

Abstract
Vascular system function involves complex interactions among the vascular endothelium, smooth muscle, the immune system, and the nervous system. The toxic metals cadmium (Cd), arsenic (As), and lead (Pb) can target the vascular system in a variety of ways, ranging from hemorrhagic injury to subtle pathogenic remodeling and metabolic changes. Acute Cd exposure results in hemorrhagic injury to the testis, although some strains of animals are resistant to this effect. A comparison of Cd-sensitive with Cd-resistant mouse strains showed that expression of the Slc39a8 gene, encoding the ZIP8 transporter, in the testis vasculature endothelium is responsible for this difference. Endogenously, ZIP8 is a Mn2+/HCO3−symporter that may also contribute to Cd damage in the kidney. Chronic Cd exposure is associated with various cardiovascular disorders such as hypertension and cardiomyopathy and it is reported to have both carcinogenic and anticarcinogenic activities. At noncytotoxic concentrations of 10–100nM, Cd can inhibit chemotaxis and tube formation of vascular endothelial cells. These angiostatic effects may be mediated through disruption of vascular endothelial cadherin, a Ca2+-dependent cell adhesion molecule. With regard to As, ingestion of water containing disease-promoting concentrations of As promotes capillarization of the liver sinusoidal endothelium. Because capillarization is a hallmark precursor for liver fibrosis and contributes to an imbalance of lipid metabolism, this As effect on hepatic endothelial cells may be a pathogenic mechanism underlying As-related vascular diseases. With regard to Pb, perinatal exposure may cause sustained elevations in adult blood pressure, and genetically susceptible animals may show enhanced sensitivity to this effect. Taken together, these data indicate that the vascular system is a critical target of metal toxicity and that actions of metals on the vascular system may play important roles in mediating the pathophysiologic effects of metals in specific target organs.
Keywords: arsenic, cadmium, lead, vasculature, endothelium, metal transporters
Toxic metals, such as cadmium (Cd), arsenic (As), and lead (Pb), pose serious risks to human health. The importance of these metals as environmental health hazards is readily evident from the fact that all three are ranked in the top 10 on the current Agency for Toxic Substances and Disease Registry Priority List of Hazardous Substances (ATSDR, 2005). As a result of the extensive use of these metals and their compounds in industry and consumer products, these agents have been widely disseminated in the environment. Because metals are not biodegradable, they can persist in the environment and produce a variety of adverse effects. Exposure to these metals can result in damage to a variety of organ systems (Hughes, 2002; Ibrahim et al., 2006; Jarup et al., 1998) and, in some cases, these metals also have the potential to be teratogenic and carcinogenic (Han et al., 2000; IARC 1987; Kitchin, 2001; Waalkes et al., 1992).
Even though the importance of metals as environmental health hazards is now widely appreciated, the specific mechanisms by which metals produce their adverse effects have yet to be fully elucidated. However, a growing volume of evidence indicates that many of the effects of metals may result from specific actions on various components of the vascular system. These recent advances in the field of metal toxicology have coincided with advances in the understanding of the intricate functioning of the vascular system. Studies over the past 25 years have revealed that the vascular system is much more than the body's “plumbing.” Rather than being a static series of pipes and tubes, the vascular system is extremely dynamic and plays a critical role in homeostasis and in regulating the function of all organs of the body. The functioning of the vascular system involves complex interactions among the vascular endothelium, vascular smooth muscle, the immune system, the nervous system, and even the local chemical/metabolic environment of individual organs (Galley and Webster, 2004; Gibbins et al., 2003; Hill et al., 2001; Triggle et al., 2003; Villar et al., 2006). Recent studies have shown that toxic metals can target the vascular system for both acute injury and disease promotion. These vascular effects contribute to a variety of pathologic conditions including edema, atherosclerosis, and hypertension. In addition, the vascular effects of the metals may play key roles in mediating the toxic actions of metals in specific organ systems (Navas-Acien et al., 2005b; Prozialeck et al., 2006) and in promoting tumor growth (Kamat et al., 2005; Liu et al., 2006; Soucy et al., 2003). In order to highlight some of the most recent work in this area, a symposium titled “The Vascular System as a Target of Metal Toxicity” was held at the 2007 Meeting of the Society of Toxicology (SOT) in Charlotte, NC. This symposium was cosponsored by the Metals, Mechanisms and Toxicologic and Exploratory Pathology Specialty Sections of the SOT. The purpose of this report is to summarize the work presented in that symposium.
The Vascular Endothelium in Cd-Induced Edema and Hemorrhagic Injury
The vascular endothelium consists of specialized epithelial-like cells that line the lumenal surface of all blood vessels and form the capillary networks that mediate the delivery of oxygen and nutrients to tissues of the body. Obviously, vascular endothelial cells would be exposed to any toxic metal(s) circulating in the blood stream and, if the metals are present at sufficiently high concentrations, the endothelial cells could be injured or killed. The resulting loss of endothelial barrier integrity would result in edema and hemorrhaging in various tissues. Indeed, acute exposure to high levels of many metals has been shown to cause hemorrhaging in tissues such as the lung. However, one metal that appears to be unique in its ability to injure vascular endothelial cells and alter vascular permeability is Cd.
The idea that the vascular endothelium is an important target of Cd toxicity stemmed from an observation by Alsberg and Schwartze (1919) almost 90 years ago when they reported that acute exposure to subcutaneously administered Cd in rats caused purple discoloration of the testes. This observation went largely unnoticed until the 1950's and 1960's when other investigators reported that Cd caused hemorrhaging of the testes in a wide variety of species (Chiquoine, 1964; Hoey, 1966; Kar and Das, 1960; Parizek and Zahor, 1956). Later studies showed that Cd produced this effect by causing the breakdown of the junctions between the endothelial cells of the testicular capillaries and venules, resulting in an increase in vascular permeability, followed by edema, hemorrhage, and testicular necrosis (Aoki and Hoffer, 1978; Fende and Niewenhuis, 1977; Gabbiani et al., 1974; Gunn and Gould, 1970; Sacerdote and Cavicchia, 1983). A great deal of morphologic and biochemical evidence indicated that these effects of Cd on microvascular permeability resulted from direct actions of Cd on the endothelial cells in this particular vascular bed. However, this also raised the intriguing question as to why the endothelial cells in the testis were sensitive to this effect of Cd, whereas the endothelial cells in most other vascular beds were not affected.
Identification of A Specific Metal Transporter that Conveys Cd Sensitivity to Vascular Endothelial Cells
Lucis and Lucis (1969) discovered that certain inbred strains of animals were resistant to Cd-induced testicular necrosis. Discovery of mutant mouse strains provided the foothold needed for solving this mystery through the use of genetics and genomics research. By screening inbred strains of mice and performing various genetic crosses, Taylor et al. (1973) defined genetically the so-called Cdm locus within a 24-centiMorgan (cM) segment of DNA that confers sensitivity versus resistance to Cd-induced testicular necrosis. Using recombinant inbred lines and several dozen microsatellite markers, Dalton et al. (2000) were able to decrease the distance of 24 cM to 0.64 cM on mouse chromosome 3. With the advent of new knowledge about the mouse genome, the 0.64 cM was further reduced to 880 kb; one of the three functional genes therein was discovered to be Slc39a8, encoding the 8-transmembrane ZIP8 transporter (Dalton et al., 2005). In retrovirally infected mouse fetal fibroblast cultures (rvZIP8 cells), the complementary DNA (cDNA)–expressed ZIP8 protein was shown to enhance Cd uptake by 10-fold and increase sensitivity to cell killing by 30-fold. By in situ hybridization, two Cd-sensitive mouse inbred strains exhibited high ZIP8 expression in vascular endothelial cells of the testis, whereas two Cd-resistant strains showed negligible expression in these cells. Endothelial cell injury results in vascular leakage, which includes red cell extravasation and platelet plugging, ultimately causing testicular ischemia, followed by necrosis. Interestingly, although striking differences in ZIP8 expression are found in endothelial cells of the testis vasculature between inbred strains of mice, ZIP8 total messenger RNA (mRNA) levels are widely distributed in many tissues and do not differ substantially between strains. This observation, in conjunction with data demonstrating no mRNA sequence alterations between sensitive and resistant inbred strains of mice, led to the hypothesis that differential endothelial cell expression of Slc39a8 in blood vessels of the testis is the consequence of a DNA variant site(s) within an intron or in a 5′- or 3′-flanking region cis to the Slc39a8 gene (Dalton et al., 2005).
In Hank's balanced-salt solution, ZIP8 in the rvZIP8 cells has a Km of 0.62mM for Cd and 2.2μM for Mn uptake. In the Madin–Darby canine kidney polarized epithelial cell line, ZIP8 is localized on the apical surface. Cd or Mn uptake is absolutely dependent on HCO3− in the medium. The ZIP8 endogenous function thus appears to be a Mn2+/HCO3− symporter, which Cd is able to highjack and thus gain entrance into cells, using a HCO3− gradient (He et al., 2006).
The Cd-sensitivity trait is dominant over the Cd-resistance trait (Dalton et al., 2000; Taylor et al., 1973). Therefore, the Slc39a8 gene was isolated on a bacterial artificial chromosome (BAC) clone, derived from a BAC library constructed from the genome of the Cd-sensitive 129S6/SvEvTac (abbreviated 129) strain. This BAC was inserted into the resistant C57BL/6J (B6) mouse genome. The BAC insertion turned out to be successful in demonstrating the accumulation of ZIP8 mRNA and protein specifically in the testicular endothelial cells of the BTZIP8-3 BAC-transgenic line (Wang et al., 2007). This means that the Cd-sensitive 129 strain's Slc39a8 gene is successfully expressed in the BAC-transgenic mouse line having > 99.8% of the Cd-resistant B6 strain's genes. Moreover, the phenotype of Cd-resistance was shown to revert to sensitivity to Cd-induced testicular necrosis, thereby proving unequivocally that the Slc39a8 gene is indeed the Cdm locus (Wang et al., 2007). It should be noted that, this genetic difference in ZIP8 expression between inbred mouse strains occurs only in these specialized vascular endothelial cells that one finds in the testis; hence, the only test for the phenotype is in the intact animal and cannot be carried out in vitro or in cell culture.
The BTZIP8-3 mouse line, having three Slc39a8 gene copies derived from the 129 mouse plus the two normal Slc39a8 gene copies from the B6 mouse, therefore has five Slc39a8 genes total and is viable and fertile. Two additional BAC-transgenic lines, one having seven, and the other eight Slc39a8 gene copies, never produced offspring. Wang et al., (2007) thus concluded that having seven or more Slc39a8 gene copies causes infertility and/or very early embryolethality. ZIP8 is highly expressed in placenta, and also contributes to Zn2+ uptake. It is therefore likely that Zn perturbation by excessive ZIP8 expression can lead to infertility and/or very early embryolethality.
Testing the BTZIP8-3 line for Cd-induced testicular necrosis led to an unexpected bonus finding: acute renal failure occurred and actually preceded damage to the testis by several hours; a high abundance of ZIP8 mRNA and protein was demonstrated on the apical surface of the renal proximal tubular epithelial cells (Wang et al., 2007). It is therefore postulated that ZIP8 might be the most important metal transporter in causing Cd-induced renal metabolic acidosis and kidney damage, conditions often seen in human populations chronically exposed to environmental Cd.
Effects of Cd on Adherens Junctions and Vascular Endothelial Cadherin-Cadherin
Although the foregoing findings with the S1c39a8/ZIP8 transporter have provided significant insights into the reason why certain vascular beds and strains of animals are sensitive to Cd-induced increases in microvascular permeability, they do not explain the mechanism by which Cd acts on the endothelial cells to produce this effect. Results of early morphologic studies suggested that this increase in microvascular permeability involved specific changes in the ultrastructure of the adhering junctional complexes that mediate adhesion between the capillary endothelial cells (Niewenhuis et al., 1997; Peereboom-Stegeman and Jongstra-Spaapen, 1979; Sacerdote and Cavicchia, 1983), although the specific molecular targets on which Cd acted to produce these effects remained unknown. However, results of recent studies suggest that these microvascular effects of Cd may involve alterations in the function of the Ca-dependent cell adhesion molecule, VE-cadherin (vascular endothelial cadherin).
VE-cadherin is a member of the cadherins superfamily of Ca-dependent cell adhesion molecules (Goodwin and Yap, 2004; Koch et al., 2004; Nollet et al., 2000). Although E-cadherin is the dominant cadherin expressed in most epithelial cells, vascular endothelial cells primarily express VE-cadherin (Dejana et al., 2001; Lampugnani et al., 1992; Vincent et al., 2004). Both E-cadherin and VE-cadherin are single pass transmembrane proteins that are usually localized at adherens-type cell–cell junctions. The extracellular domain of the cadherin contains multiple Ca-binding sites, as well as the adhesive regions of the molecule. The intracellular domain is bound to β-catenin and several associated molecules that link the junctional complex to the actin cytoskeleton.
The finding that the cadherins might be targets of Cd toxicity stemmed from a series of observations by Prozialeck and coworkers (Prozialeck, 2000; Prozialeck and Niewenhuis, 1991a,b) who found that exposing cultured renal epithelial cells to 10–20μM Cd for 1–4 h caused the cells to separate from each other and change morphologically from epithelioid to rounded. This effect coincided with the loss of E-cadherin from the cell–cell contacts and a reorganization of the actin cytoskeleton. These effects differed from those produced by other metals such Hg (Prozialeck and Niewenhuis, 1991b), but resembled those that occurred when the cells were incubated in the presence of the Ca chelator ethylene glycol-bis (β-aminoethylether) N,N,N′,N′-tetraacetic acid (EGTA) (Prozialeck, 2000). Moreover, they occurred at Cd concentrations and times of exposure that did not cause the loss of cell membrane integrity or alter cellular levels of ATP or glutathione, suggesting that they represented relatively specific toxic actions of Cd on the E-cadherin–dependent junctions between the cells (Prozialeck, 2000). The disruption of cadherin-dependent cell junctions by Cd was not cell- or cadherin-specific, as similar effects were also observed on E- and N-cadherin junctions in several other types of epithelial cells (Prozialeck, 2000). Additional studies have shown that Cd has similar effects on VE-cadherin in vascular endothelial cells in culture (Prozialeck, 2000; Prozialeck et al., 2006). In addition, studies utilizing a murine model of Cd-induced pulmonary injury have shown that Cd causes a redistribution of VE-cadherin in vascular endothelial cells of the lung (Pearson et al., 2003). Together, these findings indicate that VE-cadherin may be a key target on which Cd acts to disrupt endothelial barrier integrity. Recently, Pereira et al. (2007) have recently found that As also disrupts VE-cadherin–dependent intercellular junctions in vascular endothelial cells and they suggested that this effect may contribute to the development of atherosclerosis.
Effects of Cd and As on Angiogenesis and Vascular Remodeling
Over the past two decades the process of angiogenesis has been the subject of considerable research. In light of this large volume of work, it is somewhat surprising that relatively little has been published regarding the effects of metals on angiogenesis. However, there is a growing volume of evidence indicating that certain toxic metals, most notably Cd and As, can have profound effects on angiogenesis.
The process of angiogenesis involves several steps each of which can be studied in vitro. These steps that are shown schematically in Figure 1 include: (1) basement membrane degradation; (2) endothelial cell migration away from the vessel in response to a chemoattractant gradient; (3) endothelial cell proliferation; and (4) morphogenesis into tube-like structures (Carmeliet, 2000; Hayden and Tyagi, 2004). In the mid 1990's, Kishimoto et al.(1996a; 1996b) reported that Cd inhibited proliferation, migration and tube formation by endothelial cells in culture. However, these studies were carried out in the absence of serum and utilized relatively high concentrations of Cd that also affected cell viability. In addition, some of the results suggested that Cd produced these effects by acting on the Matrigel matrix rather than the endothelial cells themselves (Kishimoto et al., 1996b). Recent studies from the Woods and Prozialeck laboratories have shown that Cd, at noncytotoxic concentrations, has direct effects on endothelial cell migration and tube formation (Prozialeck et al., 2006). These inhibitory effects clearly resulted from direct actions on the endothelial cells and were evident when the cells were exposed to concentrations of Cd as low as 0.1–1.0μM in the presence of serum, conditions that mimic the patterns of exposure of endothelial cells in vivo. Additional studies showed that these same levels of exposure resulted in a loss of VE-cadherin from the cell–cell contacts (Prozialeck et al., 2006).
Fig. 1.
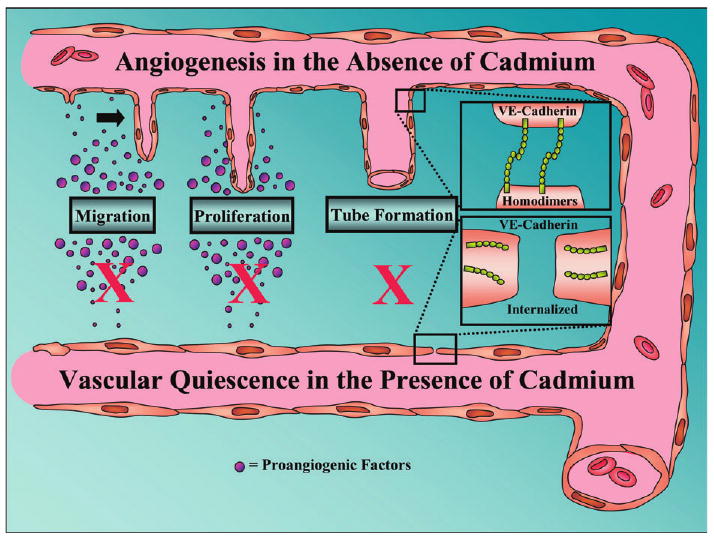
The effects of Cd on the angiogenic process. The angiogenic process can be broken down into several sequential steps, which can be examined in vitro, including endothelial cell migration, proliferation, and tube formation. Above these steps, labeled in the figure, is the normal angiogenic process occurring in the absence of Cd. Endothelial cells migrate in response to a chemoattractant, they proliferate while extending the existing blood vessel, and ultimately they differentiate into tube-like structures. The latter process is known to involve VE-cadherin, which is involved in cell–cell adhesion as well as intercellular formation of the lumen (Yang et al., 1999). Below the labels, in the presence of Cd, we summarize evidence suggesting that vascular quiescence predominates in the presence of Cd. Studies from our lab and others (Kishimoto et al., 1996a) suggest that Cd inhibits endothelial cell migration, proliferation, and the process of tube formation. Moreover, our data suggest that VE-cadherin is sequestered away from the endothelial cell surface, which may cause the inhibition of tube formation.
In light of the fact that VE-cadherin plays a critical role in endothelial tube formation, these findings suggest that Cd disrupts angiogenesis by redistributing VE-cadherin from the endothelial cell surface and inhibiting cell migration and tube formation. Recently, Kolluru et al. (2006), have reported additional evidence showing that Cd can directly inhibit endothelial cell migration and tube formation, and they presented evidence that these effects may be associated with decreased nitric oxide (NO) production by the endothelial cells. Additional studies are needed to examine the possible relationships between the Cd-induced changes in NO metabolism and the alterations in VE-cadherin expression and localization.
In contrast to Cd, low levels of As(III) promote endothelial cell tube formation and angiogenesis in both cell culture and in vivo mouse models (Kamat et al., 2005; Kao et al., 2003; Liu et al., 2006; Soucy et al., 2005). Angiogenesis is the rate limiting step in tumor growth and the in vivo angiogenic effects of low dose As(III) promote tumor growth in mouse xenograph models (Kamat et al., 2005; Liu et al., 2006). However, higher doses of As(III) are toxic to endothelial cells (Roboz et al., 2000) and inhibit angiogenesis (Kao et al., 2003; Liu et al., 2006; Soucy et al., 2003). As noted previously, angiogenesis is a complex process of endothelial cell proliferation, migration, and vessel maturation (reviewed in Carmeliet, 2000; Hayden and Tyagi, 2004). Pathological angiogenesis is usually accompanied by recruitment of inflammatory and progenitor cells that elaborate growth factors to complete remodeling of the new vessel wall (Ruiz et al., 2006). A recent examination of As(III) effects on inflammatory angiogenesis demonstrated dose-dependent increases in CD45 positive leukocytes in Matrigel plugs implanted in mice exposed to 50–500 ppb (0.3–3.3μM) of As(III) through their drinking water, relative to plugs from control unexposed mice (Straub et al., 2007b). This increase in CD45 positive cells was prominent after 5 weeks of exposure and remained significant through 20 weeks of exposure (Soucy et al., 2005). These same exposures produced highly significant increases in the number of CD31/PECAM positive blood vessels (i.e., luminal structures containing red blood cells) in the Matrigel plugs (Soucy et al., 2005). An important finding in these studies was that the threshold for the angiogenic response was between 1 and 5 ppb (6 and 33nM) (Soucy et al., 2005), which is below the current drinking water arsenic MCL of 10 ppb (66nM).
A limitation of xenograph models and the mouse Matrigel models of angiogenesis is that both are inherently inflammatory in the mouse. This inflammatory potential is appropriate for modeling potentiation of tumor angiogenesis, but it may mask direct pathogenic vascular effects that promote the range of vascular diseases caused by environmental exposure to As(III). These vascular diseases include hypertension, atherosclerosis, coronary vessel disease, noncirrhotic portal hypertension, and possibly diabetes (Navas-Acien et al., 2005a).
To examine the effects of As(III) on an endogenous vascular bed, we focused studies on the specialized vasculature of the liver sinusoids (Straub et al., 2007a,b). The liver sinusoidal endothelial cells (LSEC) are both morphologically and phenotypically unique relative to macrovascular and microvascular endothelium. Their roles as the major scavenger cells and filters in the liver are facilitated by their fenestrations and loose intracellular connections, as well as their unique expression of scavenger receptors on a specialized microtubular network (Falkowska-Hansen et al., 2007). These receptors allow the LSEC to be the primary site of removal of all major circulating biological molecules, including modified proteins, polysaccharides, lipids, and nucleic acids (Falkowska-Hansen et al., 2007) and loss of this function has significant systemic consequences (McCuskey, 2006). Exposure of mice to As(III) in their drinking water caused defenestration and capillarization of the LSEC (Straub et al., 2007a,b). Capillarization is a process in which the normally fenestrated, discontinuous LSEC become a continuous endothelium with limited transendothelial cell transport due to loss of fenestrae, formation of tight intercellular endothelial junctions, and formation of a basement membrane (Braet and Wisse, 2002; Couvelard et al., 1993; Dubuisson et al., 1995; Xu et al., 2003). Capillarization precedes vascular remodeling of other liver vessels, such as the hepatic arterioles and the peribiliary vascular plexus causing the shunting of blood flow, vascular channel formation, and eventually liver fibrosis (Couvelard et al., 1993; DeLeve et al., 2004; Li et al., 2005).
Quantitative morphometric analysis revealed that 2- to 5-week exposures to 250 ppb As(III) decreased the average size of the fenestrae and eliminated gaps between cells to decrease overall sinusoid porosity (i.e., open space per unit area), relative to unexposed mice (Straub et al., 2007a). The surface of the As(III) exposed sinusoids also showed an increase in associated detritus and projections, some of which were microvilli from the underlying hepatocytes protruding through the remaining LSEC fenestrae. There were no zonal differences along the sinusoids for the effect of As(III) on porosity (Straub et al., 2007a) suggesting that an effect directly on the endothelial cells and not dependent on the metabolic function of the underlying hepatocytes. Quantitative immunofluorescent analysis demonstrated that junctional expression of CD31/PECAM-1 protein increased as porosity decreased (Straub et al., 2007a,b). Concomitant with the increase in PECAM-1, a laminin-1 positive basement membrane formed. These protein changes confirmed that As(III) promoted the formation of tight endothelial cell junctions and underlying matrix that are characteristic of continuous endothelium.
Further studies demonstrated that the effect of As(III) was both dose and time dependent. Exposure to As(III) at the current arsenic MCL of 10 ppb (66nM) reduced porosity by 20–30% and 50 ppb exposures caused almost complete loss of porosity. These decreases were partially apparent following 1 week of exposure, but were highly significant at 2 weeks. There were no additional differences in As(III) effects on porosity between 2 and 5 weeks (Straub et al., 2007b). It is important to note that during the 5-week experiments the sinusoidal porosity in the control mice also tended to decrease. This was expected because capillarization increases with age and is thought to contribute to the age-related risk for atherosclerosis (Cogger et al., 2004; Hilmer et al., 2005). Transmission electron microscopy revealed that as porosity decreased, the hepatocytes developed microvilli that clogged the space of Disse. After 5 weeks, but not 2 weeks, exposures, caveolae were apparent on the surface of the LSEC (Straub et al., 2007b). This increase in caveolae was associated with appearance of caveolin-1 protein. These data suggest that As(III) caused time-dependent changes in LSEC function from proficient scavengers to normal endothelium that use caveolae to transcytose macromolecules. Because caveolin-1 is a major scaffold for endothelial cell signaling proteins, the data suggest that prolonged As(III) exposure changes the signaling phenotype of the cells, as well.
The cellular mechanisms for the vascular effects of arsenic remain unresolved. However, As(III) has been shown to stimulate Rac1-GTPase, which mediates both endothelial shape change and nicotinamide adenine dinucleotide phosphate (reduced) oxidase (NOX)–dependent generation of oxidants (Qian et al., 2005; Smith et al., 2001). To investigate whether LSEC Rac1 was affected by As(III) in vivo, livers were perfused with a colloidal silica solution to specifically coat the LSEC membranes immediately after euthanizing the mice (Straub et al., 2007a). The colloid-bound membranes were separated from the rest of the liver cell membranes and then probed for Rac1 protein changes relative to changes in actin content. The analysis revealed that only the LSEC cell membranes from arsenic exposed mice contained Rac1 protein. To further investigate whether As(III) has a direct functional effect on LSEC Rac1, LSEC were isolated from unexposed mice and cultured in collagen-coated dishes. Addition of 1–5μM As(III) to the cultures promoted junctional PECAM-1 expression. Incubating the cells with apocynin, a Rac1 and NOX inhibitor (Klees et al., 2006), before adding As(III) prevented the As(III)-stimulated expression of PECAM-1. DeLeve et al. (2004) demonstrated that LSEC fenestrations are maintained by vascular endotheliam growth factor (VEGF)-stimulated NO production. Thus, As(III) stimulation of Rac1-dependent NOX superoxide generation and cytoskeletal regulation provide mechanisms for defenestration through NO quenching and cell shape change. This mechanism is schematically shown in Figure 2. More definitive studies are needed to resolve how As(III) stimulates LSEC Rac1 and to prove the role of Rac1 in mediating LSEC activation and capillarization. Nonetheless, these data suggest that low, noncytotoxic levels of As(III) signal through specific cellular pathways to functionally alter the phenotype of the endothelial cells in this important metabolic vascular bed.
Fig. 2.
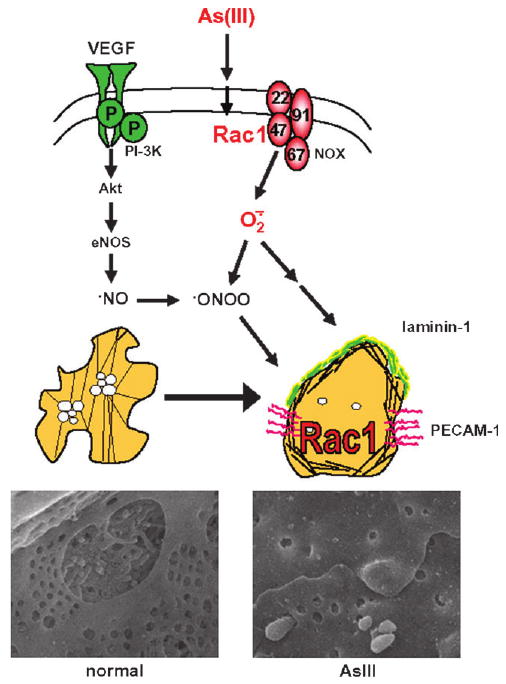
Hypothetical scheme for AS(III)-stimulated remodeling of LSEC. Fenestrations in the normal LSEC Rac1-GTPase activity are maintained by constitutive VEGF-stimulated NO production As(III) stimulates LSEC Rac1-GTPase activity which increases NOX generated superoxide and regulates cell spreading. Thus, As(III) may promote defenestration and capillarization by quenching NO through peroxynitrite formation (ONOO) and cytoskeletal rearrangement. The capillarized endothelium is stabilized by increased junctional expression of PECAM and formation of a laminin-containing basement matrix.
Cd and Pb in Hypertension
Results of a large number of epidemiologic studies suggest that exposure to metals such as Cd and Pb may play a role in the development of hypertension (for reviews see Harlan, 1988; Nakagawa and Nishijo, 1996; Navas-Acien et al., 2005b; Pirkle et al., 1985; Satarug and Moore, 2004; Schwartz, 1988). For example, many epidemiologic studies have suggested a possible association between exposure to Cd and hypertension (Luoma et al., 1995; Satarug et al., 2003, 2005; Vivoli et al., 1989), although other studies showed no direct relationship between blood pressure and blood levels of Cd (Fontana and Boulos, 1986; Staessen et al., 1996, 2000). In addition, a large number of animal studies have shown that chronic exposure to Cd can lead to elevations in blood pressure (Nechay, 1978; Ohanian et al., 1978; Oner et al., 1995; Perry et al., 1977; Revis et al., 1981).
Although the cardiovascular system is not typically viewed as a primary target of Pb toxicity, high concentrations of Pb, such as might occur during occupational exposure, are toxic to both the heart and vascular smooth muscle. Furthermore, because the kidney is a primary organ of Pb toxicity, indirect cardiovascular effects could occur secondarily to renal injury. A number of studies have found correlations among occupational Pb exposure, nephrotoxicity, and increased arterial blood pressure (Harlan, 1988; Lal et al., 1991; Pirkle et al., 1985; Schwartz, 1988). However, in these studies blood Pb levels ranged from 100 to 120 μg/dl (4.7–5.8μM) (Gross, 1981; Kehoe, 1961). At these exposures, severe kidney damage has generally been present; thus, this hypertension is probably of renal origin.
Several more recent epidemiological studies suggest that Pb concentrations below Center for Disease Control limits, and within the realm of environmental exposure, may be a risk factor for hypertension. A study in which bone Pb levels, as determined by K-shell x-ray fluorescence, were compared with development of hypertension provides one of the stronger pieces of evidence for environmental Pb exposure as a risk factor in development of hypertension (Cheng et al., 2001). Some reports suggest that even transient exposure to Pb during childhood can have a long-term and delayed hypertensive effect (Hu, 1991). Studies in experimental animals also support a correlation between low-level exposure to Pb and development of hypertension (Aviv et al., 1980; Kopp et al., 1980; Perry et al., 1988; Vaziri et al., 1999). However, whether or not low-level exposure to Pb is actually a causal factor in hypertension remains controversial, as other epidemiological studies found no definitive connection between blood pressure and blood or bone Pb levels (Staessen et al., 1999). Moreover, the increases in blood pressure associated with Pb exposure, though consistent, are modest. Vupputuri et al. (2003) found that Pb exposure (blood Pb ≥ 5 μg/dl; 0.24μM) caused a 1.67 and 1.68 mmHg increase for black males, and a 3.48 and 2.22 mmHg increase in black females. Hypertension involves interactions among the cardiovascular, nervous, renal, and endocrine systems, and is expressed primarily in adulthood, whereas environmental-level exposure to Pb is generally without obvious symptoms and typically occurs during childhood. Thus, as noted by Vaziri et al. (1988), it may be difficult to make an association of a role for Pb in a disease in which the onset is far removed from the postulated exposure, with no noticeable intervening pathophysiology.
Epidemiological data in humans also indicate that certain subgroups are especially sensitive to Pb. Men (Glenn et al., 2003) and postmenopausal women (Nash et al., 2003) are more likely to develop hypertension with occupational Pb exposure than are premenopausal, adult women. Muntner et al. (2005) found that both non-Hispanic blacks and Mexican-Americans had a higher association of Pb with hypertension. One potential common explanation for these findings is that individuals predisposed to develop hypertension may also be more prone to increased risk of hypertensive responses to Pb.
To examine the role of perinatal Pb exposure on the development of adult hypertension in a “susceptible population” studies were undertaken using the “spontaneously” hypertensive rat (SHR) as a model. SHRs were exposed to Pb acetate in the maternal drinking water (100 ppm, 263μM) perinatally or perinatally and postweaning (100 ppm in drinking water). Blood Pb levels were undetectable in rats receiving water, or Pb perinatally only, and ranged from 0 to 12.5 μg/dl (60μM) in rats exposed to Pb postweaning. SHR became markedly hypertensive over the next 6 months as expected. At this point radiotelemetry transmitters were surgically implanted and the animals were allowed 7 days of surgical recovery. Subsequently, one week of 24 h/day radiotelemetric recordings of blood pressure, heart rate, and activity were made. Continuous 24-h average blood pressure measurements revealed that neither perinatal nor perinatal plus postweaning Pb exposure affected basal blood pressure. A more comprehensive study is underway using a wider range of Pb doses, and the comparative response of Pb-treated and untreated SHR rats to several commonly used antihypertensive drugs. Nonetheless, using the most sensitive method available for measuring blood pressure in rodents, and a genetically susceptible strain of rats, we were unable to demonstrate a statistically significant increase in basal blood pressure in this initial study.
There are many reports in the literature proposing possible physiological and cellular mechanisms of metal-induced hypertension based on animal and in vitro studies. Figure 3 highlights some of the reported mechanisms by which metals such as Cd and Pb could contribute to the development of hypertension. In the case of Cd, considerable evidence suggests that hypertensive effects result from complex actions on both the vascular endothelium and vascular smooth muscle. For example, Cd causes the release of a variety of proinflammatory mediators such as tumor necrosis factor alpha, from endothelial cells, see site 1 (Kaji, 2004; Mlynek and Skoczynska, 2005; Szuster-Ciesielska et al., 2000). In addition, Cd stimulates the release of antithrombolytic agents such as plasminogen activator inhibitor-1 and facilitates the adhesion of leukocytes and platelets to the endothelium, see site 2 (Hernandez and Macia, 1996; Kaji, 2004; Yamamoto et al., 1993). Pb may cause enhanced sympathetic nerve activity with increases in circulating epinephrine and norepinephrine levels (site 3) in conjunction with decreased density of vasodilating β2 adrenergic receptors (site 4) (Chang et al., 1996, 1997; Tsao et al., 2000). Although acute exposure to Cd and Pb results in depressed plasma renin levels (Fleischer et al., 1980; Puri, 1992) chronic low-level exposure to Pb results in increased activity of angiotensin converting enzyme activity and increases in plasma renin, angiotensin II, and aldosterone levels (Boscolo and Carmignani, 1988; McAllister et al., 1971; Vaziri, 2002). Plasma kininase I and II levels are higher during Pb exposure. This can lead to decreases in plasma bradykinin levels resulting in a reduction in endothelial NO production (site 5) (Carmignani et al., 1999). In rodent models of Pb-induced hypertension there is an elevation in plasma concentration of the potent vasoconstrictor, endothelin-3, see site 6 (Khalil-Manesh et al., 1993). Furthermore, coronary microsvascular endothelial cells exposed to 2μM CdCl2 exhibit increased secretion of endothelin-1 and angiotensin II (Kusaka et al., 2000). However, Cd was also found to antagonize the actions of endothelin (Koschel et al., 1995; Wada et al., 1991). There is very strong evidence that both Cd and Pb decrease the functional availability of the potent vasodilator NO (site 7), most likely through direct or indirect mechanisms involving oxidative stress (Bilgen et al., 2003; Grabowska-Maslanka et al., 1998; Kishimoto et al., 1994; Skoczynska and Martynowicz, 2005; Vaziri, 2002). Elevations of intracellular smooth muscle Ca could lead to increased arterial tone leading to hypertension. Pb is a well-known inhibitor of Na/K ATPase. Inhibition of this enzyme leads to elevations of intracellular Na resulting in increased intracellular Ca levels, see site 8 (Piccinini et al., 1977). Another mechanism by which metals can affect vascular smooth muscle is by altering protein kinase C (PKC) activity. Both Pb and Cd exposure are associated with increased PKC activity (Hwang et al., 2001; Washington et al., 2006). Furthermore, in isolated tissue baths, PKC and L-type voltage-gated Ca channel antagonists, when applied separately, significantly diminished Pb-induced vascular smooth muscle contraction, see site 9 (Watts et al., 1995). Lastly, Cd promotes the proliferation of vascular smooth muscle cells (site 10) and enhances the production of extracellular matrix components (site 11) that increase the stiffness of blood vessels (Abraham et al., 2000; Fujiwara et al., 1998; Jeong et al., 2000; Kaji, 2004).
Fig. 3.
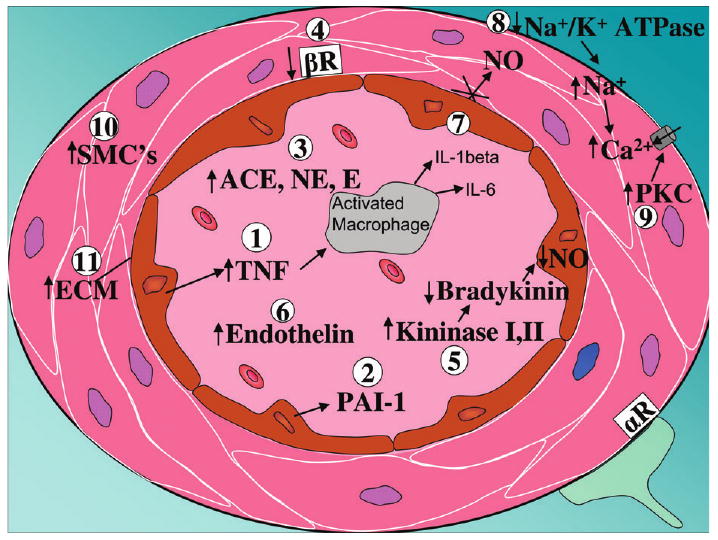
Synopsis of proposed physiological and cellular mechanism of metal-induced hypertension. On the graph, sites 1–7 reflect endocrine or paracine vaso-active mediators/receptors, sites 8–11 reflect direct changes in vascular smooth muscle physiology and associated connective tissue caused by metal exposure. Abbreviations: αR = α adrenergic receptor; ACE = angiotensin converting enzyme; βR = β adrenergic receptor; E = epinephrine; ECM = extracellular matrix; IL = interleukin; NE = norepinephrine; NO = nitric oxide; PAI-1 = plasminogen activator inhibitor -1; PKC = protein kinase C; SMC = smooth muscle cells; TNF = tumor necrosis factor α.
These mechanisms reflect the multifactorial nature of hypertension with the obvious interdependence among several organ systems, as well as the fact that Pb and Cd have many and multifarious actions. It remains to be determined how all of these various effects of metal exposure summate to influence the net regulation of blood pressure.
Although the evidence in support of a role of metal-induced hypertension is strong, it is not conclusive, nor are the mechanisms by which Pb or Cd may act on the vascular system clear. Pb exposure may be more of a risk factor for certain “susceptible populations”, and thus may not be apparent in the general population. Inasmuch as hypertension remains a significant risk factor for other forms of cardiovascular disease, the propensity for environmental exposure factors to contribute to this risk is, potentially, an important public health concern.
Conclusion
The functioning of the vascular system involves complex interactions among multiple cell types, with each one utilizing a myriad of cellular signaling pathways that allow the vascular system to respond and adapt to ever-changing environmental conditions. Recent studies have provided new insights into the mechanisms by which metals can influence vascular function. Expression of the ZIP8 metal ion transporter (Slc39a8 gene) appears to be a key factor contributing to the selective toxicity of Cd in the endothelial cells of organs such as the testes and kidneys. At the cellular level, metals such as Cd and As have profound effects on the process of angiogenesis. These effects involve alterations in the function and expression of cell adhesion molecules such as VE-cadherin and PECAM-1, although the specific signaling pathways that mediate these actions have yet to be elucidated. Further research is needed, especially in the area of metal-induced hypertension, to determine the significance and the mechanism of the adverse effects. Clearly, much work remains to be done. It is our hope that this symposium overview will be useful to investigators in this field and help to provide a conceptual framework for future studies.
Acknowledgments
Funding: National Institutes of Health grants (R01 ES006478) to W.C.P., (R01 ES007373) to A.B., (R01 ES010416) to D.W.N., (P30 ES06096) to D.W.N., and (1R21 ES123459) to W.D.A.
The authors thank Vicki Sears for her help in preparing the manuscript.
Footnotes
Symposium held in March, 2007 at the 41st Annual Meeting of the Society of Toxicology (SOT) in Charlotte, NC. Sponsored by the Metals, Mechanisms and Toxicologic and Exploratory Pathology Specialty Sections of the SOT.
References
- Abraham D, Hofbauer R, Schafer R, Blumer R, Paulus P, Miksovsky A, Traxler H, Kocher A, Aharinejad S. Selective downregulation of VEGF-A(165), VEGF-R(1), and decreased capillary density in patients with dilative but not ischemic cardiomyopathy. Circ Res. 2000;87:644–647. doi: 10.1161/01.res.87.8.644. [DOI] [PubMed] [Google Scholar]
- Alsberg CL, Schwartze EW. Pharmacological action of cadmium. J Pharmacol Exp Ther. 1919;13:504–505. [Google Scholar]
- Aoki A, Hoffer AP. Reexamination of the lesions in rat testis caused by cadmium. Biol Reprod. 1978;18:579–591. doi: 10.1095/biolreprod18.4.579. [DOI] [PubMed] [Google Scholar]
- ATSDR. CERCLA Priority List of Hazzardous Substances. 2005 Available at: http://www.atsdr.cdc.gov.cercla.
- Aviv A, John E, Bernstein J, Goldsmith DI, Spitzer A. Lead intoxication during development: Its late effects on kidney function and blood pressure. Kidney Int. 1980;17:430–437. doi: 10.1038/ki.1980.51. [DOI] [PubMed] [Google Scholar]
- Bilgen I, Oner G, Edremitlioglu M, Alkan Z, Cirrik S. Involvement of cholinoceptors in cadmium-induced endothelial dysfunction. J Basic Clin Physiol Pharmacol. 2003;14:55–76. doi: 10.1515/jbcpp.2003.14.1.55. [DOI] [PubMed] [Google Scholar]
- Boscolo P, Carmignani M. Neurohumoral blood pressure regulation in lead exposure. Environ Health Perspect. 1988;78:101–106. doi: 10.1289/ehp.8878101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp Hepatol. 2002;102(2):207–218. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- Carmignani M, Boscolo P, Poma A, Volpe AR. Kininergic system and arterial hypertension following chronic exposure to inorganic lead. Immunopharmacology. 1999;44:105–110. doi: 10.1016/s0162-3109(99)00115-0. [DOI] [PubMed] [Google Scholar]
- Chang HR, Chen SS, Chen TJ, Ho CH, Chiang HC, Yu HS. Lymphocyte beta2-adrenergic receptors and plasma catecholamine levels in lead-exposed workers. Toxicol Appl Pharmacol. 1996;139:1–5. doi: 10.1006/taap.1996.0136. [DOI] [PubMed] [Google Scholar]
- Chang HR, Chen SS, Tsao DA, Cheng JT, Ho CK, Yu HS. Reduced vascular beta-adrenergic receptors and catecholamine response in rats with lead induced hypertension. Arch Toxicol. 1997;71:778–781. doi: 10.1007/s002040050460. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Schwartz J, Sparrow D, Aro A, Weiss ST, Hu H. Bone lead and blood lead levels in relation to baseline blood pressure and the prospective development of hypertension: The Normative Aging Study. Am J Epidemiol. 2001;153:164–171. doi: 10.1093/aje/153.2.164. [DOI] [PubMed] [Google Scholar]
- Chiquoine AD. Observations on the early events of cadmium necrosis of the testis. Anat Rec. 1964;149:23–35. doi: 10.1002/ar.1091490104. [DOI] [PubMed] [Google Scholar]
- Cogger VC, Muller M, Fraser R, McLean AJ, Khan J, LeCouteur DG. The effects of oxidative stress on the liver sieve. J Hepatol. 2004;41:370–376. doi: 10.1016/j.jhep.2004.04.034. [DOI] [PubMed] [Google Scholar]
- Couvelard A, Scoazec JY, Feldmann G. Expression of cell-cell and cell-matrix adhesion proteins by sinusoidal endothelial cells in the normal and cirrhotic human liver. Am J Pathol. 1993;143:738–752. [PMC free article] [PubMed] [Google Scholar]
- Dalton TP, He L, Wang B, Miller ML, Jin L, Stringer KF, Chang X, Baxter CS, Nebert DW. Identification of mouse SLC39A8 as the transporter responsible for cadmium-induced toxicity in the testis. Proc Natl Acad Sci USA. 2005;102:3401–3406. doi: 10.1073/pnas.0406085102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton TP, Miller ML, Wu X, Menon A, Cianciolo E, McKinnon RA, Smith PW, Robinson LJ, Nebert DW. Refining the mouse chromosomal location of Cdm, the major gene associated with susceptibility to cadmium-induced testicular necrosis. Pharmacogenetics. 2000;10(2):141–151. doi: 10.1097/00008571-200003000-00006. [DOI] [PubMed] [Google Scholar]
- Dejana E, Spagnuolo R, Bazzoni G. Interendothelial junctions and their role in the control of angiogenesis, vascular permeability and leukocyte transmigration. Thromb Haemost. 2001;86:308–315. [PubMed] [Google Scholar]
- DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G757–G763. doi: 10.1152/ajpgi.00017.2004. [DOI] [PubMed] [Google Scholar]
- Dubuisson L, Boussarie L, Bedin CA, Balabaud C, Bioulac-Sage P. Transformation of sinusoids into capillaries in a rat model of selenium-induced nodular regenerative hyperplasia: An immunolight and immunoelectron microscopic study. Hepatology. 1995;21:805–814. [PubMed] [Google Scholar]
- Falkowska-Hansen B, Falkowski M, Metharom P, Krunic D, Goerdt S. Clathrin-coated vesicles form a unique net-like structure in liver sinusoidal endothelial cells by assembling along undisrupted microtubules. Exp Cell Res. 2007;313:1745–1757. doi: 10.1016/j.yexcr.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Fende PL, Niewenhuis RJ. An electron microscopic study of the effects of cadmium chloride on cryptorchid testes of the rat. Biol Reprod. 1977;16:298–305. doi: 10.1095/biolreprod16.3.298. [DOI] [PubMed] [Google Scholar]
- Fleischer N, Mouw R, Vander AJ. Chronic effects of lead on renin and renal sodium excretion. J Lab Clin Med. 1980;95:759–770. [PubMed] [Google Scholar]
- Fontana SA, Boulos BM. Lifestyle/environmental factors and blood cadmium levels in hypertensive and normotensive individuals. J Hypertens Suppl. 1986;4:S361–S363. [PubMed] [Google Scholar]
- Fujiwara Y, Watanabe S, Kaji T. Promotion of cultured vascular smooth muscle cell proliferation by low levels of cadmium. Toxicol Lett. 1998;94:175–180. doi: 10.1016/s0378-4274(98)00005-8. [DOI] [PubMed] [Google Scholar]
- Gabbiani G, Badonnel MC, Mathewson SM, Ryan GB. Acute cadmium intoxication. Early selective lesions of endothelial clefts. Lab Invest. 1974;30:686–695. [PubMed] [Google Scholar]
- Galley HF, Webster NR. Physiology of the endothelium. Br J Anaesth. 2004;93:105–113. doi: 10.1093/bja/aeh163. [DOI] [PubMed] [Google Scholar]
- Gibbins IL, Jobling P, Morris JL. Functional organization of peripheral vasomotor pathways. Acta Physiol Scand. 2003;177:237–245. doi: 10.1046/j.1365-201X.2003.01079.x. [DOI] [PubMed] [Google Scholar]
- Glenn BS, Stewart WF, Links JM, Todd AC, Schwartz BS. The longitudinal association of lead with blood pressure. Epidemiology. 2003;14:30–36. doi: 10.1097/00001648-200301000-00011. [DOI] [PubMed] [Google Scholar]
- Goodwin M, Yap AS. Classical cadherin adhesion molecules: Coordinating cell adhesion, signaling and the cytoskeleton. J Mol Histol. 2004;35:839–844. doi: 10.1007/s10735-004-1833-2. [DOI] [PubMed] [Google Scholar]
- Grabowska-Maslanka A, Janik A, Chlap Z, Szuperska-Ocetkiewicz A, Slawinski M, Grylewski RJ, Korbut R. Influence of cadmium intoxication on thromboresistance of vascular endothelium in rabbits. J Physiol Pharmacol. 1998;49:61–69. [PubMed] [Google Scholar]
- Gross SB. Human oral and inhalation exposures to lead: Summary of Kehoe balance experiments. J Toxicol Environ Health. 1981;8:333–377. doi: 10.1080/15287398109530075. [DOI] [PubMed] [Google Scholar]
- Gunn SA, Gould TC. Cadmium and other mineral elements. In: Gomes WR, Van Demark NL, editors. The Testis. Academic Press; New York: 1970. pp. 377–481. [Google Scholar]
- Han S, Pfizenmaier DH, Garcia E, Eguez ML, Ling M, Kemp FW, Bogden JD. Effects of lead exposure before pregnancy and dietary calcium during pregnancy on fetal development and lead accumulation. Environ Health Perspect. 2000;108:527–531. doi: 10.1289/ehp.00108527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan WR. The relationship of blood lead levels to blood pressure in the U.S. population. Environ Health Perspect. 1988;78:9–13. doi: 10.1289/ehp.88789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC. Vasa vasorum in plaque angiogenesis, metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: A malignant transformation. Cardiovasc Diabetol. 2004;3(1):1–16. doi: 10.1186/1475-2840-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Girijashanker K, Dalton TP, Reed J, Li H, Soleimani M, Nebert DW. ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: Characterization of transporter properties. Mol Pharmacol. 2006;70:171–180. doi: 10.1124/mol.106.024521. [DOI] [PubMed] [Google Scholar]
- Hernandez M, Macia M. Free peripheral sulfhydryl groups, CD11/CD18 integrins, and calcium are required in the cadmium and nickel enhancement of human-polymorphonuclear leukocyte adherence. Arch Environ Contam Toxicol. 1996;30:437–443. doi: 10.1007/BF00213393. [DOI] [PubMed] [Google Scholar]
- Hill CE, Phillips JK, Sandow SL. Heterogeneous control of blood flow amongst different vascular beds. Med Res Rev. 2001;21:1–60. doi: 10.1002/1098-1128(200101)21:1<1::aid-med1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Hilmer SN, Cogger VC, Fraser R, McLean AJ, Sullivan D, Le Couteur DG. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology. 2005;42:1349–1354. doi: 10.1002/hep.20937. [DOI] [PubMed] [Google Scholar]
- Hoey MJ. The effects of metallic salts on the histology and functioning of the rat testis. J Reprod Fertil. 1966;12:461–472. doi: 10.1530/jrf.0.0120461. [DOI] [PubMed] [Google Scholar]
- Hu H. A 50-year follow-up of childhood plumbism. Hypertension, renal function, and hemoglobin levels among survivors. Am J Dis Child. 1991;145:681–687. doi: 10.1001/archpedi.1991.02160060099029. [DOI] [PubMed] [Google Scholar]
- Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002;133:1–16. doi: 10.1016/s0378-4274(02)00084-x. [DOI] [PubMed] [Google Scholar]
- Hwang KY, Schwartz BS, Lee BK, Strickland PT, Todd AC, Bressler JP. Associations of lead exposure and dose measures with erythrocyte protein kinase C activity in 212 current Korean lead workers. Toxicol Sci. 2001;62:280–288. doi: 10.1093/toxsci/62.2.280. [DOI] [PubMed] [Google Scholar]
- IARC. International Agency for Research on Cancer Suppl. 7th. World Health Organization; Lyon, France: 1987. Monographs on the evaluation of the carcinogenic risk of chemicals to humans: Arsenic and arsenic compounds (Group 1) pp. 100–106. [Google Scholar]
- Ibrahim D, Froberg B, Wolf A, Rusyniak DE. Heavy metal poisoning: Clinical presentations and pathophysiology. Clin Lab Med. 2006;26:67–97. doi: 10.1016/j.cll.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Jarup L, Berglund M, Elinder CG, Nordberg G, Vahter M. Health effects of cadmium exposure—A review of the literature and a risk estimate. Scand J Work Environ Health. 1998;24(Suppl 1):1–51. [PubMed] [Google Scholar]
- Jeong SH, Habeebu SS, Klaassen CD. Cadmium decreases gap junctional intercellular communication in mouse liver. Toxicol Sci. 2000;57:156–166. doi: 10.1093/toxsci/57.1.156. [DOI] [PubMed] [Google Scholar]
- Kaji T. Cell biology of heavy metal toxicity in vascular tissue. Yakugaku Zasshi. 2004;124:113–120. doi: 10.1248/yakushi.124.113. [DOI] [PubMed] [Google Scholar]
- Kamat CD, Green DE, Curilla S, Warnke L, Hamilton JW, Sturup S, Clark C, Ihnat MA. Role of HIF signaling on tumorigenesis in response to chronic low-dose arsenic administration. Toxicol Sci. 2005;86:248–257. doi: 10.1093/toxsci/kfi190. [DOI] [PubMed] [Google Scholar]
- Kao YH, Yu CL, Chang LW, Yu HS. Low concentrations of arsenic induce vascular endothelial growth factor and nitric oxide release and stimulate angiogenesis in vitro. Chem Res Toxicol. 2003;16:460–468. doi: 10.1021/tx025652a. [DOI] [PubMed] [Google Scholar]
- Kar AB, Das RP. Testicular changes in rats after treatment with cadmium chloride. Acta Biol Med Ger. 1960;5:153–173. [PubMed] [Google Scholar]
- Kehoe RA. The Harben Lectures, 1960: The metabolism of lead in man in health and disease. 3. Present hygienic problems relating to the absorption of lead. J R Inst Public Health. 1961;24:177–203. [PubMed] [Google Scholar]
- Khalil-Manesh F, Gonick HC, Weiler EW, Prins B, Weber MA, Purdy RE. Lead-induced hypertension: Possible role of endothelial factors. Am J Hypertens. 1993;6:723–729. doi: 10.1093/ajh/6.9.723. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Oguri T, Ohno M, Matsubara K, Yamamoto K, Tada M. Effect of cadmium (CdCl2) on cell proliferation and production of EDRF (endothelium-derived relaxing factor) by cultured human umbilical arterial endothelial cells. Arch Toxicol. 1994;68:555–559. doi: 10.1007/s002040050113. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Oguri T, Yamabe S, Tada M. Effect of cadmium injury on growth and migration of cultured human vascular endothelial cells. Hum Cells. 1996a;9:43–48. [PubMed] [Google Scholar]
- Kishimoto T, Ueda D, Isobe M, Tada M. Cadmium injures tube formation by cultured human vascular endothelial cells. Hum Cells. 1996b;9:244–250. [PubMed] [Google Scholar]
- Kitchin KT. Recent advances in arsenic carcinogenesis: Modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- Klees RF, De Marco PC, Salasznyk RM, Ahuja D, Hogg M, Antoniotti S, Kamath L, Dordick JS, Plopper GE. Apocynin derivatives interrupt intracellular signaling resulting in decreased migration in breast cancer cells. J Biomed Biotechnol. 2006;2006:87246. doi: 10.1155/JBB/2006/87246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch AW, Manzur KL, Shan W. Structure-based models of cadherin-mediated cell adhesion: The evolution continues. Cell Mol Life Sci. 2004;61:1884–1895. doi: 10.1007/s00018-004-4006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolluru GK, Tamilarasan KP, Geetha PS, Durgha NP, Chatterjee S. Cadmium induced endothelial dysfunction: Consequence of defective migratory pattern of endothelial cells in association with poor nitric oxide availability under cadmium challenge. Cell Biol Int. 2006;30:427–438. doi: 10.1016/j.cellbi.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kopp SJ, Perry M, Jr, Glonek T, Erlanger M, Perry EF, Barany M, D'Agrosa LS. Cardiac physiologic-metabolic changes after chronic low-level heavy metal feeding. Am J Physiol. 1980;239:H22–H30. doi: 10.1152/ajpheart.1980.239.1.H22. [DOI] [PubMed] [Google Scholar]
- Koschel K, Meissner NN, Tas PW. Influence of cadmium ions on endothelin-1 binding and calcium signaling in rat glioma C6 cells. Toxicol Lett. 1995;81:189–195. doi: 10.1016/0378-4274(95)03434-x. [DOI] [PubMed] [Google Scholar]
- Kusaka Y, Kelly RA, Williams GH, Kifor I. Coronary microvascular endothelial cells cosecrete angiotensin II and endothelin-1 via a regulated pathway. Am J Physiol Heart Circ Physiol. 2000;279:H1087–H1096. doi: 10.1152/ajpheart.2000.279.3.H1087. [DOI] [PubMed] [Google Scholar]
- Lal B, Murthy RC, Anand M, Chandra SV, Kumar R, Tripathi O, Srimal RC. Cardiotoxicity and hypertension in rats after oral lead exposure. Drug Chem Toxicol. 1991;14:305–318. doi: 10.3109/01480549109002192. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco LP, Dejana E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol. 1992;118:1511–1522. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Niu JZ, Wang JF, Li Y, Tao XH. Pathological mechanisms of alcohol induced hepatic portal hypertension in early stage fibrosis rat model. World J Gastroenterol. 2005;11:6483–6488. doi: 10.3748/wjg.v11.i41.6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Pan SG, Dong XS, Qiao HQ, Jiang HC, Krissansen GW, Sun XY. Opposing effects of arsenic trioxide on hepatocellular carcinomas in mice. Cancer Sci. 2006;97:675–681. doi: 10.1111/j.1349-7006.2006.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucis OJ, Lucis R. Distribution of cadmium 109 and zinc 65 in mice of inbred strains. Arch Environ Health. 1969;19:334–336. doi: 10.1080/00039896.1969.10666853. [DOI] [PubMed] [Google Scholar]
- Luoma PV, Nayha S, Pyy L, Hassi J. Association of blood cadmium to the area of residence and hypertensive disease in Arctic Finland. Sci Total Environ. 1995;160–161:571–575. doi: 10.1016/0048-9697(95)04391-d. [DOI] [PubMed] [Google Scholar]
- McAllister RG, Jr, Michelakis AM, Sandstead HH. Plasma renin activity in chronic plumbism. Effect of treatment. Arch Intern Med. 1971;127:919–923. [PubMed] [Google Scholar]
- McCuskey RS. Sinusoidal endothelial cells as an early target for hepatic toxicants. Clin Hemorheol Microcirc. 2006;34:5–10. [PubMed] [Google Scholar]
- Mlynek V, Skoczynska A. The proinflammatory activity of cadmium. Postepy Hig Med Dosw. 2005;59:1–8. Online. [PubMed] [Google Scholar]
- Muntner P, Menke A, DeSalvo KB, Rabito FA, Batuman V. Continued decline in blood lead levels among adults in the United States: The National Health and Nutrition Examination Surveys. Arch Intern Med. 2005;165:2155–2161. doi: 10.1001/archinte.165.18.2155. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Nishijo M. Environmental cadmium exposure, hypertension and cardiovascular risk. J Cardiovasc Risk. 1996;3:11–17. [PubMed] [Google Scholar]
- Nash D, Magder L, Lustberg M, Sherwin RW, Rubin RJ, Kaufmann RB, Silbergeld EK. Blood lead, blood pressure, and hypertension in perimenopausal and postmenopausal women. JAMA. 2003;289:1523–1532. doi: 10.1001/jama.289.12.1523. [DOI] [PubMed] [Google Scholar]
- Navas-Acien A, Sharrett AR, Silbergeld EK, Schwartz BS, Nachman KE, Burke TA, Guallar E. Arsenic exposure and cardiovascular disease: A systematic review of the epidemiologic evidence. Am J Epidemiol. 2005a;162:1037–1049. doi: 10.1093/aje/kwi330. [DOI] [PubMed] [Google Scholar]
- Navas-Acien A, Silbergeld EK, Sharrett R, Calderon-Aranda E, Selvin E, Guallar E. Metals in urine and peripheral arterial disease. Environ Health Perspect. 2005b;113:164–169. doi: 10.1289/ehp.7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechay BR. Increased vascular response to adrenergic stimulation in rats exposed to cadmium. J Toxicol Environ Health. 1978;4:559–567. doi: 10.1080/15287397809529679. [DOI] [PubMed] [Google Scholar]
- Niewenhuis RJ, Dimitriu C, Prozialeck WC. Ultrastructural characterization of the early changes in intercellular junctions in response to cadmium (Cd2+) exposure in LLCPK1 cells. Toxicol Appl Pharmacol. 1997;142:1–12. doi: 10.1006/taap.1996.8026. [DOI] [PubMed] [Google Scholar]
- Nollet F, Kools P, van Roy F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J Mol Biol. 2000;299:551–572. doi: 10.1006/jmbi.2000.3777. [DOI] [PubMed] [Google Scholar]
- Ohanian EV, Iwai J, Leitl G, Tuthill R. Genetic influence on cadmium-induced hypertension. Am J Physiol. 1978;235:H385–H391. doi: 10.1152/ajpheart.1978.235.4.H385. [DOI] [PubMed] [Google Scholar]
- Oner G, Senturk UK, Uysal NI. The role of atrial natriuretic peptide in cadmium induced hypertension. J Trace Elem Exp Med. 1995;8:147–153. [Google Scholar]
- Parizek J, Zahor K. Effect of cadmium salts on testicular tissue. Nature. 1956;177:1036. doi: 10.1038/1771036b0. [DOI] [PubMed] [Google Scholar]
- Pearson CA, Lamar PC, Prozialeck WC. Effects of cadmium on E-cadherin and VE-cadherin in mouse lung. Life Sci. 2003;72:1303–1320. doi: 10.1016/s0024-3205(02)02379-2. [DOI] [PubMed] [Google Scholar]
- Peereboom-Stegeman JH, Jongstra-Spaapen EJ. The effect of a single sublethal administration of cadmium chloride on the microcirculation in the uterus of the rat. Toxicology. 1979;13:199–213. [PubMed] [Google Scholar]
- Pereira FE, Coffin JD, Beall HD. Activation of protein kinase C and disruption of endothelial monolayer integrity by sodium arsenite—Potential mechanism in the development of atherosclerosis. Toxicol Appl Pharmacol. 2007;220:164–177. doi: 10.1016/j.taap.2006.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry HM, Jr, Erlanger M, Perry EF. Hypertension following chronic, very low dose cadmium feeding (39900) Proc Soc Exp Biol Med. 1977;156:173–176. doi: 10.3181/00379727-156-39900. [DOI] [PubMed] [Google Scholar]
- Perry HM, Jr, Erlanger MW, Perry EF. Increase in the blood pressure of rats chronically fed low levels of lead. Environ Health Perspect. 1988;78:107–111. doi: 10.1289/ehp.8878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinini F, Favalli L, Chiari MC. Experimental investigations on the contraction induced by lead in arterial smooth muscle. Toxicology. 1977;8:43–51. doi: 10.1016/0300-483x(77)90022-1. [DOI] [PubMed] [Google Scholar]
- Pirkle JL, Schwartz J, Landis JR, Harlan WR. The relationship between blood lead levels and blood pressure and its cardiovascular risk implications. Am J Epidemiol. 1985;121:246–258. doi: 10.1093/oxfordjournals.aje.a113995. [DOI] [PubMed] [Google Scholar]
- Prozialeck WC. Evidence that E-cadherin may be a target for cadmium toxicity in epithelial cells. Toxicol Appl Pharmacol. 2000;164:231–249. doi: 10.1006/taap.2000.8905. [DOI] [PubMed] [Google Scholar]
- Prozialeck WC, Edwards JR, Woods JM. The vascular endothelium as a target of cadmium toxicity. Life Sci. 2006;79:1493–1506. doi: 10.1016/j.lfs.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Prozialeck WC, Niewenhuis RJ. Cadmium (Cd2+) disrupts Ca(2+)-dependent cell-cell junctions and alters the pattern of E-cadherin immunofluorescence in LLC-PK1 cells. Biochem Biophys Res Commun. 1991a;181:1118–1124. doi: 10.1016/0006-291x(91)92054-n. [DOI] [PubMed] [Google Scholar]
- Prozialeck WC, Niewenhuis RJ. Cadmium (Cd2+) disrupts intercellular junctions and actin filaments in LLC-PK1 cells. Toxicol Appl Pharmacol. 1991b;107:81–97. doi: 10.1016/0041-008x(91)90333-a. [DOI] [PubMed] [Google Scholar]
- Puri VN. Acute effects of cadmium on the renin angiotensin system in rats. Biochem Pharmacol. 1992;44:187–188. doi: 10.1016/0006-2952(92)90056-o. [DOI] [PubMed] [Google Scholar]
- Qian Y, Liu KJ, Chen Y, Flynn DC, Castranova V, Shi X. Cdc42 regulates arsenic-induced NADPH oxidase activation and cell migration through actin filament reorganization. J Biol Chem. 2005;280:3875–3884. doi: 10.1074/jbc.M403788200. [DOI] [PubMed] [Google Scholar]
- Revis NW, Zinsmeister AR, Bull R. Atherosclerosis and hypertension induction by lead and cadmium ions: An effect prevented by calcium ion. Proc Natl Acad Sci U S A. 1981;78:6494–6498. doi: 10.1073/pnas.78.10.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roboz GJ, Dias S, Lam G, Lane WJ, Soignet SL, Warrell RP, Jr, Rafii S. Arsenic trioxide induces dose- and time-dependent apoptosis of endothelium and may exert an antileukemic effect via inhibition of angiogenesis. Blood. 2000;96:1525–1530. [PubMed] [Google Scholar]
- Ruiz DA, Luttun A, Carmeliet P. An SDF-1 trap for myeloid cells stimulates angiogenesis. Cell. 2006;124:18–21. doi: 10.1016/j.cell.2005.12.023. [DOI] [PubMed] [Google Scholar]
- Sacerdote FL, Cavicchia JC. Ultrastructural effects of cadmium on the rat epididymis. Int J Androl. 1983;6:533–540. doi: 10.1111/j.1365-2605.1983.tb00344.x. [DOI] [PubMed] [Google Scholar]
- Satarug S, Baker JR, Urbenjapol S, Haswell-Elkins M, Reilly PE, Williams DJ, Moore MR. A global perspective on cadmium pollution and toxicity in nonoccupationally exposed population. Toxicol Lett. 2003;137:65–83. doi: 10.1016/s0378-4274(02)00381-8. [DOI] [PubMed] [Google Scholar]
- Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect. 2004;112:99–103. doi: 10.1289/ehp.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satarug S, Nishijo M, Ujjin P, Vanavanitkun Y, Moore MR. Cadmium induced nephropathy in the development of high blood pressure. Toxicol Lett. 2005;157:57–68. doi: 10.1016/j.toxlet.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Schwartz J. The relationship between blood lead and blood pressure in the NHANES II survey. Environ Health Perspect. 1988;78:15–22. doi: 10.1289/ehp.887815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoczynska A, Martynowicz H. The impact of subchronic cadmium poisoning on the vascular effect of nitric oxide in rats. Hum Exp Toxicol. 2005;24:353–361. doi: 10.1191/0960327105ht536oa. [DOI] [PubMed] [Google Scholar]
- Smith KR, Klei LR, Barchowsky A. Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am J Physiol. 2001;280:L442–L449. doi: 10.1152/ajplung.2001.280.3.L442. [DOI] [PubMed] [Google Scholar]
- Soucy NV, Ihnat MA, Kamat CD, Hess L, Post MJ, Klei LR, Clark C, Barchowsky A. Arsenic stimulates angiogenesis and tumorigenesis in vivo. Toxicol Sci. 2003;76:271–279. doi: 10.1093/toxsci/kfg231. [DOI] [PubMed] [Google Scholar]
- Soucy NV, Mayka D, Klei LR, Nemec AA, Bauer JA, Barchowsky A. Neovascularization and angiogenic gene expression following chronic arsenic exposure in mice. Cardiovasc Toxicol. 2005;5:29–42. doi: 10.1385/ct:5:1:029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staessen JA, Buchet JP, Ginucchio G, Lauwerys RR, Lijnen P, Roels H, Fagard R. Public health implications of environmental exposure to cadmium and lead: An overview of epidemiological studies in Belgium. Working Groups. J Cardiovasc Risk. 1996;3:26–41. [PubMed] [Google Scholar]
- Staessen JA, Kuznetsova T, Roels HA, Emelianov D, Fagard R. Exposure to cadmium and conventional and ambulatory blood pressures in a prospective population study. Public Health and Environmental Exposure to Cadmium Study Group. Am J Hypertens. 2000;13:146–156. doi: 10.1016/s0895-7061(99)00187-9. [DOI] [PubMed] [Google Scholar]
- Staessen JA, Kuznetsova T, Wang JG, Emelianov D, Vlietinck R, Fagard R. M235T angiotensinogen gene polymorphism and cardiovascular renal risk. J Hypertens. 1999;17:9–17. doi: 10.1097/00004872-199917010-00003. [DOI] [PubMed] [Google Scholar]
- Straub AC, Stolz DB, Ross MA, Hernandez-Zavala A, Soucy NV, Klei LR, Barchowsky A. Arsenic stimulates sinusoidal endothelial cell capillarization and vessel remodeling in mouse liver. Hepatology. 2007a;45:205–212. doi: 10.1002/hep.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub AC, Stolz DB, Vin H, Ross MA, Soucy NV, Klei LR, Barchowsky A. Low level arsenic promotes progressive inflammatory angiogenesis and liver blood vessel remodeling in mice. Toxicol Appl Pharmacol. 2007b;222:327–336. doi: 10.1016/j.taap.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuster-Ciesielska A, Lokaj I, Kandefer-Szerszen M. The influence of cadmium and zinc ions on the interferon and tumor necrosis factor production in bovine aorta endothelial cells. Toxicology. 2000;145:135–145. doi: 10.1016/s0300-483x(00)00147-5. [DOI] [PubMed] [Google Scholar]
- Taylor BA, Heiniger HJ, Meier H. Genetic analysis of resistance to cadmium induced testicular damage in mice. Proc Soc Exp Biol Med. 1973;143:629–633. doi: 10.3181/00379727-143-37380. [DOI] [PubMed] [Google Scholar]
- Triggle CR, Hollenberg M, Anderson TJ, Ding H, Jiang Y, Ceroni L, Wiehler WB, Ng ES, Ellis A, Andrews K, et al. The endothelium in health and disease—A target for therapeutic intervention. J Smooth Muscle Res. 2003;39:249–267. doi: 10.1540/jsmr.39.249. [DOI] [PubMed] [Google Scholar]
- Tsao DA, Yu HS, Cheng JT, Ho CK, Chang HR. The change of beta adrenergic system in lead-induced hypertension. Toxicol Appl Pharmacol. 2000;164:127–133. doi: 10.1006/taap.1999.8871. [DOI] [PubMed] [Google Scholar]
- Vaziri ND. Pathogenesis of lead-induced hypertension: Role of oxidative stress. J Hypertens Suppl. 2002;20(3):S15–S20. [PubMed] [Google Scholar]
- Vaziri ND, Alikhani S, Patel B, Nguyen Q, Barton CH, Gonzales EV. Increased levels of protein C activity, protein C concentration, total and free protein S in nephrotic syndrome. Nephron. 1988;49:20–23. doi: 10.1159/000184980. [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Liang K, Ding Y. Increased nitric oxide inactivation by reactive oxygen species in lead-induced hypertension. Kidney Int. 1999;56:1492–1498. doi: 10.1046/j.1523-1755.1999.00670.x. [DOI] [PubMed] [Google Scholar]
- Villar IC, Francis S, Webb A, Hobbs AJ, Ahluwalia A. Novel aspects of endothelium-dependent regulation of vascular tone. Kidney Int. 2006;70:840–853. doi: 10.1038/sj.ki.5001680. [DOI] [PubMed] [Google Scholar]
- Vincent PA, Xiao K, Buckley KM, Kowalczyk AP. VE-cadherin: Adhesion at arm's length. Am J Physiol Cell Physiol. 2004;286:C987–C997. doi: 10.1152/ajpcell.00522.2003. [DOI] [PubMed] [Google Scholar]
- Vivoli G, Bergomi M, Borella P, Fantuzzi G, Caselgrandi E. Cadmium in blood, urine and hair related to human hypertension. J Trace Elem Electrolytes Health Dis. 1989;3:139–145. [PubMed] [Google Scholar]
- Vupputuri S, He J, Muntner P, Bazzano LA, Whelton PK, Batuman V. Blood lead level is associated with elevated blood pressure in blacks. Hypertension. 2003;41:463–468. doi: 10.1161/01.HYP.0000055015.39788.29. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Coogan TP, Barter RA. Toxicological principles of metal carcinogenesis with special emphasis on cadmium. Crit Rev Toxicol. 1992;22:175–201. doi: 10.3109/10408449209145323. [DOI] [PubMed] [Google Scholar]
- Wada K, Fujii Y, Watanabe H, Satoh M, Furuichi Y. Cadmium directly acts on endothelin receptor and inhibits endothelin binding activity. FEBS Lett. 1991;285:71–74. doi: 10.1016/0014-5793(91)80727-k. [DOI] [PubMed] [Google Scholar]
- Wang B, Schneider SN, Dragin N, Girijashanker K, Dalton TP, He L, Miller ML, Stringer KF, Soleimani M, Richardson DD, et al. Enhanced cadmium-induced testicular necrosis and renal proximal tubule damage caused by gene-dose increase in a Slc39a8-transgenic mouse line. Am J Physiol Cell Physiol. 2007;292:C1523–C1535. doi: 10.1152/ajpcell.00409.2006. [DOI] [PubMed] [Google Scholar]
- Washington B, Williams S, Armstrong P, Mtshali C, Robinson JT, Myles EL. Cadmium toxicity on arterioles vascular smooth muscle cells of spontaneously hypertensive rats. Int J Environ Res Public Health. 2006;3:323–328. doi: 10.3390/ijerph2006030040. [DOI] [PubMed] [Google Scholar]
- Watts SW, Chai S, Webb RC. Lead acetate-induced contraction in rabbit mesenteric artery: Interaction with calcium and protein kinase C. Toxicology. 1995;99:55–65. doi: 10.1016/0300-483x(94)03003-k. [DOI] [PubMed] [Google Scholar]
- Xu B, Broome U, Uzunel M, Nava S, Ge X, Kumagai-Braesch M, Hultenby K, Christensson B, Ericzon BG, Holgersson J, et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am J Pathol. 2003;163:1275–1289. doi: 10.1016/S0002-9440(10)63487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto C, Kaji T, Sakamoto M, Kozuka H. Cadmium stimulation of plasminogen activator inhibitor-1 release from human vascular endothelial cells in culture. Toxicology. 1993;83:215–223. doi: 10.1016/0300-483x(93)90103-y. [DOI] [PubMed] [Google Scholar]
- Yang S, Graham J, Kahn JW, Schwartz EA, Gerritsen ME. Functional roles for PECAM-1 (CD31) and VE-cadherin (CD144) in tube assembly and lumen formation in three-dimensional collagen gels. Am J Pathol. 1999;155:887–895. doi: 10.1016/S0002-9440(10)65188-7. [DOI] [PMC free article] [PubMed] [Google Scholar]