

Abstract
Motivation: LC-MS allows for the identification and quantification of proteins from biological samples. As with any high-throughput technology, systematic biases are often observed in LC-MS data, making normalization an important preprocessing step. Normalization models need to be flexible enough to capture biases of arbitrary complexity, while avoiding overfitting that would invalidate downstream statistical inference. Careful normalization of MS peak intensities would enable greater accuracy and precision in quantitative comparisons of protein abundance levels.
Results: We propose an algorithm, called EigenMS, that uses singular value decomposition to capture and remove biases from LC-MS peak intensity measurements. EigenMS is an adaptation of the surrogate variable analysis (SVA) algorithm of Leek and Storey, with the adaptations including (i) the handling of the widespread missing measurements that are typical in LC-MS, and (ii) a novel approach to preventing overfitting that facilitates the incorporation of EigenMS into an existing proteomics analysis pipeline. EigenMS is demonstrated using both large-scale calibration measurements and simulations to perform well relative to existing alternatives.
Availability: The software has been made available in the open source proteomics platform DAnTE (Polpitiya et al., 2008)) (http://omics.pnl.gov/software/), as well as in standalone software available at SourceForge (http://sourceforge.net).
Contact: yuliya@stat.tamu.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Spectral peaks from LC-MS can be used to identify and quantify proteins in complex biological samples (Nesvizhskii et al., 2007). However, LC-MS experiments are susceptible to many sources of systematic bias, including non-constant instrument calibration, imperfect sample preparation, sample run order, etc. (Fig. 1; Callister et al., 2006, Jaitly et al., 2006, Petyuk et al., 2008). Here, we characterize typical biases in LC-MS peak intensities using large-scale calibration measurements with known internal controls. We compare several existing normalization methods and propose an algorithm for estimating and removing systematic biases using the singular value decomposition (SVD) of residual peak intensities. Existing methods are found to either be incapable of capturing complex bias trends or to overfit the data, resulting in invalid downstream statistical inference. The proposed algorithm, called EigenMS, is demonstrated to capture biases with great accuracy without overfitting (Tables 2 and 3).
Fig. 1.

Heatmaps of peak intensities for raw Salmonella (A) and EigenMS-normalized peptides (B). Sample indices are shown on the x-axis; the first five rows correspond to batches 1–5 of the first concentration group, and so on. The y-axis shows the residual eigenpeptides; the plot of the first residual eigenpeptide shows the entries in the first column of the V0 matrix (see Section 2.4), etc. Arrows indicate samples for which systematic biases are apparent; black arrows correspond to samples from batch 4, gray to batch 5. For each highlighted sample, note the dark horizontal bands in the raw Salmonella peptides. These features are thought to be due to the use of a different LC column in the last two batches. The bands have been removed after normalization.
Table 2.
Comparison of the number of significant peptides by FDR in the Salmonella data (left) and simulation data (right)
|
Salmonella typhi |
Simulation |
|||||
|---|---|---|---|---|---|---|
| FDR | 0.01 | 0.05 | 0.10 | 0.01 | 0.05 | 0.10 |
| Raw | 266 | 399 | 473 | 0 | 58 | 77 |
| EigenMS | 444 | 476 | 476 | 80 | 82 | 87 |
| Global | 211 | 312 | 372 | 96 | 150 | 172 |
| Smoothing | 245 | 345 | 412 | 200 | 200 | 200 |
| ANOVA | 462 | 476 | 476 | 60 | 79 | 80 |
In the simulation, there were 200 peptides, of which only 80 were differentially expressed.
Table 3.
Distribution of the null P-values for the simulated data before and after normalization.
| Quantile | 0.01 | 0.05 | 0.10 | 0.25 | 0.50 |
|---|---|---|---|---|---|
| Raw | 0 | 0 | 0 | 0 | 0 |
| EigenMS | 0 | 0.04 | 0.06 | 0.19 | 0.45 |
| Global | 0.34 | 0.69 | 0.83 | 0.96 | 1.00 |
| Smoothing | 0.35 | 0.68 | 0.78 | 0.89 | 0.99 |
| ANOVA | 0 | 0 | 0 | 0 | 0.03 |
Since none of the 120 peptides included in this comparison were differentially expressed, the distribution of P-values should be uniform between 0 and 1. Hence, for a given quantile, we expect that percentage of the 120 null peptides to be called significant. The raw data have null P-values that are extremely skewed left, due to strong unmodeled sources of bias. ANOVA normalization adjusts for batch effects and misses the strong additional technical features included in the simulation, resulting in a P-value distribution skewed like that in the raw data. Scatterplot smoothing and TIC normalization result in underestimated P-values and anticonservative inferences. EigenMS is approximately uniformly distributed as expected, being slightly conservative.
Calibration datasets are extremely valuable for assessing the performance of computational methods, since they permit comparisons between the results of a method and known characteristics of the experimental design (Tseng et al., 2001). In order to address the normalization of peak intensities in LC-MS, we obtained a large-scale calibration dataset prepared in-house. Samples of Salmonella (S.typhi) proteins under two biologically distinct conditions were mixed together in five different concentrations, with equal concentrations of a mixture of quality control (QC) proteins added to each. Five replicates of each of the five concentration groups were obtained in five batches using a randomized block design. With this design, we are able to definitively separate biological signal from technical bias. In particular, the QC proteins can be used to evaluate a normalization method's ability to remove bias, and the Salmonella proteins can be used to evaluate the method's ability to preserve biological signal. We note, however, that maintaining perfect equality of control protein concentrations across samples is difficult in practice, an issue that we encountered here.
We examine several standard normalization methods for LC-MS peak intensities, including global scaling, peptide-specific ANOVA models (Kerr et al., 2000) and scatterplot smoothing techniques like lowess (Yang et al., 2002) and quantile normalization (Bolstad et al., 2003). Global scaling cannot capture complex bias trends like those commonly seen in high-throughput genomic or proteomic experiments. When the sources of bias are known exactly, ANOVA models can effectively estimate and remove systematic biases (Hill et al., 2008). However, the use of peptide-specific models for preprocessing may overfit the data and invalidate downstream statistical analysis (Dabney and Storey 2007a). Furthermore, it will not generally be possible to identify all of the relevant sources of bias to sufficiently model biases with ANOVA. Scatterplot smoothing techniques are widely used in the microarray literature, but they do not necessarily address all sources of systematic bias, and they can actually introduce additional biases in common settings (Dabney and Storey 2007b).
We present an algorithm, called EigenMS, that adapts the surrogate variable analysis (SVA) method (Leek and Storey 2007) to the problem of normalization of LC-MS peak intensities. SVA uses SVD on model residuals to find trends that are responsible for significant variation that is not explained by the experimental factors of interest. In our context, these ‘residual eigenpeptides’ can be used to flexibly capture and remove complex biases in LC-MS peak intensities. Our adaptations of SVA result in several beneficial features of EigenMS for the proteomics setting. First, EigenMS is applicable to data with widespread missing measurements, as is common in MS-based proteomics. Second, the EigenMS algorithm is well-suited for inclusion in an existing proteomics analysis pipeline, as it does not require any special downstream steps or housekeeping. EigenMS is shown to perform well relative to existing alternatives, based on the calibration data as well as simulations. The algorithm is available as part of the open source proteomics analysis platform DAnTE (Polpitiya et al., 2008). EigenMS is generally applicable in bottom-up MS-based experiments based on either tandem MS (Nesvizhskii et al., 2007), high-resolution LC-MS or hybrids of the two (Zimmer et al., 2006).
2 METHODS
2.1 Experiments
2.1.1 Calibration dataset
Salmonella samples grown under two biologically distinct conditions (Log—logarithmic phase cultures grown in a rich medium and MgM—acidic, magnesium-depleted minimal nutrients medium) were combined in five different concentrations, with 25 QC proteins added in equal concentrations to each as internal controls. The Log phase is what is typically achieved by Salmonella cells in rich medium, whereas MgM is thought to mimic the virulent conditions created within host organisms. The QC proteins are derived from a mixture of organisms, including horse, rabbit and cow. Five replicates of each of the five concentration groups were obtained in five batches. Concentration run order was randomized within each batch. Table 1 details the concentrations for each of the Log, MgM and QC proteomes by experiment number. We removed the first concentration group from our analysis due to its overall low intensity across all batches. The Figure 1A shows a heatmap of the raw peak intensities (see Fig. 5 in Supplementary Material for the corresponding picture of the QC peptides). There are 228 peptides identified from the 25 QC proteins, and 3627 Salmonella peptides, corresponding to 686 unique Salmonella proteins. Overall, about 50% of all peak intensities are missing, due to peptides that were identified in some samples but not in others.
Table 1.
Concentration percentages accounted for by the different proteomes in each sample from the calibration data
| Experiment | Concentration percentage |
|||
|---|---|---|---|---|
| Total S.typhi. | Log | MgM | QC | |
| 1 | 100 | 100 | 0 | 15 |
| 2 | 100 | 75 | 25 | 15 |
| 3 | 100 | 50 | 50 | 15 |
| 4 | 100 | 25 | 75 | 15 |
| 5 | 100 | 0 | 100 | 15 |
While the peptides derived from the QC proteins should be identical, on average, across samples, there are practical difficulties (e.g. pipetting errors) in ensuring exactly equal concentrations. Another technical feature to note is that batches 4 and 5 were run using a different liquid chromatography column than batches 1, 2 and 3; differences between the data from the two columns are apparent in the heatmap of the raw intensities. Furthermore, as is the case with many high-throughput experiments, replication in these samples is technical in nature, rather than biological. This has the potential to underestimate the sources of variability present in the resulting samples, producing invalid P-values (Churchill 2002, Dabney and Storey 2006). Nevertheless, the data from the calibration measurements considered here proved to be valuable for building and validating our normalization algorithm. The data are included in the software download at SourceForge, for reference; simply search for ‘EigenMS’ at http://sourceforge.net.
2.1.2 Simulation
We simulated data to mimic the calibration data described above, using the following model:
| (1) |
where yijk is the log-transformed peak intensity for peptide i, comparison group j and batch k; μi is the overall mean intensity for peptide i; C, B and T represent mean differences between comparison groups, batches and arbitrary additional sources of technical variation, respectively; and the ϵ represent random error. Usual sum-to-zero constraints apply to the C, B and T terms. We generated five comparison groups, five batches within each group and m = 200 peptides, of which 80 were differentially expressed across groups. The group effects were chosen as monotonic step functions, similar to what is expected in the calibration dataset. The batch effects were constructed to reflect the LC column effect expected in the calibration dataset, with batches 4 and 5 differing from batches 1–3. Finally, the T terms in the simulation model were generated completely randomly, which meant to reflect systematic differences between samples that are not related to any known aspect of the experimental design. For full details of the simulation, see the Supplementary Material. The Figure 4A shows a heatmap of the raw intensities.
Fig. 4.

Heatmaps of peak intensities for raw (A) and EigenMS-normalized (B) simulated peptides. The first 80 peptides are differentially expressed, as can be seen from the decreasing intensities from top to bottom, while the last 120 peptides are not.
2.2 Existing normalization approaches
Most widely used normalization techniques in high-throughput genomic or proteomic studies involve some variation of global scaling, scatterplot smoothing or ANOVA (Quackenbush 2002). Global scaling generally involves shifting all the measurements for a single sample by a constant amount, so that the means, medians or (in mass spectrometry) total ion currents (TICs) of all samples are equivalent. Scatterplot smoothing techniques are widely used in microarray studies (Bolstad et al., 2003, Yang et al., 2002). ‘M versus A’ plots (or MA plots, for short) compare two samples by forming a scatterplot with the averages of intensities on the x-axis and the differences of intensities on the y-axis; these are often used for highlighting intensity-dependent biases. As an example, in terms of model (1), an MA plot comparing batches k = 1 and k = 2 from group j = 1 would plot Ai = ( yi11 + yi12)/2 versus Mi = yi12 − yi11, i = 1, 2,…, m. Under certain conditions, any systematic deviations in the scatterplot away from the horizontal zero line can be interpreted as bias. MA smoothing would proceed by fitting a smooth curve to the relationship between the Ai and Mi, then shifting the points to remove the fitted values of that curve, forcing the scatter to follow the horizontal zero line. In the results that follow, we apply scatterplot smoothing of MA plots, arbitrarily using the first sample as the baseline to normalize all other samples too. ANOVA-based normalization can be carried out by fitting gene- or peptide-specific ANOVA models, with specific model terms included for expected sources of bias, then subtracting off the estimated biases from the model (Kerr et al., 2000). In the calibration dataset, we employ peptide-specific models that include batch effects:
| (2) |
similar to model (1), but only including terms that represent known aspects of the experimental design.
2.3 Surrogate variable analysis
The EigenMS algorithm is an adaptation of the SVA method for detecting significant unmodeled factors in the gene expression microarray setting (Leek and Storey 2007). The basic idea of SVA is to (i) use knowledge of the experimental design to estimate effects of the experimental factors of interest, (ii) carry out SVD on the model residuals to capture remaining systematic trends, then (iii) use the estimated residual trends as additional factors to be adjusted for in the downstream, inferential model. This captures the cumulative effect of technical features without requiring knowledge of their exact sources. The SVA algorithm estimates ‘surrogate variables’ as projections of gene expression trends due to technical factors and requires that downstream statistical inference include the surrogate variables as covariates. In so doing, normalization is carried out simultaneously with inference, resulting in a clever work-around to the common problem of overfitting due to complex preprocessing.
2.4 EigenMS
EigenMS follows the general approach of SVA with a few modifications. Because EigenMS is intended to be implemented within a pipeline of informatics tools, we prefer to avoid requiring the user to keep track of surrogate variables in downstream analysis. Instead, we follow the general SVA approach of estimating systematic residual trends using SVD, then subtracting off those estimates from the raw data. To prevent overfitting, we employ a rescaling trick to the normalized data, resulting in valid downstream inference and no qualitative difference from the full SVA results. We adapt the SVA algorithm as follows:
For peptide i, i = 1, 2,…, m, estimate the model yi = Xβi + ϵi, where yi is the vector of n intensities for peptide i, X is the model matrix including only the experimental factors of interest, βi is a vector of model coefficients and ϵi is random error. Let R be the m × n matrix of residuals, with i-th row containing the vector
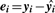 , i = 1, 2,…, m.
, i = 1, 2,…, m.Compute the SVD of R as R = UDV′, where U is m × n, and both D and V are n × n.
Identify the number, H, of eigenvalues that account for a significant amount of residual variation. This is done by bootstrap significance analysis as in the original SVA paper. The first H columns of V are then taken to represent bias. Let V0 be the corresponding n × H matrix. The columns of V0 are referred to as ‘residual eigenpeptides’.
To estimate peptide-specific bias, perform the regression R = BV0 + ϵ, where B is a matrix of coefficients that relate the residual eigenpeptides to each peptide's vector of residuals. Estimate B by least squares:
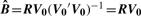 (since V0 is an orthonormal matrix).
(since V0 is an orthonormal matrix).Normalize the raw intensities by subtracting off the estimated bias:
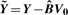 . Here, Y is the m × n matrix of raw intensities with i-th row containing yi, i = 1, 2,…, m.
. Here, Y is the m × n matrix of raw intensities with i-th row containing yi, i = 1, 2,…, m.
The normalized intensities are then contained in 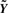 , a m × n matrix of the same dimensions as the raw data. Our intention is that the users be able to explore and analyze the normalized data without considering the steps taken in preprocessing. However, as detailed in the SVA paper, a substantial number of degrees of freedom have been used up by the above algorithm. Thus, downstream inference on the normalized data without account for this will proceed as though there are more available degrees of freedom than there really are, resulting in underestimated P-values and anticonservative inference. With this in mind, we describe next a rescaling trick that avoids this problem.
, a m × n matrix of the same dimensions as the raw data. Our intention is that the users be able to explore and analyze the normalized data without considering the steps taken in preprocessing. However, as detailed in the SVA paper, a substantial number of degrees of freedom have been used up by the above algorithm. Thus, downstream inference on the normalized data without account for this will proceed as though there are more available degrees of freedom than there really are, resulting in underestimated P-values and anticonservative inference. With this in mind, we describe next a rescaling trick that avoids this problem.
2.4.1 Rescaling to prevent overfitting
To maintain our ability to compute valid P-values on the normalized data, we incorporate a rescaling algorithm that replaces the systematic variability removed by EigenMS with just enough random variability to approximately achieve the correct number of degrees of freedom. We first compute P-values for experimental factors of interest (‘group effects’ in the examples considered here) using a model that also includes the residual eigenpeptides computed by EigenMS, as would be done if carrying out normalization and inference simultaneously. We then compute P-values using the already-normalized data and a model that only includes the experimental factors of interest, as would be done if carrying out normalization in a preprocessing step, then inference on the already-normalized data, without any special steps to address the degrees of freedom used up in normalization. The rescaling algorithm then adds residual variability to the second model by spreading out its residuals, until its resulting P-values are indistinguishable from the first model's.
Let yij be the intensity for peptide i in sample j, i = 1, 2,…, m, j = 1, 2,…, n. Consider the model
| (3) |
where the wkj are the primary variables of interest, and the glj are secondary factors whose effects we would like to remove from downstream analysis. Significance analysis could be based on the null hypotheses H0i : α1i = … = αKi, i = 1, 2,…, m. A natural test statistic for peptide i is the ANOVA F-statistic, Fi = MSTri/MSEi, computed under model (3). Under standard ANOVA model assumptions, Fi follows the F(K−1),(n−K−L) distribution if the null hypothesis is true, and the corresponding P-value can be computed as P(ℱ(K−1),(n−K−L) ≥ Fi), where ℱ(K−1),(n−K−L) is a random variable from the F(K−1),(n−K−L) distribution.
For the purposes of normalization, we would like to estimate and remove the effects of the glj to produce normalized data
| (4) |
Subsequent significance analysis on the normalized data would be conveniently carried out by computing F-statistics 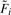 under the model assuming only primary factors are present:
under the model assuming only primary factors are present:
| (5) |
However, without acknowledging the preprocessing steps taken to estimate and remove bias, the assumed null distributions for the resulting test statistics will be incorrect. For example, if model (5) were assumed to apply to the data after normalization, the null distribution for a resulting F-statistics would be assumed to be F(K−1),(n−K), instead of F(K−1),(n−K−L). This overestimate of the number of degrees of freedom would result in underestimated P-values.
Various approaches are available for avoiding this problem, including (i) Storing the estimated secondary factor effects and plugging them in as covariates in the significance-testing model, and (ii) manually adjusting the number of degrees of freedom in the null distribution. Both of these require the user to keep track of the preprocessing steps taken and incorporate them into any software used for downstream inference. To avoid this requirement, we instead consider adding random variability to the model residuals to effectively remove the appropriate number of degrees of freedom. This allows for significance analysis post-normalization with no special steps required on by the user; that is, no additional covariates need to be added to the inferential model, and no adjustment to the null sampling distribution of test statistics are required. The specific algorithm employed for rescaling is as follows:
For peptide i, i = 1, 2,…, m, estimate the model yi = W ϕi + ϵi, where W = [XV0], and ϕi is the vector of coefficients relating W to yi. Compute the P-values for the experimental factors of interest, and call these p1, p2,…, pm. These correspond to the analysis that carries out inference and normalization simultaneously.
For peptide i, i = 1, 2,…, m, estimates the model
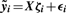 , where ζi is the vector of coefficients relating X to
, where ζi is the vector of coefficients relating X to 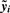 , and call the resulting P-values
, and call the resulting P-values 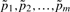 ; also, let ei denote the vector of model residuals for the i-th peptide, i = 1, 2,…, m. These P-values correspond to the analysis that carries out inference in a separate step, after normalization.
; also, let ei denote the vector of model residuals for the i-th peptide, i = 1, 2,…, m. These P-values correspond to the analysis that carries out inference in a separate step, after normalization.- For peptide i, i = 1, 2,…, m, and for each of a range of scale factors γr ∈ (0, S), r = 1, 2,…, nγ:
- Define rescaled residuals eir as the ei whose elements have been shifted further from zero by the selected scale factor γr: the v-th residual eiv becomes eivr = eiv + sign(eiv) × γr, v = 1, 2,…, n.
- Compute the corresponding rescaled version of the normalized data as
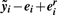 .
. - Compute the P-value based on the rescaled, normalized data
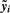 and a model that only includes the experimental factors (the regression of
and a model that only includes the experimental factors (the regression of 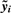 on X). Call this
on X). Call this 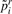 .
. - Record the absolute difference between pi and
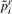 :
: 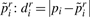 .
. - Choose the scale factor
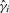 that minimizes the absolute difference:
that minimizes the absolute difference: 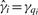 , where qi = argminr (dir: r = 1, 2,…, nγ).
, where qi = argminr (dir: r = 1, 2,…, nγ).
Compute the rescaled, normalized data by applying the scale factors for each peptide that were selected as above.
The upper boundary S for the range of the scale factors is chosen such that the rescaled residual SDs are not permitted to exceed the residual standard deviations estimated from model (3). In our analyses, we have set nγ = 100.
By using the selected residual eigenpeptides to carry out inference and normalization simultaneously, we minimize issues with overfitting. Note, however, that the above procedure does not address the uncertainty associated with the computation and selection of the residual eigenpeptides themselves, although simulation results suggest this is a minor omission. P-values for the normalized data are computed without any need for inclusion of surrogate variables as model covariates, or adjustments to the degrees of freedom used in the null sampling distribution. Thus, for example, the complete peptides from a two-class problem with n samples in each comparison group could be tested for differential expression using a standard two-sample t-statistic and t2(n−1) null distribution (assuming equal variances) after EigenMS normalization; note that we define ‘EigenMS normalization’ to include both the SVD-based adjustment and the rescaling algorithm.
2.4.2 Dealing with missing values
Peptides with missing measurements cannot be included in the matrix algebra required for SVD. However, since all peptides in the same sample were subjected to identical experimental conditions, complete peptides from that sample can be used to identify the residual eigenpeptides required for normalization by EigenMS. Normalization of the complete peptides proceeds exactly as described above. Incomplete peptides are treated similarly, but (i) the residual eigenpeptides found using the complete data are used, and (ii) the subsequent normalization and rescaling steps proceed using only the complete measurements. For an incomplete peptide with intensity vector y, let k0 = (k01, k02,…, k0n0) be the vector of indices for the n0 entries in y with missing measurements. Let y(k0) be the vector containing only the n − n0 complete measurements. Similarly, let e(k0) be the vector of n − n0 residuals from using y(k0) to fit the model in step one of the EigenMS algorithm, V0(k0) be the (n − n0) × H version of V0 after removal of rows corresponding to k0 and X(k0) be the similarly modified version of X. Normalization proceeds with e(k0) and V0(k0) in steps 4 and 5 of the EigenMS algorithm, then using y(k0), V0(k0), and X(k0) in the rescaling algorithm.
3 RESULTS
3.1 Calibration data
We applied EigenMS separately to both the Salmonella and QC peptides in the calibration dataset; in both cases, we fit the model with only concentration group included:
| (6) |
since concentration group is the only experimental factor of interest. Under the assumption that the QC peptides originated from proteins of the same concentration in all samples, identification and removal of any systematic trends present in their profiles across samples will reduce bias. As noted below, however, there are indications of variations from sample preparation that may have resulted in slightly unequal QC protein concentrations across concentration groups. Furthermore, the use of technical replication may have the effect here of underestimating sources of variability. While EigenMS is applicable to incomplete peptides (see Section 2.4.2), we only consider complete peptides in what follows, since: (i) our performance assessments are largely based on the validity of downstream statistical inference, and (ii) for peptides with missing values, special statistical methods are required to obtain unbiased model estimates and valid inference (Karpievitch et al., 2009), and this is not the focus of the present article. In the calibration data, this left us with 66 QC peptides and 425 Salmonella peptides.
In the Salmonella peptides, EigenMS identified three-residual eigenpeptides that explained a significant portion of residual variation in intensities. These eigenpeptides are shown in theFigure 2A (see Fig. 6 in Supplementary Material for the corresponding picture for the QC peptides). On the x-axis is sample index, with the first five ticks corresponding to batches 1–5 of concentration one, the second five ticks corresponding to concentration two and so on. The first residual eigenpeptide explains 49% of the variance and is evidently due to the different amounts of the Salmonella proteins in the mixture, as expected. Note that, while it is difficult to pull a coherent trend out of the remaining pictures, batch 4 appears to differ in all concentrations in eigenpeptide 2, as does batch 5 in eigenpeptide 3. These differences in eigenpeptides for batches 4 and 5 may be due to the use of a different LC column for these measurements. Note that each residual eigenpeptide has the same interpretation if reflected about the horizontal zero line. Thus, an eigenpeptide with an apparent negative effect of batch 4, for example, is equivalent to that with a corresponding positive effect. In the Salmonella peptides, for example, the step-function appearance of the first eigenpeptide reflects the experimental design. However, it is expected that some peptides will become more abundant and some less abundant as the concentration combinations are changed. The one step-function trend reflects both possibilities.
Fig. 2.

Top three eigenpeptides with first one at the top, second in the middle and third at the bottom for raw (A) and EigenMS-normalized (B) Salmonella peptides from the calibration dataset. Sample indices are shown on the y-axis; the first five rows correspond to batches 1–5 of the first concentration group, and so on. Percentages show the percent of total variability in the raw data that is explained by each eigenpeptide. EigenMS proceeds by first removing signal due to the experimental design (here reflected by the first eigenpeptide), then using the remaining trends deemed to be statistically significant (called residual eigenpeptides, and here reflected by eigenpeptides 2–4, the fourth not shown in the figure) to normalize the data.
EigenMS removed the effects of the first three residual eigenpeptides (corresponding to eigenpeptides 2–4, the fourth of which is not shown in Fig. 2) in Salmonella, as described in Section 2. The Figure 1B shows a heatmap of the normalized Salmonella peptides, and it is qualitatively evident that much of the systematic within-group features in the raw data have been removed; a similar picture is seen for the QC peptides in the Supplementary Materials. We also carried out SVD analysis of the normalized Salmonella data, with the results shown in Figure 2B. We observed an increase in the relative variance explained by the first eigenpeptide from 49% in raw Salmonella peptides to 62% and a decrease in relative variance explained by the rest of the top eigenpeptides. Since the first eigenpeptide evidently reflects the pattern across samples expected by the experimental design, we take this as evidence that EigenMS has enhanced our ability to discern real concentration differences, an important goal of any normalization procedure.
As discussed in Section 2, EigenMS should produce valid P-values and, in particular, null P-values that are approximately uniformly distributed. We checked this by examining the distribution of P-values for the QC peptides. Figure 3 shows histograms of the P-values for the raw QC peptides, as well as for data normalized by EigenMS and ANOVA. The P-values for the raw QC intensities are not uniformly distributed, indicating that either the actual concentrations of the QC peptides changed from group to group, or technical replication substantially underestimated the variation that led to the observed data; either mechanism (or both) could lead to apparent differential expression in the QC peptides (Churchill 2002, Dabney and Storey 2006). The distribution of the P-values after EigenMS normalization does not look uniform either, although the P-values have been shifted in the right direction; see Figure 9 in Supplementary Data for a comparison of P-values before and after rescaling. This highlights the fact that normalization cannot undo systematic biases across comparison groups, cases in which bias and biological signal are confounded. The ANOVA normalization method produces extremely right-skewed P-value distributions, an indication of overfitting. The scatterplot smoothing normalization method, as well as global scaling, produce more uniformly distributed P-values than does ANOVA, but visual examination of the heatmaps confirm that these normalization methods are unable to capture the systematic biases present in the calibration data. Furthermore, global scaling or scatterplot smoothing to a single reference sample, as is often done, actually obscures the concentration differences (see Fig. 7 in Supplementary Material and the left portion of Table 2).
Fig. 3.

Histogram of null P-values for raw (A), EigenMS-normalized (B) and ANOVA-normalized (C) QC peptides from the calibration dataset; each x-axis goes from 0 to 1, and the y-axis is the frequency. Since the design of the calibration experiment called for the QC proteins to be kept constant across Salmonella concentration groups, the null hypothesis of no differential expression should be true for all QC peptides, and their associated P-values should be uniformly distributed between 0 and 1. It is clear that ANOVA overfits and underestimates the null P-values. While the post-EigenMS P-values are not uniform, they are shifted in the correct direction as compared with the raw distribution. As mentioned in the text, the imperfect null distribution after normalization may be due to slight pipetting errors or technical replication. In the absence of these issues, EigenMS correctly produces uniform null P-values (see the right portion of Table 3).
We then compared the normalization methods in terms of the number of Salmonella peptides selected as statistically significant over a range of false discovery rate (FDR) cutoffs. To estimate the FDR associated with a particular significance cutoff, we used the Q-value, an FDR analog of the P-value (Storey and Tibshirani 2003). At a Q-value cutoff of 5%, corresponding to an expected FDR of 5% among the selected peptides, the number of Salmonella peptides called significant increased by 77 over the raw data (see the left portion of Table 2). Furthermore, EigenMS results in a greater increase in the number of selected peptides than all other normalization methods considered, except ANOVA, although the performance of ANOVA is in large part due to its tendency to underestimate P-values.
3.2 Simulation
We applied EigenMS to the simulated data, again based on model (6).Figure 4B shows the EigenMS-normalized intensities for the simulated peptides, and it is qualitatively evident that much of the systematic within-group trends have been successfully removed. EigenMS identified one-residual eigenpeptide that explained 80% of the variation in intensities after accounting for the concentration differences (Fig. 8 in Supplementary Material). This residual eigenpeptide incorporated the effects of both the B and T factors in the simulation model (1). The right portion of Table 2 shows the number of significant peptides by FDR cutoff for the different normalization methods. EigenMS consistently identifies all of the 80 truly differentially expressed peptides with high confidence. ANOVA normalization fails to identify as many as EigenMS. Scatterplot smoothing and TIC normalization select more peptides than EigenMS, but they underestimate their FDR levels. This is due to the fact that these methods underestimate P-values in the simulation, as seen in Table 3. Table 3 shows the distribution of P-values for the null peptides for the different methods. In the simulation, the T factors, representing technical features in addition to batch effects in Equation (1), were created in such a way that they account for much of the experimental variation. Thus, analysis of the raw data, failing to account for the sources of bias, results in underfitting and a left-skewed null distribution. ANOVA normalization only adjusts for batch effects and hence fails to capture the additional strong technical factors, again resulting in left-skewed null P-values. Scatterplot smoothing and TIC normalization are sample-specific and hence are able to remove much of the bias, but they underestimate P-values, resulting in many more type I errors than expected. The null P-values for EigenMS are approximately uniform as expected, being slightly conservative.
4 DISCUSSION
Normalization is an important, but difficult problem in high-throughput proteomics or any other -omics data. Failing to properly account for biases that result from uncontrolled technical aspects of an experiment can have serious adverse effects on the conclusions that can be drawn from the resulting data. On the other hand, overly aggressive normalization routines can easily do more harm than good by overfitting and thus invalidating downstream inferential decisions. In all cases, it is necessary to consider normalization in the larger context of the entire analysis of a dataset, as the steps taken to preprocess data will inevitably affect all subsequent analysis steps. EigenMS has been constructed with these issues in mind, and has been demonstrated to be able to flexibly capture complex biases while preserving the validity of downstream statistical inference.
The approach taken by EigenMS to the normalization of MS peak intensities could be applied to a wide variety of problems in MS-based proteomics, such as the normalization of mass measurements (Petyuk et al., 2008) or elution times (Jaitly et al., 2006). Similar approaches may be applicable to the alignment of LC-MS data, where elution time and mass images are warped across samples to maximize overlap of LC-MS feature clusters (Finney et al., 2008). More generally, systematic errors in alignment on additional dimensions, incorporating ion mobility drift times, for example, could be approached using similar strategies to those employed here.
Supplementary Material
ACKNOWLEDGEMENTS
Portions of this work were performed in the Environmental Molecular Science Laboratory, a United States Department of Energy (DOE) national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, WA.
Funding: NIH R25-CA-90301 training grant in biostatistics and bioinformatics at TAMU; the National Institute of Allergy and Infectious Diseases NIH/DHHS through interagency agreement Y1-AI-4894-01; National Center for Research Resources (NCRR grant no. RR 18522); PNNL is operated for the DOE Battelle Memorial Institute (contract DE-AC05-76RLO01830).
Conflict of Interest: none declared.
REFERENCES
- Bolstad BM, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Callister SJ, et al. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res. 2006;5:277–286. doi: 10.1021/pr050300l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA. Fundamentals of experimental design for cDNA microarrays. Nat. Genet. 2002;32:490–495. doi: 10.1038/ng1031. [DOI] [PubMed] [Google Scholar]
- Dabney AR, Storey JD. A reanalysis of a published Affymetrix GeneChip control dataset. Genome Biol. 2006;7:401. doi: 10.1186/gb-2006-7-3-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney AR, Storey JD. A new approach to intensity-dependent normalization of two-channel microarrays. Biostatistics. 2007a;8:128–139. doi: 10.1093/biostatistics/kxj038. [DOI] [PubMed] [Google Scholar]
- Dabney AR, Storey JD. Normalization of two-channel microarrays accounting for experimental design and intensity-dependent relationships. Genome Biol. 2007b;8:R44. doi: 10.1186/gb-2007-8-3-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney GL, et al. Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high resolution μLC-MS data. Anal. Chem. 2008;80:961–971. doi: 10.1021/ac701649e. [DOI] [PubMed] [Google Scholar]
- Hill EG, et al. A statistical model for iTRAQ data analysis. J. Proteome Res. 2008;7:3091–3101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitly N, et al. Robust algorithm for alignment of liquid chromatography-mass spectrometry analyses in an accurate mass and time tag data analysis pipeline. Anal. Chem. 2006;78:7397–7409. doi: 10.1021/ac052197p. [DOI] [PubMed] [Google Scholar]
- Karpievitch YV, et al. A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics. 2009 doi: 10.1093/bioinformatics/btp362. [Epub ahead of print, doi: 10.1093/bioinformatics/btp362, June 17, 2009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MK, et al. Analysis of variance for gene expression microarray data. J. Comput. Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, et al. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- Petyuk VA, et al. Elimination of systematic mass measurement errors in liquid chromatography-mass spectrometry based proteomics using regression models and a priori partial knowledge of the sample content. Anal. Chem. 2008;80:693–706. doi: 10.1021/ac701863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polpitiya AD, et al. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics. 2008;24:1556–1558. doi: 10.1093/bioinformatics/btn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Microarray normalization and transformation. Nat. Genet. 2002;32:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, et al. Issues in cDNA microarray analysis: Quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001;29:2540–2557. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer JS, et al. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 2006;23:450–482. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.