

Abstract
Small molecule dimer disruptors that inhibit an essential dimeric protease of human Kaposi’s sarcoma-associated herpesvirus (KSHV) were identified by screening an α-helical mimetic library. Subsequently, a second generation of low micromolar inhibitors with improved potency and solubility was synthesized. Complementary methods including size exclusion chromatography and 1H-13C HSQC titration using selectively labeled 13C-Met samples revealed that monomeric protease is enriched in the presence of inhibitor. 1H-15N-HSQC titration studies mapped the inhibitor binding-site to the dimer interface, and mutagenesis studies targeting this region were consistent with a mechanism where inhibitor binding prevents dimerization through the conformational selection of a dynamic intermediate. These results validate the interface of herpesvirus proteases and other similar oligomeric interactions as suitable targets for the development of small molecule inhibitors.
Background
Targeting protein-protein interactions for a therapeutic purpose is an attractive idea that has proved to be extremely challenging in practice. This is due to the large and flat landscape of most contact surfaces that make them less amenable to intervention by a small molecule1-4. However, in recent years a growing body of evidence has demonstrated that small molecules can disrupt such large and complex protein interactions by binding to interface “hotspots” with drug-like potencies5. Examples include interactions involving cytokine interleukin-2 (IL-2), with the IL-2 receptor α-chain (IL-2Rα); B-cell lymphoma protein (Bcl-XL), with pro-apoptotic molecule Bcl-2 antagonist of cell death (BAK); the oncogene human protein double minute 2 (HDM2), with tumor-suppressor protein p53; and the human papilloma virus transcription factor E2 complex with viral helicase E15. Notably, an inhibitor of Bcl-XL /BAK interaction has entered phase I/II clinical trials6. In this study we have identified the first of such small molecule protein interaction inhibitors for the family of herpesvirus proteases.
Herpesviruses make up one of the most prevalent viral families including eight human types that cause a variety of devastating illnesses including mononucleosis (Epstein-Barr virus, EBV), shingles (varicella zoster virus, VZV), genital herpes (herpes simplex virus, HSV), retinitis (cytomegalovirus, CMV), and cancer (Kaposi’s sarcoma-associated herpesvirus, KSHV)7. The standard course of treatment for common herpesviral infections, a class of broad-acting viral DNA replication inhibitors such as ganciclovir and foscarnet, though widely used, exhibit undesirable toxicity, poor oral bioavailability, and in some cases inadequate efficacy8. With the incidents of drug resistance also on the rise and no other specific antivirals available, there is a need for the identification of new human herpesvirus (HHV) therapeutic targets.
All herpesviruses express a structurally and functionally conserved dimeric serine protease that plays an essential role in capsid assembly during the lytic stage9-13. Efforts by pharmaceutical companies to specifically target the active site of human cytomegalovirus protease with small molecule inhibitors has lead to either covalent inhibitors or molecules with non-ideal pharmacological properties8,14-18. In light of evidence supporting a strong linkage between the dimer interface and the protease active site, we have focused our efforts on the dimer interface for identifying novel allosteric inhibitors.
Although HHV proteases are initially expressed as a monomer, studies with recombinant protease have demonstrated the dimerization dependence of enzyme activity19-26. An array of structural studies has suggested a model where protease dimerization induces folding of both the interfacial and the C-terminal α-helices, which positions the oxyanion loop within the active site and activates the enzyme19,23,27,28. Interestingly, the non-cannonical Ser-His-His catalytic triad of each monomer is located 15Å away from the dimer interface and acts independently (Fig. 1a)25,29-34. Considering the extensive mechanistic knowledge of the human Kaposi’s sarcoma-associated herpesvirus protease (KSHV Pr), we chose it as a candidate for the development of dimerization inhibitors for modulating the function of this enzyme family.
Figure 1.
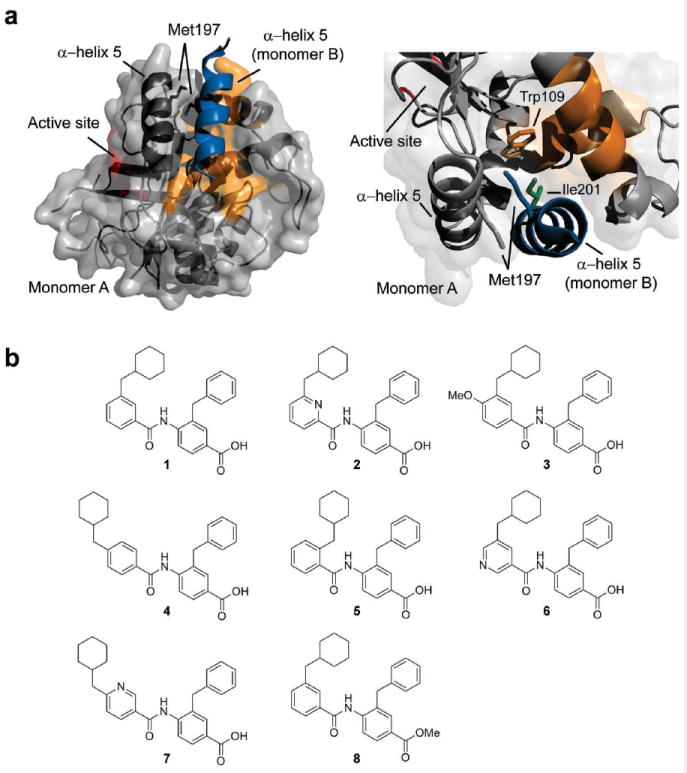
KSHV Pr dimer interface and helical mimetic inhibitors of KSHV Pr activity. (a) The interface of monomer A (gray) and the α-helix 5 of monomer B (blue) is shown from two viewpoints. The α-helix 5 (dark gray) and other interface residues (orange) on monomer A form key contacts with α-helix 5 of monomer B. Interfacial residues Met197 (gray and blue), Trp109 (orange), and Ile201 (green) are highlighted throughout the paper. The active site (red) is located 15Å away from the dimer interface. (b) Chemical structures of KSHV Pr activity inhibitors identified by screening an α-helical mimetic library. Compounds 2-8 are structural analogues of initial hit compound 1.
The KSHV Pr dimer interface covers approximately 2500 Å2, and includes the α-helix 5 of each monomer as the major constituent (Fig. 1a)25. Mutagenesis studies with KSHV and other HHV proteases have shown that the dimer interface is very sensitive to genetic perturbation where a single point mutation leads to loss of dimerization and enzyme activity19,24. Importantly, this dimerization affinity is weak with a reported in vitro KD of 1.7 μM for KSHV Pr24,35. Hence, the protease is thought to be regulated by concentration-driven zymogen activation, where initially it exists as an inactive monomer in the host cytosol, but achieves dimerization and activation upon reaching a high local concentration inside the pre-capsid. These observed characteristics define the dimer interface as a critical allosteric regulatory site and a potential candidate for inhibition by small molecule dimer disruptors.
Recently we reported evidence for sterically disrupting the dimer interface of a herpesvirus protease36. Avian pancreatic polypeptide, aPP, a 30-amino acid peptide containing an internally stabilized α-helix, is used as a scaffold to design a KSHV Pr α-helix 5 mimetic. The resulting peptide fully disrupts KSHV Pr dimerization and inhibits activity, demonstrating that in addition to the active site, the protease dimer interface is a potential target for the development of small molecule inhibitors.
In this study we report a small molecule inhibitor of KSHV Pr dimerization. Inhibitors of KSHV Pr activity were obtained by screening a helical mimetic compound library. The top hits were subsequently assayed for dimer disruption and characterized for mode of binding. Our data supports a mechanism of inhibition where inhibitor binding to critical interfacial residues on the protease monomer prevents dimerization. These studies illustrate that the dimer interface of HHV proteases is amenable to targeting by small molecule inhibitors.
Results
Identification of inhibitors by high throughput screening
Given that two α-helices are a major component of the KSHV Pr dimer interface, we screened a library of small molecule helical mimetics. This library of 182 compounds was originally developed by computational design to disrupt the interacting α-helix of p53 tumor suppressor protein with oncoprotein MDM237. Our experimental design was based on the knowledge that interactions in both complexes are dominated by an α-helical motif.
Screening was performed in a 96-well plate format using a fluorogenic activity assay. The substrate used is an optimized peptide sequence attached to 7-amino-4-carbamoylmethyl coumarin (ACC), where cleavage at the scissile bond releases the ACC group resulting in increased fluorescence38,39. We identified six molecules that inhibited KSHV Pr activity by at least 50%, the most potent compound being DD1, (1), with an IC50 of 8.8 ± 0.3 μM (Fig. 1b and Supplementary Table 1 online). DD1 was not found to inhibit the interaction of p53-MDM2, suggesting these proteomimetic molecules can potentially discriminate between different intermolecular interactions involving an α-helix37. A series of DD1 analogs was designed to explore modifications around the 4-benzoylamino-benzoic acid scaffold using previously reported synthetic methods (Supplementary Scheme 1 online)37.
Further screening of eight DD1 structural analogues yielded additional hits, the most potent being (2) with an IC50 of 3.1 ± 0.2 μM, and (3) with an IC50 of 16.4 ± 0.7 μM (Fig. 1b and Supplementary Table 1 online). The improved IC50 of the top inhibitor, DD2, (2) is possibly due to the hydrogen bonding capabilities of the added pyridine nitrogen to residues on the protease. Additionally, the structure-activity relationship (SAR) for this series indicated that correct positioning of the cyclohexylmethyl side chain is critical, where substitution from its original meta position to either the para (4) or the ortho (5) positions diminished inhibition (Fig. 1b and Supplementary Table 1 online). This effect is consistent with the original design rationale of the helical mimetic molecules, where the spatial and angular arrangement of the side chains on the scaffold were fixed to mimic the i and i+4 residues of an α-helix. It is also in agreement with a specific mechanism of inhibition whereby the cyclohexylmethyl side chain makes critical hydrophobic interactions with KSHV Pr residues.
Compounds were tested for non-specific aggregation by assessing their behavior in the presence of a non-ionic detergent40. KSHV Pr was substituted with an unrelated serine protease Granzyme B, which unlike KSHV Pr, tolerates detergents in assays for biochemical activity. In a spectrophotometric activity assay monitoring Granzyme B activity, DD2 failed to show evidence of inhibition in the presence or absence of detergent, while a known aggregator, hexachlorophene, inhibited Granzyme B activity only in the absence of detergent (Supplementary Fig. 1 online). DD2 also did not show evidence of particle formation when analyzed by dynamic light scattering. Collectively, this data suggests that DD2 acts by a specific mechanism to inhibit KSHV Pr activity.
Helical mimetic inhibitors disrupt protease dimerization
To assess the dimerization state of KSHV Pr in the presence of DD2, we took two complementary approaches. The first approach used a size exclusion chromatography assay where the dimeric and monomeric protease populations are monitored by their distinct elution profiles24,28. At a concentration of 5 μM, KSHV Pr eluted as two separate peaks corresponding to the dimeric and monomeric species, respectively (Fig. 2a). As expected for a sample concentration above the recorded KD of 1.7 μM, a larger fraction of the protease appeared in the dimer peak. However, when pre-incubated with 30 μM DD2, KSHV Pr eluted predominantly as a monomer (Fig. 2a). The enrichment of monomeric KSHV Pr strongly suggests that DD2 acts as a dimer disruptor.
Figure 2.

DD2 disrupts the KSHV Pr dimer. (a) Wt KSHV Pr (5 μM) was incubated with either DMSO (dotted) or 30 μM DD2 (solid). Mixtures were loaded onto a size exclusion chromatography column, pre-equilibrated with running buffer containing either DMSO or 30 μM DD2 respectively. (b) The elution profile of KSHV Pr variant W109A (5 μM) in buffer alone indicated the absence of the dimeric species. (c) The overlaid 1H-13C HSQC spectra of selectively labeled 13C methionine wt KSHV Pr (23 μM) in the presence of 0 μM (red), 6 μM (yellow), 12 μM (green) and, 24 μM (blue) DD2. The spectra of apo protease (red) exhibited individual resonances for the N-terminus Met1 (M1), as well as the interfacial Met197 in the dimeric and monomeric states.
As an alternate approach to evaluating protease dimerization, we performed 2-D nuclear magnetic resonance (NMR) assays using selectively isotope-labeled KSHV Pr28. In this experiment, methionine, which occurs in the sequence only at the N-terminus (Met1) and at the dimer interface (Met197) was used as a probe to evaluate monomer-dimer transitions (Fig. 1a). Heteronuclear single quantum coherence (HSQC) spectra of selectively labeled 13C-Met KSHV Pr exhibits resonances corresponding to the methyl group of Met1 as well as Met197 in both the monomeric and dimeric conformations (Fig. 2c). Overlaid spectra of the titration data reveal that increasing DD2 concentrations induces a chemical shift perturbation and increase in the peak volume of the Met197-monomer resonance. Concomitantly, we observed a decrease in the peak volume of the M197-dimer peak, which broadens beyond detection at 1 molar equivalence inhibitor (24 μM). Inhibitor concentrations up to 2 molar equivalence resulted in no additional change. The Met1 resonance remained unchanged throughout the titrations and served as an internal control. The data further confirms that DD2 disrupts the protease dimer and highlights changes in the local conformational environment of the interfacial Met197. DD1 produced similar results in both the size exclusion chromatography as well as the HSQC experiments (data not shown). These results are consistent with a model of helical mimetic inhibitors of KSHV Pr activity acting via a mechanism of dimer disruption.
DD2 shows characteristics of mixed inhibition
The activation mechanism of KSHV Pr involves a series of allosteric conformational changes that link the dimer interface to positioning of the oxyanion loop in the active site 15Å away. Binding of a covalent and irreversible peptide phosphonate inhibitor to the KSHV Pr active site promotes enzyme dimerization, illustrating communication between the two sites28. In such a complex system, an inhibitor acting as a dimer disruptor is expected to show properties of mixed-type inhibition, with a characteristic change in both KM for substrate and maximum reaction velocity, Vmax. We performed kinetic assays to measure the Michaelis-Menten constants for substrate in the presence of increasing concentrations of DD2, and plotted the initial reaction velocities as a function of substrate concentration in Lineweaver–Burk format (Fig. 3a). A 1 -10 μM concentration range of DD2, caused a 2.5 fold increase in KM and a 3 fold decrease in Vmax (Supplementary Table 2 online). This effect on both the KM and the Vmax is conceivable since dimer dissociation dismantles the structural framework responsible for forming the KSHV Pr active site. Based on this data, DD2 is a dimer disruptor that acts as a mixed-type inhibitor by preventing the formation of a conformationally stable active site.
Figure 3.

Kinetic studies with DD2 show evidence of mixed-type inhibition. (a) A standard enzyme assay was performed using 1 μM KSHV Pr with final substrate (P6) concentration range of 80 - 2 μM, across inhibitor concentrations of 10 (orange), 7 (brown), 5 (purple), 3 (green), 1 (blue), and 0 (red) μM. Double-reciprocal plots of initial reaction velocities are shown. (b) IC50 of inhibition by DD2 is higher for the tighter-KD KSHV Pr variant. Activities of wt and M197L KSHV protease at 1 μM were monitored in the presence of increasing concentrations of DD2 (~0.14μM – 100μM). Data is plotted as the ratio of experimental end-point fluorescence (F), and un-inhibited end-point fluorescence (F0), as a function of increasing inhibitor concentration. The IC50 of inhibition were 2.6 ± 0.4 μM and 7.3 ± 0.8 μM for wt (dotted) and M197L (solid) KSHV Pr respectively. Data represents mean values ± s.d. (n=3).
DD2 is weaker against a tighter- KD KSHV Pr variant
In order to verify that DD2 acts at the dimer interface, we tested its inhibitory activity against a KSHV Pr variant with a lower KD. Previously we showed that replacing the interfacial Met197 with a leucine results in approximately 4 fold improvement in binding affinity between KSHV Pr monomers (KD = 0.4 μM)36. In a comparative activity assay with wild-type and M197L KSHV Pr, DD2 exhibited IC50 values of 2.6 ± 0.4 μM and 7.3 ± 0.8 μM, respectively (Fig. 3b). Size exclusion chromatography studies with KSHV Pr M197L confirmed that DD2 still acts by dimer dissociation but less efficiently (Supplementary Fig. 2 online). The observation that DD2 was proportionally less active against a tighter-binding dimer interface, suggests that both DD2 and a KSHV Pr monomer are competing for overlapping binding sites on a second KSHV Pr monomer. We observed similar properties in previous studies with a peptidic α-helix 5 mimetic that disrupts the KSHV Pr dimer36. Consistent with earlier experimetns, the finely tuned potency of DD2 and the varied slopes of the IC50 curves suggest an inhibitory mechanism that is not aggregation-based, where minute alterations are not expected to alter the overall kinetics of inhibition. The fact that DD2 remained active against the M197L variant also shows that Met197 is not a critical residue for inhibitor binding. We conclude that DD2 is a specific inhibitor of KSHV Pr that disrupts protease dimerization by binding to interfacial residues on the KSHV monomer, excluding Met197.
Residues interacting with DD2 map to the dimer interface
To obtain a higher resolution map of the DD2 binding site on KSHV Pr we utilized the technique of chemical shift perturbation mapping. Importantly, NMR was our method of choice as the partially disordered state of KSHV protease presents a challenge for crystallographic studies41. In the absence of significant conformational perturbations, this method detects changes in the electronic environment of protein backbone amides that occur as a consequence of small molecule binding, and translates them into altered chemical shifts on a 1H-15N-HSQC spectrum. For this experiment we used an obligate monomer variant of KSHV Pr, M197D, for which over 90% of the backbone chemical shifts are assigned27. Based on data showing that DD2 remained active against KSHV Pr M197L, we hypothesized that the M197D point mutation would cause little interference with inhibitor binding. The overlaid 1H-15N-HSQC spectra of KSHV Pr M197D in the absence and presence of ~1 molar equivalence (170 μM) DD2 displayed extensive conformational peak broadening in the backbone amides of interface residues (Fig. 4a and Supplementary Fig. 3 online). The observed peak-broadening phenomenon is due to conformational exchange on an intermediate NMR time scale. The chemical shift of residues in the saturated bound state cannot be observed because the solubility limit of DD2 does not allow for collecting spectra with a 5-10 fold excess of inhibitor concentrations under the current experimental conditions.
Figure 4.

1H-15N-HSQC titration data map the DD2 binding site to the dimer interface. (a) The overlaid 1H-15N-HSQC spectra of the monomeric variant, KSHV Pr M197D (170 μM) in the absence (red) and presence (blue) of 170 μM DD2 is shown. The zoomed region highlights the Trp109 side-chain indole NH (boxed). The full 1H-15N-HSQC spectra is included in Supplementary Fig. 3 online. (b) Perturbed resonances from the titration study are mapped onto a monomeric unit of the KSHV Pr crystal structure. Residues are color coded based on the extent of peak intensity loss observed at the 1 molar equivalence titration point with DD2.
When mapped to the monomeric unit of the KSHV Pr crystal structure39, the most significantly perturbed residues were clustered at the dimer interface, where both the resident α-helix 5 and the α-helix 5 of the second monomer form key interactions (Fig. 4b). Importantly, the protease active site residues, Ser114, His46, His134, and Arg143, did not participate in inhibitor binding. The peak broadening of active site residue Arg142 that was observed at saturating DD2 concentrations is likely attributed to allosteric conformational effects propagating from the primary binding site. Notably, the Trp109 indole side chain NH signal exhibited the greatest conformational peak broadening, with over 90% loss of peak intensity in the presence of 1 molar equivalence DD2 (Fig. 4b). A similar high response was observed with concentrations as low as 0.1 molar equivalence DD2 (Supplementary Fig. 4 online). Trp109 resides in close proximity to the resident α-helix 5 and engages in key intermolecular interactions across the dimer interface (Fig. 1a). The titration data with KSHV Pr M197D also provides conclusive evidence that DD2 binds to a monomeric unit of KSHV Pr. We hypothesize that DD2 inhibits KSHV Pr by strongly associating with Trp109 and other interface residues on the monomer, thus preventing dimerization.
A point mutation on Trp109 disrupts KSHV Pr dimerization
The results of the 1H-15N-HSQC titration experiments demonstrate that Trp109 plays a critical role in KSHV Pr dimerization. To test this hypothesis, we monitored protease activity and dimerization on a variant with an alanine substitution of Trp109. KSHV Pr W109A was completely inactive when tested at concentrations up to 30 μM, and eluted as a monomer by size exclusion chromatography (Fig. 2b). This data suggests that Trp109 participates in essential dimer-promoting interactions, which are disrupted upon DD2 binding.
DD2 inhibits the activity of a related herpesvirus protease
All known HHV proteases are structurally and functionally conserved. Therefore it is conceivable to identify a broad-acting inhibitor against this family of enzymes. To examine this notion, we tested DD2 for inhibition of human cytomegalovirus protease (CMV Pr) using the KSHV Pr fluorogenic assay discussed previously. DD2 showed activity inhibition with an IC50 of 5.0 ± 0.5 μM (Supplementary Table 1 online). We conclude that while DD2 is an inhibitor of KSHV Pr, it is also active against the related CMV Pr.
Discussion
Proteases are attractive drug targets, but conventional approaches aimed at the active site can be challenging due to structural similarities in the substrate-binding pockets of related family members. We have reported a small molecule inhibitor of the herpesvirus protease family that acts at a novel allosteric site by a mechanism of dimer dissociation. Additional work is needed to improve the drug-like properties of these compounds, but they have been instrumental in revealing this regulatory binding site. To our knowledge DD2 is the first small molecule inhibitor of a herpesvirus protease to not only act outside the active site, but also select for the partially unfolded zymogen. This illustrates how the inherent conformational dynamics of proteins that are critical to their function also offer opportunity for allosteric regulation.
A dimerization driven disorder-to-order conformational switch regulates the activity of KSHV Pr. The two α-helices at the dimer interface became our rationale for screening a helical mimetic library for inhibitors. We optimized our initial hit, DD1, to produce our top inhibitor, DD2, with an IC50 of 3.1 ± 0.2 μM. Using a combination of gel filtration chromatography, 2D NMR spectroscopy, mutagenesis, and kinetic methods we demonstrated that DD2 acts at the interface, to inhibit protease dimerization, and in turn prevent formation of the active site.
Trp109 is a critical residue in the formation of the dimer, as well as its dissociation by DD2. A survey of the dimeric KSHV Pr X-ray crystal structure placed Trp109 behind the resident α-helix 5 with its indole ring extended towards the hydrophobic side chains of Met197 and Ile201 on the second interfacial α-helix 5 (Fig. 1a). Therefore, we hypothesize that DD2 disrupts protease dimerization by associating with interface residues, including Trp109, and interrupting critical dimer-stabilizing interactions. Interestingly, Met197 and Ile201 assume the i and i+4 positions on α-helix 5. In following with the design of the helical mimetic compounds, we speculate the hydrophobic side chains on DD2 mimic these residues and interact with Trp109 in a similar fashion. A homologous binding pocket does not exist in the p53-MDM2 interface, which could explain why this subset of helical mimetic compounds failed to inhibit that interaction42.
Based on these studies we propose a “monomer trap” model of inhibition by DD2 (Fig. 5). In the absence of inhibitor, inactive KSHV Pr monomer is in equilibrium with active dimer with a weak KD of 1.2 μM. DD2 competes with this process and binds to one of the pre-existing zymogen conformations, thus interfering with interactions critical for dimerization and trapping the monomer in an inactive partially disordered state.
Figure 5.

Monomer Trap model of inhibition by compound DD2. Inhibitor binding to a monomer of KSHV Pr directly competes with the process of dimerization, which is a required step in enzyme activation. When bound to interface residues, DD2 shifts the equilibrium towards a pre-existing folding intermediate, hence trapping the protease in an inactive monomeric state.
An intriguing possibility is a broad-acting inhibitor that targets the protease dimer interface in all eight known HHVs in the alpha, beta, and gamma sub-families. While in all cases the protease is structurally and functionally conserved, the highest amino acid identity observed across sub-families is at 37% between (γ) KSHV Pr and (β) CMV Pr. In our initial studies with CMV Pr, DD2 showed inhibitory activity with an IC50 of 5.0 ± 0.5 μM. Close analysis of the CMV Pr structure reveals interface residues engaged in similar hydrophobic interactions involving an aromatic residue. In this case Leu222, Val226, and Tyr128 of CMV Pr are homologous to Met197, Ile201, and Trp109 in KSHV Pr33,43. The more distant relative (α) VZV Pr, with 30% KSHV Pr identity, also reveals a similar interaction with Leu203, Val207, and Tyr115, while the closest relative (γ) EBV Pr, with 44% KSHV Pr identity, virtually replicates the interaction with Ile202, Ile206, and Trp111. We hypothesize that DD2 inhibits both KSHV Pr and CMV Pr by the same mechanism, and suggest that this allosteric site could be targeted across all HHV protease sub-families to develop a novel and broad-spectrum drug.
From a drug-design viewpoint, drugs targeted to a protein-protein interaction as opposed to an enzyme active site could be less prone to drug resistant mutations. This is because acquiring a drug-resistant point mutation, unaccompanied by the right compensatory mutations, is less likely to be tolerated by a complex protein-protein interface. This adaptability is highly dependant on the natural prevalence of amino acid variation across protease residues. For the extensively catalogued Human Immunodeficiency Virus Type 1 (HIV-1) protease the rate of natural variation at its β-sheet dimer interface is highly uncommon44. Furthermore, given the past challenges of developing therapies aimed at the active site of HHV proteases, our findings introduce a new approach to this previously validated target. In terms of drug accessibility, targeting the inactive protease monomer is especially attractive for HHVs. While the active dimer acts on capsid formation in the host nucleus, the inactive monomer, which exhibits a high in vitro KD, is believed to exist in the host cytosol as a mechanism for concentration-dependant zymogen activation24,35.
By identifying allosteric regulatory sites in KSHV Pr we have gained a better understanding of its structure and discovered additional avenues for inhibition. In other enzyme targets such as caspase-3 and caspase-7, the disulfide trapping approach was used to identify allosteric ligands that bind to the intact dimer interface and trap the enzymes in their zymogen conformation45,46. Similarly, in the case of HIV-1 protease, crosslinked interface peptides and derivatives were developed that bind to the antiparallel β-sheet interface, sterically inhibiting dimerization and thereby formation of the neighboring active site47,48. We have identified a novel inhibitor of HHV proteases that acts by combining both mechanisms. This approach can potentially be used for developing novel inhibitors for HHV proteases as well as other systems involving a similar protein-protein interaction motif.
Methods
Protein Expression and Purification
Wild-type and M197L KSHV Pr were prepared as described previously35,49. All protease constructs carry the S204G mutation, which prevents autolysis while maintaining other proteolytic functions. The W109A construct was created with the QuikChange® Multi Site-Directed Mutagenesis Kit (Stratagene). KSHV Pr wild-type (S204G) construct was used as the parent template for mutagenesis using primer: 5’-ggagatactccacacggcgctcccggggctgtc-3’ (Integrated DNA Technologies, Inc.). Protein expression and purification was carried out following the same protocol as that of wild-type protease. CMV Pr protease was expressed following a previously described protocol50. All kinetic assays were performed using KSHV Pr assays and reagents.
Selectively 13C-methionine labeled wild-type KSHV Pr and uniformly 15N-labeled monomeric KSHV Pr M197D was expressed in M9 minimal media and purified as previously described27. In the former, 250 mg 13C-labeled methionine (Cambridge Isotope Laboratories) was added to 1 L minimal media 1 hour prior to IPTG induction. Final sample concentrations were quantified by measuring UV absorbance at 280 nm using a predicted extinction coefficient of 23950 M-1 cm-1 (http://ca.expasy.org/tools/protparam.html).
Kinetic Assays
Helical mimetic screen
Protease was diluted to a final concentration of 2 μM in assay buffer (25 mM potassium phosphate pH 8, 150 mM KCl, 0.1 mM EDTA, and 1mM DTT) and 198 μl was dispensed in each well of a black round-bottom 96-well plate (Corning). 1 μl of compound, provided as 2 mM DMSO stocks, was transferred to each well for a final compound concentration of 10 μM. Following a 30-minute incubation at 30°C, enzyme activity was initiated by adding 1 μl fluorogenic hexapeptide substrate to a final concentration of 160 μM. The substrate (also referred to as P6) 7-amino-4-carbamoylmethyl coumarin (ACC) coupled to Ac-Pro-Val-Tyr-tert-butylglycine-Gln-Ala (Ac-PVYtQA-ACC), was prepared as described previously38,39. Enzyme activity was monitored for 1 hour at 30°C on the Spectra MAX Gemini EM Fluorescence Microplate Reader (Molecular Devices) using excitation and emission wavelength of 380 nm and 460 nm, respectively. Hits were identified as compounds that reduced the initial reaction velocity by at least 50% when compared to control (uninhibited) reaction. The final reaction volume and DMSO concentration were 200 μl and 1% respectively.
IC50 determination
For each compound a 3-fold dilution series DMSO stock was prepared in a concentration range of 10 mM – 0.014 mM. 1 μl of each compound dilution was added to 98ul of 1 μM KSHV Pr (wild-type or M197L) in assay buffer to give a final compound concentration of 100 μM – 0.14 μM. Following a 30-minute incubation at 30°C, enzyme activity was initiated by adding P6 substrate to a final concentration of 100 μM. Endpoint fluorescence was recorded after a 1 hour incubation at 30°C (reaction progress curves were linear at the time of measurement). The final reaction volume and DMSO concentration were 100 μl and 2% respectively. The ratio F/F0 was plotted as a function of inhibitor concentration and fitted to equation (1) using KaleidaGraph® (Synergy Software). F and F0 correspond to the endpoint fluorescence measurement of the experimental reaction and the endpoint fluorescence measurement of the control (uninhibited) reaction, respectively. Y, M0, M1, M2, M3, and M4 correspond to F/F0, compound concentration, maximum endpoint fluorescence, minimum endpoint fluorescence, IC50, and Hill slope, respectively. All IC50 values represents mean values ± s.d. (n=3).
Inhibition kinetics
A standard 100 μl enzyme assay was performed using 1 μM KSHV Pr in assay buffer. Final substrate concentrations of 80, 40, 30, 20, 10, 6, 4, and 2 μM were tested across inhibitor concentrations of 10, 7, 5, 3, 1, and 0 μM (48 total reactions). Initial reaction velocity V0 in units of rfu/sec was converted to μM substrate/sec using the conversion factor (2364 rfu) / (μM substrate). Double-reciprocal plots were fit to a linear equation to determine the KM and Vmax. The ratio Vmax / [dimer] was used to calculate kcat, where the dimer concentration was determined to be 0.26 μM using equation (2). KD is the dimerization constant for KSHV Pr and Et is the total enzyme concentration. Data represents mean values ± s.d. (n=3).
Gel Filtration
Size exclusion chromatography was performed using a ÄKTAexplorer™ (GE Healthcare) equipped with Superdex®75 column. KSHV Pr diluted to 5 μM in assay buffer (25 mM potassium phosphate pH 8, 150 mM KCl, 0.1 mM EDTA, and 1mM DTT) was combined with 30 μM compound and incubated at 30°C for 30 minutes. The final reaction volume and DMSO concentration were 200 μl and 1% respectively. Sample was then loaded onto a gel filtration column pre-equilibrated with 30 μM compound in assay buffer at room temperature. In control experiments, DMSO was used instead of compound.
NMR data acquisition
All spectra were acquired on cryoprobe-equipped Varian Inova 600 MHz or Bruker Avance 800 MHz spectrometers at 27 °C. Typical NMR samples used for the 1H-13C-HSQC dimer disruption assays consisted of 0.02 – 0.03 mM selectively labeled with 13C-methionine wild-type KSHV Pr in 0.45 – 0.50 mL buffer. Standard assay buffer consisted of 25 mM phosphate buffer (pH 8.0), 150 mM NaCl, 0.1 mM EDTA, 5 mM DTT, and 10% (v/v) 2H2O. Powdered inhibitor was first dissolved in d6-DMSO (Cambridge Isotope Laboratories) to create a 10 mM solution, then serially diluted to 5 mM, 1 mM and 0.5 mM stocks. Titrations were performed by adding variable μL aliquots of inhibitor stocks in a step-wise manner until 2 – 4 molar equivalents were reached.
Sample concentrations used for the 1H-15N-HSQC backbone mapping of the monomeric KSHV Pr M197D-dimer disruptor interactions were 0.15 – 0.20 mM uniformly 15N-labeled protease in 0.45 – 0.50 mL buffer. Due to significant solvent exchange peak broadening observed at pH 8.0, NMR samples were buffer exchanged to 25 mM phosphate buffer (pH 7.0) using Amicon Ultra 10K Centrifugal Filter Devices (Millipore Corp.). Buffer conditions and titration methodologies were otherwise identical to those used for the 13C-methionine HSQC assays. Additional experimental details are described in Supplementary Methods online.
Supplementary Material
Acknowledgments
The authors thank W. Gibson, Department of Pharmacology and Molecular Sciences at The Johns Hopkins University School of Medicine, for providing us with the CMV protease expression plasmid. This work was funded by NIH grants T32 GMO7810, AIO67423 (C.S.C.), and by the American Lebanese and Syrian Associated Charities and St Jude Children’s Research Hospital (R.K.G.). We also thank UCSF-Gladstone Institute for Virology & Immunology/Center for AIDS Research Clinical Science Pilot Award P30-AI027763 (G.M.L.).
Footnotes
Author Contributions T.S., G.M.L., A.L., C.S.C., and R.K.G. designed research; T.S., G.M.L., and A.L. carried out research; L.A.A., P.V., and C.M.R. performed chemical synthesis; T.S., G.M.L., A.L., C.S.C., and R.K.G. analyzed and interpreted data; and T.S., C.S.C. and R.K.G. prepared the manuscript.
Competing Financial Interests: Authors declare no conflict of interest.
References
- 1.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 3.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–98. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 4.Berg T. Modulation of protein-protein interactions with small organic molecules. Angew Chem Int Ed Engl. 2003;42:2462–81. doi: 10.1002/anie.200200558. [DOI] [PubMed] [Google Scholar]
- 5.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–9. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 6.Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 7.Fields BN, et al. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2006. p. 3177. [Google Scholar]
- 8.Gopalsamy A, et al. Design and syntheses of 1,6-naphthalene derivatives as selective HCMV protease inhibitors. J Med Chem. 2004;47:1893–9. doi: 10.1021/jm030540h. [DOI] [PubMed] [Google Scholar]
- 9.Gao M, et al. The protease of herpes simplex virus type 1 is essential for functional capsid formation and viral growth. J Virol. 1994;68:3702–12. doi: 10.1128/jvi.68.6.3702-3712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preston VG, Coates JA, Rixon FJ. Identification and characterization of a herpes simplex virus gene product required for encapsidation of virus DNA. J Virol. 1983;45:1056–64. doi: 10.1128/jvi.45.3.1056-1064.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheaffer AK, et al. Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J Virol. 2000;74:6838–48. doi: 10.1128/jvi.74.15.6838-6848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinheimer SP, et al. Autoproteolysis of herpes simplex virus type 1 protease releases an active catalytic domain found in intermediate capsid particles. J Virol. 1993;67:5813–22. doi: 10.1128/jvi.67.10.5813-5822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welch AR, Woods AS, McNally LM, Cotter RJ, Gibson W. A herpesvirus maturational proteinase, assemblin: identification of its gene, putative active site domain, and cleavage site. Proc Natl Acad Sci U S A. 1991;88:10792–6. doi: 10.1073/pnas.88.23.10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borthwick AD, et al. Design and synthesis of pyrrolidine-5,5-trans-lactams (5-oxohexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 2. Potency and chirality. J Med Chem. 2002;45:1–18. doi: 10.1021/jm0102203. [DOI] [PubMed] [Google Scholar]
- 15.Borthwick AD, et al. Pyrrolidine-5,5-trans-lactams as novel mechanism-based inhibitors of human cytomegalovirus protease. Part 3: potency and plasma stability. Bioorg Med Chem Lett. 2002;12:1719–22. doi: 10.1016/s0960-894x(02)00294-9. [DOI] [PubMed] [Google Scholar]
- 16.Borthwick AD, et al. Design and synthesis of monocyclic beta-lactams as mechanism-based inhibitors of human cytomegalovirus protease. Bioorg Med Chem Lett. 1998;8:365–70. doi: 10.1016/s0960-894x(98)00032-8. [DOI] [PubMed] [Google Scholar]
- 17.Ogilvie W, et al. Peptidomimetic inhibitors of the human cytomegalovirus protease. J Med Chem. 1997;40:4113–35. doi: 10.1021/jm970104t. [DOI] [PubMed] [Google Scholar]
- 18.Waxman L, Darke PL. The herpesvirus proteases as targets for antiviral chemotherapy. Antivir Chem Chemother. 2000;11:1–22. doi: 10.1177/095632020001100101. [DOI] [PubMed] [Google Scholar]
- 19.Batra R, Khayat R, Tong L. Molecular mechanism for dimerization to regulate the catalytic activity of human cytomegalovirus protease. Nat Struct Biol. 2001;8:810–7. doi: 10.1038/nsb0901-810. [DOI] [PubMed] [Google Scholar]
- 20.Buisson M, et al. Functional determinants of the Epstein-Barr virus protease. J Mol Biol. 2001;311:217–28. doi: 10.1006/jmbi.2001.4854. [DOI] [PubMed] [Google Scholar]
- 21.Darke PL, et al. Active human cytomegalovirus protease is a dimer. J Biol Chem. 1996;271:7445–9. doi: 10.1074/jbc.271.13.7445. [DOI] [PubMed] [Google Scholar]
- 22.Margosiak SA, Vanderpool DL, Sisson W, Pinko C, Kan CC. Dimerization of the human cytomegalovirus protease: kinetic and biochemical characterization of the catalytic homodimer. Biochemistry. 1996;35:5300–7. doi: 10.1021/bi952842u. [DOI] [PubMed] [Google Scholar]
- 23.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. Induced structure of a helical switch as a mechanism to regulate enzymatic activity. Nat Struct Mol Biol. 2005;12:1019–20. doi: 10.1038/nsmb1006. [DOI] [PubMed] [Google Scholar]
- 24.Pray TR, Reiling KK, Demirjian BG, Craik CS. Conformational change coupling the dimerization and activation of KSHV protease. Biochemistry. 2002;41:1474–82. doi: 10.1021/bi011753g. [DOI] [PubMed] [Google Scholar]
- 25.Reiling KK, Pray TR, Craik CS, Stroud RM. Functional consequences of the Kaposi’s sarcoma-associated herpesvirus protease structure: regulation of activity and dimerization by conserved structural elements. Biochemistry. 2000;39:12796–803. doi: 10.1021/bi001019h. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt U, Darke PL. Dimerization and activation of the herpes simplex virus type 1 protease. J Biol Chem. 1997;272:7732–5. doi: 10.1074/jbc.272.12.7732. [DOI] [PubMed] [Google Scholar]
- 27.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. One functional switch mediates reversible and irreversible inactivation of a herpesvirus protease. Biochemistry. 2006;45:3572–9. doi: 10.1021/bi0523658. [DOI] [PubMed] [Google Scholar]
- 28.Marnett AB, Nomura AM, Shimba N, Ortiz de Montellano PR, Craik CS. Communication between the active sites and dimer interface of a herpesvirus protease revealed by a transition-state inhibitor. Proc Natl Acad Sci U S A. 2004;101:6870–5. doi: 10.1073/pnas.0401613101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buisson M, et al. The crystal structure of the Epstein-Barr virus protease shows rearrangement of the processed C terminus. J Mol Biol. 2002;324:89–103. doi: 10.1016/s0022-2836(02)01040-9. [DOI] [PubMed] [Google Scholar]
- 30.Qiu X, et al. Unique fold and active site in cytomegalovirus protease. Nature. 1996;383:275–9. doi: 10.1038/383275a0. [DOI] [PubMed] [Google Scholar]
- 31.Qiu X, et al. Crystal structure of varicella-zoster virus protease. Proc Natl Acad Sci U S A. 1997;94:2874–9. doi: 10.1073/pnas.94.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shieh HS, et al. Three-dimensional structure of human cytomegalovirus protease. Nature. 1996;383:279–82. doi: 10.1038/383279a0. [DOI] [PubMed] [Google Scholar]
- 33.Tong L, et al. A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature. 1996;383:272–5. doi: 10.1038/383272a0. [DOI] [PubMed] [Google Scholar]
- 34.Hoog SS, et al. Active site cavity of herpesvirus proteases revealed by the crystal structure of herpes simplex virus protease/inhibitor complex. Biochemistry. 1997;36:14023–9. doi: 10.1021/bi9712697. [DOI] [PubMed] [Google Scholar]
- 35.Pray TR, Nomura AM, Pennington MW, Craik CS. Auto-inactivation by cleavage within the dimer interface of Kaposi’s sarcoma-associated herpesvirus protease. J Mol Biol. 1999;289:197–203. doi: 10.1006/jmbi.1999.2791. [DOI] [PubMed] [Google Scholar]
- 36.Shimba N, Nomura AM, Marnett AB, Craik CS. Herpesvirus protease inhibition by dimer disruption. J Virol. 2004;78:6657–65. doi: 10.1128/JVI.78.12.6657-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu F, et al. Proteomimetic libraries: design, synthesis, and evaluation of p53-MDM2 interaction inhibitors. J Comb Chem. 2006;8:315–25. doi: 10.1021/cc050142v. [DOI] [PubMed] [Google Scholar]
- 38.Backes BJ, Harris JL, Leonetti F, Craik CS, Ellman JA. Synthesis of positional-scanning libraries of fluorogenic peptide substrates to define the extended substrate specificity of plasmin and thrombin. Nat Biotechnol. 2000;18:187–93. doi: 10.1038/72642. [DOI] [PubMed] [Google Scholar]
- 39.Lazic A, Goetz DH, Nomura AM, Marnett AB, Craik CS. Substrate modulation of enzyme activity in the herpesvirus protease family. J Mol Biol. 2007;373:913–23. doi: 10.1016/j.jmb.2007.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1:550–3. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–5. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kussie PH, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–53. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 43.Chen P, et al. Structure of the human cytomegalovirus protease catalytic domain reveals a novel serine protease fold and catalytic triad. Cell. 1996;86:835–43. doi: 10.1016/s0092-8674(00)80157-9. [DOI] [PubMed] [Google Scholar]
- 44.Wu TD, et al. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–47. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardy JA, Lam J, Nguyen JT, O’Brien T, Wells JA. Discovery of an allosteric site in the caspases. Proc Natl Acad Sci U S A. 2004;101:12461–6. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheer JM, Romanowski MJ, Wells JA. A common allosteric site and mechanism in caspases. Proc Natl Acad Sci U S A. 2006;103:7595–600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bannwarth L, Reboud-Ravaux M. An alternative strategy for inhibiting multidrug-resistant mutants of the dimeric HIV-1 protease by targeting the subunit interface. Biochem Soc Trans. 2007;35:551–4. doi: 10.1042/BST0350551. [DOI] [PubMed] [Google Scholar]
- 48.Lee SG, Chmielewski J. Rapid synthesis and in situ screening of potent HIV-1 protease dimerization inhibitors. Chem Biol. 2006;13:421–6. doi: 10.1016/j.chembiol.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 49.Unal A, et al. The protease and the assembly protein of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) J Virol. 1997;71:7030–8. doi: 10.1128/jvi.71.9.7030-7038.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brignole EJ, Gibson W. Enzymatic activities of human cytomegalovirus maturational protease assemblin and its precursor (pPR, pUL80a) are comparable: [corrected] maximal activity of pPR requires self-interaction through its scaffolding domain. J Virol. 2007;81:4091–103. doi: 10.1128/JVI.02821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.