DS, Bennett ER, Laskowitz DT. Apolipoprotein E protects against NMDA excitotoxicity. Neurobiol Dis. 2002;11:214–220. doi: 10.1006/nbdi.2002.0541. [DOI] [PubMed] [Google Scholar]
- Aono M, Bennett ER, Kim KS, Lynch JR, Myers J, Pearlstein RD, Warner DS, Laskowitz DT. Protective effect of apolipoprotein E-mimetic peptides on N-methyl-D-aspartate excitotoxicity in primary rat neuronal-glial cell cultures. Neuroscience. 2003;116:437–445. doi: 10.1016/s0306-4522(02)00709-1. [DOI] [PubMed] [Google Scholar]
- Arendt T, Schindler C, Bruckner MK, Eschrich K, Bigl V, Zedlick D, Marcova L. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. J Neurosci. 1997;17:516–529. doi: 10.1523/JNEUROSCI.17-02-00516.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27:1616–1622. doi: 10.1161/01.str.27.9.1616. [DOI] [PubMed] [Google Scholar]
- Bonita R. Epidemiology of stroke. Lancet. 1992;339:342–344. doi: 10.1016/0140-6736(92)91658-u. [DOI] [PubMed] [Google Scholar]
- Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, Fish JD, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y. Neuronspecific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Ma TR, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Brown CM, Cook D, Needham LK, Xu Q, Czapiga M, Saunders AM, Schmechel DE, Rasheed K, Vitek MP. APOE and the regulation of microglial nitric oxide production: a link between genetic risk and oxidative stress. Neurobiol Aging. 2002;23:777–785. doi: 10.1016/s0197-4580(02)00016-7. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE. Protective effects of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis: APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- Ferro MM. Neuroprotective effect of ketamine/xylazine on two rat models of Parkinson’s disease. Braz J Med Biol Res. 2007;40:89–96. doi: 10.1590/s0100-879x2007000100012. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG. Neuroprotective effect of ketamine administered after status epilepticus onset. Epilepsia. 1995;36:186–195. doi: 10.1111/j.1528-1157.1995.tb00979.x. [DOI] [PubMed] [Google Scholar]
- Fullerton SM, Shirman GA, Strittmatter WJ, Matthew WD. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein E knockout mice. Exp Neurol. 2001;169:13–22. doi: 10.1006/exnr.2001.7631. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A. 2001;98:8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowitz DT, Goel S, Bennett ER, Matthew WD. Apolipoprotein E-deficient mice have increased susceptibility to focal cerebral ischemia. J Neuroimmunol. 1997;1-2:70–74. [Google Scholar]
- Laskowitz DT, Horsburg K, Roses AD. Apolipoprotein E and the CNS response to injury. J Cereb Blood Flow Metab. 1998;18:465–471. doi: 10.1097/00004647-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, Bennett ER. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol. 2001;167:74–85. doi: 10.1006/exnr.2001.7541. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Fillet H, Yeung N, Toku K, Vitek MP. Apolipoprotein E-derived peptides reduce CNS inflammation: Implications for therapy of neurological disease. Acta Neurol Scand. 2006;114:15–20. doi: 10.1111/j.1600-0404.2006.00680.x. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, McKenna SE, Wang H, Durham L, Yeung N, Christensen D, Vitek MP. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24:1093–1107. doi: 10.1089/neu.2006.0192. [DOI] [PubMed] [Google Scholar]
- Lebedev SV, Petrov SV, Blinov DV, Lazarenko IP, Chekhonin VP. Ketamine-induced rotational asymmetry in evaluation of motor disturbances in rats with middle cerebral artery occlusion. Bull Exp Biol Med. 2003;135:424–427. doi: 10.1023/a:1024946821482. [DOI] [PubMed] [Google Scholar]
- Linton M, Gish R, Hubl S, Butler E, Esquivel C, Bry W, Boyles J, Wardell M. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;81:270–281. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomnitski L, Chapman S, Hochman A, Kohen R, Shohami E, Chen Y, Trembovler V, Michaelson DM. Antioxidant mechanisms in apolipoprotein E deficient mice prior to and following closed head injury. Biochim Biophys Acta. 1999;1453:359–368. doi: 10.1016/s0925-4439(99)00010-1. [DOI] [PubMed] [Google Scholar]
- Lopez-Martin E, Rozas G, Rodriguez J, Guerra MJ, Labandeira-Garcia JL. The corticostriatal system mediates the paradoxical contraversive rotation but not the striatal hyperexpression of Fos induced by amphetamine early after 6-hydroxydopamine lesion of the nigrostriatal pathway. Exp Brain Res. 1998;120:153–163. doi: 10.1007/s002210050389. [DOI] [PubMed] [Google Scholar]
- Lyden PD, Lu M, Levine SR, Brott TG, Broderick J, NINDS rtPA Stroke Study Group A modified National Institutes of Health Stroke Scale for use in stroke clinical trials: preliminary reliability and validity. Stroke. 2001;32:1310–1317. doi: 10.1161/01.str.32.6.1310. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT. Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol. 2001;114:107–113. doi: 10.1016/s0165-5728(00)00459-8. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, Warner DS, Laskowitz DT. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278:48529–48533. doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Wang H, Mace B, Leinenweber S, Warner DS, Bennett ER, Vitek MP, McKenna S, Laskowitz DT. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. 2005;192:109–116. doi: 10.1016/j.expneurol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- McAdoo JD, Warner DS, Goldberg RN, Vitek MP, Pearlstein R, Laskowitz DT. Intrathecal administration of a novel apoE-derived therapeutic peptide improves outcome following perinatal hypoxic-ischemic injury. Neurosci Lett. 2005;381:305–308. doi: 10.1016/j.neulet.2005.02.036. [DOI] [PubMed] [Google Scholar]
- Methia N, Andre P, Hafezi-Moghadam A, Economopoulos M, Thomas KL, Wagner DD. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. 2001;7:810–815. [PMC free article] [PubMed] [Google Scholar]
- Misra UK, Adlakha CL, Gawdi G, McMillian MK, Pizzo SV, Laskowitz DT. Apolipoprotein E and mimetic peptide initiate a calcium-dependent signaling response in macrophages. J Leukocyte Biol. 2001;70:677–683. [PubMed] [Google Scholar]
- Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta amyloid peptides. Nat Genet. 1996;14:55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- Scandinavian Stroke Study Group Multicenter trial of hemodilution in ischemic stroke—background and study protocol. Stroke. 1985;16:885–890. doi: 10.1161/01.str.16.5.885. [DOI] [PubMed] [Google Scholar]
- Spilker J, Kongable G, Barch C, Braimah J, Brattina P, Daley S, Donnarumma R, Rapp K, Sailor S, The NINDS rt-PA Stroke Study Group Using the NIH Stroke Scale to assess stroke patients. J Neurosci Nurs. 1997;29:384–392. doi: 10.1097/01376517-199712000-00008. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53–77. doi: 10.1146/annurev.ne.19.030196.000413. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- The World Health Report. World Health Organization; Geneva: 1999. p. 119. [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]