

Abstract
The unique energy demands of neurons require well-orchestrated distribution and maintenance of mitochondria. Thus, dynamic properties of mitochondria, including fission, fusion, trafficking, biogenesis, and degradation, are critical to all cells, but may be particularly important in neurons. Dysfunction in mitochondrial dynamics has been linked to neuropathies and is increasingly being linked to several neurodegenerative diseases, but the evidence is particularly strong, and continuously accumulating, in Parkinson's disease (PD). The unique characteristics of neurons that degenerate in PD may predispose those neuronal populations to susceptibility to alterations in mitochondrial dynamics. In addition, evidence from PD-related toxins supports that mitochondrial fission, fusion, and transport may be involved in pathogenesis. Furthermore, rapidly increasing evidence suggests that two proteins linked to familial forms of the disease, parkin and PINK1, interact in a common pathway to regulate mitochondrial fission/fusion. Parkin may also play a role in maintaining mitochondrial homeostasis through targeting damaged mitochondria for mitophagy. Taken together, the current data suggests that mitochondrial dynamics may play a role in PD pathogenesis, and a better understanding of mitochondrial dynamics within the neuron may lead to future therapeutic treatments for PD, potentially aimed at some of the earliest pathogenic events.
Keywords: Parkinson disease, Parkinson's disease, mitochondria, mitochondrial dynamics, mitochondrial fission, mitochondrial fusion, parkin, PINK1, mitophagy
Parkinson's disease and mitochondrial involvement
Parkinson disease (PD) is the second most common neurodegenerative disorder, and the most common neurodegenerative movement disorder. Clinically, it consists of progressive rigidity, bradykinesia, tremor, and gait abnormalities, as well as symptoms involving non-motor brain functions, including cognitive and autonomic functions (Barbas, 2006; Pallone, 2007; Weintraub et al., 2008; Weintraub and Stern, 2005). Pathologically, it is classically characterized by the loss of pigmented dopaminergic neurons in the substantia nigra in the midbrain and the presence of proteinaceous cytoplasmic inclusions called Lewy bodies (see Samii et al., 2004). While loss of the nigral dopaminergic neurons and terminals are responsible for the movement disorders associated with PD, it has become clear that additional neuronal populations throughout the brain are also affected in the disease (Braak et al., 2004).
Over the last several decades, evidence has accumulated that mitochondrial dysfunction is associated with PD. This evidence has been detailed in numerous comprehensive reviews (e.g. see Banerjee et al., 2008; Beal, 2003; Beal, 2007; Orth and Schapira, 2002; Schapira, 2008), and will only be briefly summarized here. A mild deficiency in mitochondrial respiratory electron transport chain NADH dehydrogenase (Complex I) activity was first found in the substantia nigra of patients with PD (Mann et al., 1994; Schapira et al., 1989), followed by studies identifying a similar Complex I deficit in platelets (Blandini et al., 1998; Haas et al., 1995; Krige et al., 1992; Parker et al., 1989), lymphocytes (Barroso et al., 1993; Yoshino et al., 1992), and, less consistently, in muscle tissue (Penn et al., 1995; Taylor et al., 1994) from PD patients. These results suggest there is a systemic, low-grade inhibition of Complex I activity associated with PD. Consistent with this, chronic systemic administration of rotenone, a specific Complex I inhibitor and pesticide, results in neuropathologic and behavioral changes in rats that is similar to human PD (Alam and Schmidt, 2002; Betarbet et al., 2000; Fleming et al., 2004; Sherer et al., 2003). MPTP, a meperidine analog found to cause parkinsonism in humans, has been found to exert its toxic effects through metabolism to 1-methyl-4-phenylpyridinium (MPP+), another Complex I inhibitor (Langston et al., 1999; Dauer and Przedborski, 2003). These compounds have long been used as animal models of PD.
In addition, oxidative stress has been linked to the neurodegeneration of PD, and alterations in levels of antioxidants and oxidized targets have been reported in PD (see Jenner, 2003). As the mitochondrial electron transport chain is a known source of reactive oxygen species generation, and inhibition of mitochondrial complexes can increase production of free radicals (Fiskum et al., 2003; Orth and Schapira, 2002; Turrens, 2003), this has lent further evidence to a role for mitochondrial dysfunction in PD. Mitochondrial protein expression and/or abundance have also been found to be altered in PD. A recent proteomic analysis of mitochondria-enriched fractions from post mortem PD substantia nigra revealed differential expression of multiple mitochondrial proteins in PD brain as compared to control, including subunits of Complex I, mitochondrial creatine kinase, and the chaperone mortalin/GRP75/mtHSP70 (Jin et al., 2006). Also, decreased immunostaining for mitochondrial alpha-ketoglutarate, a protein of the tricarboxylic acid cycle, was noted in post mortem PD brain (Mizuno et al., 1994). While cause and effect is difficult to determine in post mortem studies, these findings are suggestive that mitochondrial function may be altered in PD.
More recently, the identification of familial forms of PD caused by genetic mutations has further supported a critical role for mitochondrial function. PD-linked mutations have been identified in genes encoding both mitochondrially targeted proteins and proteins involved in mitochondrial function and/or oxidative stress responses, including mutations in PINK-1, DJ-1, parkin, and possibly Omi/HtrA2 (see Thomas and Beal, 2007, and discussed in more detail below). Together, the body of evidence suggests an important role for mitochondria in PD pathogenesis, yet detailed exploration of the mechanisms involved is still ongoing.
Mitochondrial dynamics and neurodegeneration
Mitochondrial dynamics
Mitochondria are dynamic organelles, and these dynamic functions are emerging as critical to neuronal function and neurodegeneration. Mitochondria actively divide (fission), fuse with one another (fusion) and, in neurons, are actively transported throughout axons and dendrites (Hollenbeck and Saxton, 2005; Okamoto and Shaw, 2005). Mitochondrial fission and fusion are important in maintaining integrity of mitochondria, electrical and biochemical connectivity, in turnover of mitochondria, and in segregation, stabilization, and protection of mitochondrial DNA (mtDNA) (Westermann, 2002). In addition to normal mitochondrial maintenance functions, mitochondrial fission, as well as mitochondrial fusion factors, have been found to play a critical role in apoptotic mechanisms as well (reviewed in (Suen et al., 2008).
Mediators of mitochondrial fission and fusion are best described in yeast, but many yeast mediators have mammalian homologues (Okamoto and Shaw, 2005). In mammalian cells, the GTPase Dynamin-related protein 1 (Drp1; yeast Dnm1) translocates to the outer mitochondrial membrane, and in conjunction with outer membrane proteins such as hFis1 as well as others, mediates mitochondrial fission (Hoppins et al., 2007). Mitochondrial fusion is mediated in mammalian cells in part by the outer membrane dynamin-like GTPases mitofusin-1 and -2 (Mfn1 and Mfn2) and the inner membrane optic atrophy protein (Opa1) (Cerveny et al., 2007; Detmer and Chan, 2007).
Importance of mitochondrial dynamics in neurons
Whereas mitochondrial fission and fusion have critical functions in all cells, these dynamic processes may be of particular importance in neurons, perhaps due to unique features such as their post-mitotic state and their long processes with higher energy requirements. Along these lines, specific mutations in fusion genes result in human neuropathies (Alexander et al., 2000; Delettre et al., 2000; Zuchner et al., 2004), while mutations in both fission and fusion genes have been associated with central nervous system disease (Brockmann et al., 2008; Waterham et al., 2007). In neurons, the mitochondrial fission/fusion machinery is intimately and critically involved in formation of synapses and dendritic spines. Preventing mitochondrial fission leads to a loss of mitochondria from dendritic spines and a reduction of synapse formation, whereas increasing fission increases synapse formation (Li et al., 2004). In addition, the absence of the mitochondrial fission protein Drp1 has been shown to prevent mitochondria from distributing to synapses and to lead to synaptic dysfunction (Verstreken et al., 2005).
In addition, maintenance of mtDNA cannot take place without mitochondrial fusion (Rapaport et al., 1998), and fusion of mitochondria has been directly implicated in preventing the accumulation of damaged mtDNA (Nakada et al., 2001; Ono et al., 2001). More recently, mitochondrial fission has also been shown to be critical to mtDNA maintenance (Parone et al., 2008), suggesting that fission and fusion may function in a coordinated manner to maintain and protect mtDNA. Since mtDNA mutations accumulate in the brain with age (Corral-Debrinski et al., 1992), these functions may have particular importance in neurons and neurodegenerative disease.
Also important in proper mitochondrial localization, maintenance, and function, and specific to neurons, is the active transport of mitochondria through axons and dendrites to sites of increased energy requirement. Mitochondria are transported in both anterograde and retrograde directions, by a process regulated in part by cytoskeletal and mitochondrial proteins and by extrinsic signaling mechanisms, including growth factor signaling (Hollenbeck and Saxton, 2005). In addition, mitochondrial movement is affected by several intrinsic components of mitochondrial bioenergetics, including ATP synthesis and changes in mitochondrial membrane potential (Rintoul et al., 2003; Miller and Sheetz, 2004), suggesting links between mitochondrial bioenergetic functions and mitochondrial transport and distribution. In Drosophila, specific genetic disruption of mitochondrial transport has been shown not only to lead to the absence of mitochondria at nerve terminals but also to result in synaptic dysfunction (Stowers et al., 2002). Disruption of mitochondrial movement has been noted in excitotoxicity and oxidative stress (Rintoul et al., 2003), two factors implicated in cellular stress and neuronal death.
Mitochondrial dynamics in neurodegenerative diseases other than PD
As briefly mentioned above, mutations in the GTPase proteins comprising the mitochondrial fusion machinery are directly linked to neuropathy and neurodegeneration. Mutations in OPA1 result in autosomal dominant optic atrophy, or ADOA, a progressive degeneration of the optic nerve and retinal ganglion (Alexander et al., 2000; Delettre et al., 2000), whereas mfn-2 mutations are linked to the peripheral neuropathy Charcot-Marie-Tooth subtype 2A, which involves axonal degeneration of motor and sensory neurons (Zuchner et al., 2004). In addition, mfn-2 mutations may cause central nervous system white matter disease (Brockmann et al., 2008). Recently, a mutation in the fission gene drp1 was found in an infant with lethal abnormal brain development, emphasizing the importance of the fission machinery in neuronal maintenance (Waterham et al., 2007).
In addition to the associations of genetic diseases of the fission/fusion machinery with neurodegeneration, evidence is suggesting possible involvement of mitochondrial dynamics in the pathogenesis of several chronic neurodegenerative diseases of aging. While this review focuses on PD, mitochondrial dysfunction has also been associated with Alzheimer's disease (AD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) (see Beal, 2007; Kwong et al., 2006), and there is evidence that the dynamic functions of mitochondria may be involved.
Much of the evidence in AD, HD, and ALS relies on observations of morphologic changes in mitochondria. In an animal model of ALS, for example, expression of the mutant (G93A) human SOD1 in NSC-34 motoneuronal-like cells resulted in a fragmented mitochondrial network and mitochondrial swelling, though the mechanism is unknown (Raimondi et al., 2006).
In AD models, the amyloid beta peptide (Aβ), a component of AD-related neurodegenerative plaques, has been shown to induce increased mitochondrial fragmentation in rat cortical neurons (Barsoum et al., 2006). Also, overexpression of the amyloid precursor protein (APP) and the familial AD-causing APPsw mutation can lead to increased fragmentation and perinuclear aggregation of mitochondria in M17 cells (Wang et al., 2008b). APP and an Aβ-derived diffusible ligand had similar effects on the mitochondria of primary hippocampal neurons (Wang et al., 2008b).
With regard to HD, a neurodegenerative disorder of striatal neurons caused by a triplication repeat mutation in the huntington gene (Htt), aggregates of mutant Htt protein were found to impair trafficking and mobility of mitochondria in rat primary cortical neurons (Chang et al., 2006), possibly by blocking trafficking pathways in the neuron. Recent findings also suggest that mutant Htt can impact mitochondrial fission and fusion. Wang et al. (2008b) demonstrated that HeLa cells expressing mutant Htt exhibited mitochondria with reduced movement and increased Drp1-dependent fragmentation. Using a transgenic C. elegans model of HD, the authors found that RNAi-mediated knock-down of Drp1 rescued the mutant Htt-induced motility defect in the worms, suggesting Htt-mediated dysfunction can be rescued by preventing fragmentation of mitochondria (Wang et al., 2008a).
Mitochondrial dynamics and PD: Why might mitochondrial dynamics be particularly important in PD?
Although there is increasing evidence linking mitochondrial dynamics to many neurodegenerative diseases, we will detail below that the evidence is particularly strong, and rapidly accumulating, in PD. There are several features of the pathogenesis of PD that may help to explain this.
Selective vulnerability in PD
One key question is why only specific sets of neurons die in PD. Interestingly, several features of PD neurodegeneration suggest that mitochondrial dynamics could be important. It has become clear that the degeneration of PD affects not only dopaminergic neurons, but many other neurons in the CNS, including populations in the brainstem as well as in subcortical and cortical regions (Braak et al., 2003). What makes these particular sets of disparate neurons selectively vulnerable in PD is not known, but may be key to understanding the underlying mechanisms of neurodegeneration in PD. Strikingly, they have in common two features: they have long and thin axons, and the axons have little or no myelination (Braak et al., 2004). The high energy requirement of these selectively vulnerable neurons, and the long distance between axon terminals and cell bodies, is likely to provide clues to the underlying mechanisms involved. Neurons with these features are likely to be particularly dependent on proper mitochondrial dynamics. In addition, Liang et al. (2007) recently found that the cytoplasmic area occupied by mitochondria in the dopaminergic neurons in the substantia nigra (susceptible to PD degeneration) is lower than in neighboring non-dopaminergic neurons or in dopaminergic neurons of the ventral tegmental area (resistant in PD), suggesting the possibility that the vulnerable neurons may be more susceptible to subtle changes in mitochondrial maintenance.
It is also generally believed that the degenerative process in PD begins at the terminals rather than the cell body (e.g., see Braak et al., 2004). Whether ‘classic’ apoptotic programmed cell death mechanisms occur in PD is still controversial, and there is evidence that axonal/dendritic degenerative mechanisms may be different than the classic mechanisms of programmed cell death (Finn et al., 2000; Raff et al., 2002; Whitmore et al., 2003). Given the importance of mitochondrial dynamics for mitochondrial distribution to synapses, synaptic function, and overall mitochondrial function, dysfunction in these dynamic processes could be an early step in PD neurodegeneration.
Additionally, as detailed above, mitochondrial fusion and fission maintains mtDNA stability and perhaps helps to reduce the burden of damaged mtDNA. PD is a disease of aging brain, and mtDNA damage has been found to increase with age (Corral-Debrinski et al., 1992). Evidence regarding mtDNA damage in the etiology of PD is still conflicting, but several studies have suggested an association with mtDNA mutations and PD (e.g. Parker and Parks, 2005; Winkler-Stuck et al., 2005). In addition, cybrid cell lines with normal nuclear genomes but mtDNA from PD patients have been reported to exhibit a Complex I deficit, a higher sensitivity to MPP+ (the toxic metabolite of MPTP), and generation of Lewy body-like inclusions, suggesting that possible genetic defects in mtDNA genes encoding Complex I subunits are associated with PD pathogenesis (Sheehan et al., 1997; Swerdlow et al., 1996; Trimmer et al., 2004). Thus, it is possible that mitochondrial fusion-related stabilization of mtDNA integrity is an important modulator in pathogenesis of PD.
Toxin models of PD and mitochondrial dynamics
Evidence from toxin-induced PD models supports a role for mitochondrial fission/fusion and transport. Exposure to sublethal concentrations of the PD-related toxin rotenone, the mitochondrial electron transport chain Complex I inhibitor, resulted in profound mitochondrial morphologic changes in HeLa cells, causing mitochondria to become more ‘donut’-shaped (Benard et al., 2007). In human fibroblasts, acute exposure to high-dose rotenone resulted in decreased mitochondrial membrane potential and caused fragmented morphology (Mortiboys et al., 2008). In cultured rat cortical neurons, acute rotenone exposure was found to cause rapid mitochondrial fragmentation and cell death in a manner dependent on the fission protein Drp1 (Barsoum et al., 2006). Similar results were obtained from rat-derived dopaminergic N27 cells exposed to rotenone or MPP+ (Barsoum et al., 2006). These studies suggest that rotenone influences maintenance of mitochondrial morphology, and that, at least for acute toxicity, this may involve the mitochondrial fission machinery.
Using real-time microscopy, rotenone-induced swelling and decreased motility of mitochondria in fibroblasts was also observed (Pham et al., 2004). In addition, chronic exposure to rotenone was reported to reduce mitochondrial movement in differentiated dopaminergic SH-SY5Y cells (Borland et al., 2008). Together, these results suggest that PD-related toxins exert effects on fission/fusion and transport.
Genetics evidence of mitochondrial involvement in PD: PINK1 and parkin
The strongest evidence of a role for mitochondrial dynamics in PD, however, is rapidly emerging from studies of several genes whose mutated forms are associated with familial PD.
Parkin and mitochondrial function and morphology
Mutations in the parkin gene (PARK2) are associated with autosomal recessive inheritance of juvenile-onset PD (Kitada et al., 1998). The gene codes for parkin, a cytosolic ubiquitin E3 ligase protein that plays a role in the ubiquitin-dependent proteasome pathway (Shimura et al., 2000). Parkin loss-of-function mutants in Drosophila exhibited increased sensitivity to oxygen radical stress, dopaminergic cell loss, and severe mitochondrial damage in muscle and germline tissues that included swollen mitochondria and fragmented cristae (Greene et al., 2003; Pesah et al., 2004). It was later found that functional parkin was necessary for proper mitochondrial organization and morphology throughout spermatid development in Drosophila (Riparbelli and Callaini, 2007). Further, Drosophila overexpressing one pathogenic parkin mutation, R275W, were found to exhibit dopaminergic degeneration and mitochondrial abnormalities similar to parkin knockout flies, including mitochondria with large vacuoles, concentric membranous structures, disorganized cristae, or degenerated membranes (Wang et al., 2007). These studies suggest parkin plays a key role in maintaining mitochondrial stability in certain cell types.
Additionally, mitochondrial respiratory defects and morphological abnormalities have been noted in brains of parkin-knockout and parkin-mutant transgenic mice (Palacino et al., 2004; Stichel et al., 2007) and in leukocytes from PD patients with parkin mutations (Mortiboys et al., 2008; Muftuoglu et al., 2004). Primary fibroblasts from patients carrying mutations in parkin, or control fibroblasts treated with siRNA against parkin, exhibited lower mitochondrial membrane potential, Complex I activity, lower ATP levels, decreased complex I-dependent ATP production, and increased susceptibility to rotenone toxicity (Mortiboys et al., 2008). The fibroblasts also exhibited mitochondrial morphological abnormalities, revealing mitochondria that were longer and more highly branched. Interestingly, there was a correlation between decreased Complex I-dependent ATP production and increased length and branching, as well as between Complex I activity and branching. Further, parkin-mutant cells were more susceptible to rotenone toxicity.
As parkin is not specifically a mitochondrial protein and must be translocated to the organelle, its effects on mitochondrial morphology are likely tied into a larger pathway mediating mitochondrial maintenance. Recent studies have not only begun to elucidate such a pathway, but have collectively pointed to mitochondrial dynamics as a critical target, as will be detailed below.
PINK1 and mitochondrial function and morphology
The PTEN-induced putative kinase 1 protein, PINK1, is a nuclear-expressed mitochondrially-targeted kinase. Mutations in the PINK1 (PARK6) gene are associated with autosomal recessive inheritance of PD (Valente et al., 2004), directly linking a mitochondrial protein with PD pathogenesis. In PINK1 knock-out mice, loss of PINK1 significantly impairs mitochondrial respiration selectively in the striatum (Gautier et al., 2008). While no gross changes in the ultrastructure or the total number of mitochondria were observed in the striatum, the number of larger mitochondria was found to be selectively increased.
Several independent research groups developed transgenic Drosophila fruit fly lines that either expressed loss-of-function PINK1 mutants or were PINK1-deficient. In all cases, the flies exhibited increased susceptibility to stress, decreased cellular ATP levels, reduced mtDNA content, and mitochondrial morphological defects (Clark et al., 2006; Park et al., 2006; Yang et al., 2006; Yang et al., 2008). Interestingly, the mitochondrial morphologies and tissues in which they were exhibited were noted to be remarkably similar to those of parkin mutant Drosophila. Specifically, PINK1 mutant flies, like parkin mutant flies, exhibited disorganized indirect flight muscle fibers, swollen mitochondria with disorganized cristae in muscle tissue and dopaminergic neurons, and dopaminergic neuron degeneration (Clark et al., 2006; Park et al., 2006; Yang et al., 2006).
Linking PINK1 and parkin together and to mitochondrial dynamics
Noting the mitochondrial morphologic similarities, the studies mentioned above tested whether ectopic expression of parkin would influence the PINK1 mutant phenotype. In all cases, parkin overexpression completely rescued the effect of the loss of PINK1 activity (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). Interestingly, PINK1 overexpression did not rescue the phenotype of parkin knockout flies, while double PINK1/parkin knockouts exhibited the same level of deficits as either model alone. These studies suggest that the PD-related proteins parkin and PINK1 participate in a related pathway to affect mitochondrial function and stability, in which parkin is downstream of PINK1.
Mitochondrial effects were also observed in HeLa cells in which PINK1 expression was knocked down via siRNA (Exner et al., 2007). In these cells, PINK1 deficiency resulted in mitochondrial fragmentation, disorganized cristae, and functional deficiencies. As in the transgenic Drosophila models, wildtype parkin overexpression, but not PD-related parkin mutants, rescued the phenotype. The added observation of mitochondrial fragmentation suggests that mitochondrial fission/fusion machinery may be involved in the effects of the PINK1/parkin pathway on mitochondria. These results suggest that mutations in parkin may be adversely influencing mitochondrial stability, downstream of PINK1, and may be a pathway by which these proteins contribute to PD pathogenesis.
Reasoning that the mitochondrial morphological deficits may be the result of dysfunctional fission and/or fusion, several research teams investigated the relationship of these two proteins further in the Drosophila transgenic lines. By modulating expression of fission and fusion machinery in the fly lines, it was found that the PINK1/parkin pathway promotes fission and/or inhibits fusion of mitochondria (Deng et al., 2008; Park et al., 2009; Poole et al., 2008; Yang et al., 2008). Specifically, overexpression of Drp1, the mitochondrial fission protein, was protective against the PINK1 or parkin mutant phenotypes, but not overexpression of the inactive form of Drp1. The independent groups also found that loss of fusion machinery, Opa1 or Mfn, (knockdown or mutation) also partially rescued either parkin or PINK1 mutant phenotypes. Park et al. (2009) also showed that overexpression of Opa1 or Mfn alone, presumably upregulating mitochondrial fusion, resulted in mitochondrial swelling, and did not rescue the PINK1 mutant phenotype. However, parkin overexpression did rescue the Opa1 overexpression phenotype. Interestingly, Yang et al (2008) showed that the PINK1/parkin interaction with mitochondrial fusion and fission occurs not only non-neuronal tissues but in dopaminergic neurons as well. Together, these findings illustrate a role for the PINK1/parkin pathway promoting fission and/or inhibiting fusion in Drosophila muscle and neuronal mitochondria.
However, the above findings also create a notable discrepancy between findings in flies and some studies in mammalian cells. As mentioned above, siRNA-knockdown of PINK1 in HeLa cells was observed to increase mitochondrial fragmentation, or presumably promote fission (Exner et al., 2007). Similar results were observed in neuroblastoma cells with siRNA knockdown of PINK1 and parkin (Park et al., 2009) and primary cells cultured from patients carrying PINK1 mutations (Exner et al., 2007; Wood-Kaczmar et al., 2008). Similarly, preliminary work from (Dagda et al., 2008) found that stable PINK1 expression in SH-SY5Y cells increased interconnectivity of mitochondria, while PINK1 RNAi knockdown or expression of kinase-inactive PINK1 increased mitochondrial fragmentation. Although mitochondrial morphology alone does not necessarily predict the expected effects on fission and fusion (Berman et al., 2009), these results would suggest that the PINK1/parkin pathway could also promote fusion and/or inhibit fission in mammalian cells, the exact opposite of the findings from Drosophila models. Conversely, Mortiboys et al. (2008) recently demonstrated that cultured fibroblasts from patients carrying parkin mutations exhibited mitochondrial morphological abnormalities, being longer and more branched than controls. In addition, overexpression of PINK1 in COS-7 cells resulted in punctuate mitochondria, whereas suppression of PINK1 with shRNA resulted in long, tubular mitochondria, a phenotype inhibited by the overexpression of the fission proteins hFis1 or Drp1 (Yang et al., 2008). Thus, these observations would support the findings from the Drosophila models. While clearly, further studies are necessary to understand these conflicts, it is entirely possible that given the dynamic and interrelated regulation between fission and fusion, PINK1/parkin effects may differ depending on the cell type and/or cellular conditions.
What potential mechanisms could explain the effects of PINK1 and parkin on mitochondrial dynamics?
What has been consistently found is that parkin acts downstream of PINK1 in effects on mitochondrial morphology and function. Although the mechanisms of the interaction between PINK1 and parkin and the actions of parkin on mitochondrial dynamics are far from clear, some lines of evidence suggest intriguing possibilities (Summarized in Figure 1).
Figure 1. The emerging role for parkin and PINK1 in mitochondrial dynamics and homeostasis.
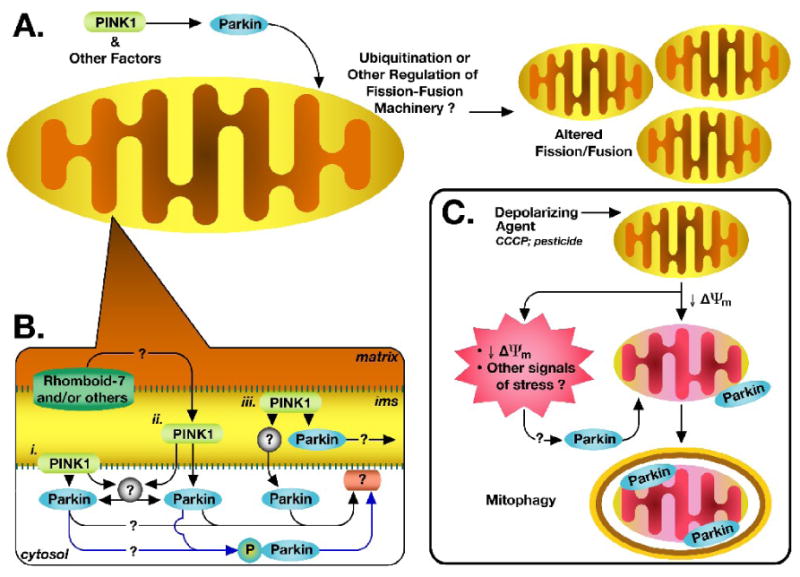
A. Mounting evidence suggests that parkin operates downstream of PINK1 in a common pathway to regulate mitochondrial dynamics. In many cells, it appears to promote fission and/or inhibit fusion, though fission/fusion regulatory effects may differ by cell type or cellular environment. Such regulation may involve parkin-mediated regulation of mitochondrial fission/fusion machinery, either directly or indirectly, as through protein ubiquitination. Regulation may also involve some as yet unidentified parkin-mediated regulatory or protein-interaction pathway. B. How and through what pathway PINK1 and parkin interact is not known. The specific localization of both proteins is subject to controversy. Given current evidence, PINK1 may directly interact with and recruit parkin either, i. at the mitochondrial outer membrane, ii. in the cytosol, following cleavage and/or translocation of PINK1 out of the mitochondria, or iii. within the mitochondria. Alternatively, PINK1 may act through an intermediary (grey circles) to modify and/or recruit parkin to the mitochondria. PINK1 was also shown to directly phosphorylate cytosolic parkin, recruiting it to the mitochondria (blue pathways). However, where and how parkin exerts its effects on the mitochondria, whether inside the mitochondria, at the surface, or by interacting with surface proteins (pink ovoid), is unknown. C. Parkin was recently shown to localize to depolarized mitochondria, targeting them for mitophagy. The signals that target parkin to the damaged mitochondria, however, are not known. Whether this pathway is related to the fission/fusion regulatory functions of the PINK1/parkin pathway is also not known, though mitochondrial fission was previously shown to be an apparent prerequisite for mitophagy. See text for related references. ims = intermembrane space
PINK1 influencing parkin localization?
Because in most cases, parkin appears to be largely cytosolic under basal conditions (Darios et al., 2003; Shimura et al., 1999), it would seem that parkin effects on mitochondrial fission and fusion might occur by 1) parkin translocating to mitochondria and exerting its actions there, or 2) acting in the cytosol on other proteins that then translocate to mitochondria (Figure 1B). There is some evidence that PINK1 might influence parkin localization. In neuroblastoma cells, it was found that overexpression of parkin alone resulted in cytosolic localization, but that co-expression with PINK1 resulted in parkin translocation to mitochondria (Kim et al., 2008). This was dependent on PINK1's kinase activity, since a kinase-dead mutant did not cause parkin translocation. Parkin localization to the mitochondria appeared to be mediated by phosporylation at a linker-region threonine, apparently directly phosphorylated by PINK1 (Kim et al., 2008). Whether this occurs under physiologic conditions is as yet unknown.
For the suggested direct interaction or occur, however, the two proteins must be in proximity to one another. The subcellular localization of PINK1, as well as parkin, is a subject of controversy. There is some evidence that parkin interacts with the outer mitochondrial membrane (Darios et al., 2003), and in some cases is found inside the mitochondria (Kuroda et al., 2006b). PINK1 is a mitochondrially targeted protein possessing a transmembrane domain, and it has been found localized to the inner mitochondrial membrane (Gandhi et al., 2006; Muqit et al., 2006; Pridgeon et al., 2007; Silvestri et al., 2005), the intermembrane space (Plun-Favreau et al., 2007; Pridgeon et al., 2007; Silvestri et al., 2005), and spanning the outer membrane (Gandhi et al., 2006; Zhou et al., 2008), with its kinase domain facing the cytosol (Zhou et al., 2008). PINK1 has also been described in the cytosol (Beilina et al., 2005; Haque et al., 2008; Takatori et al., 2008; Weihofen et al., 2008). There is also the possibility that the two interact via intermediary protein targets. Thus, there are multiple ways parkin and PINK1 may potentially interact, both at the mitochondria and in the cytosol (Figure 1B).
Role of ubiquitin ligase activity of parkin?
Parkin, as an E3 ubiquitin ligase (Shimura et al., 2000), could affect mitochondrial dynamics either by ubiquitination-induced proteasomal degradation or by ubiquitination-mediated cell signaling. Although many potential protein targets of parkin's ubiquitin ligase activity have been identified (Lee and Liu, 2008), no obvious direct connection with factors influencing mitochondrial fission and fusion has been identified. However, other ubiquitin ligases play a critical role in regulating mitochondrial fission, fusion and trafficking (Neutzner et al., 2008). In yeast, an inactive mutant of a homologue of the NEDD4/NEDD-like ubiquitin ligase family induced abnormal mitochondrial morphology, distribution, and inheritance, which was rescued by overexpression of ubiquitin (Fisk and Yaffe, 1999). In addition, outer mitochondrial membrane-located E3 ubiquitin ligases, MARCH-V/MITOL, MULAN, and MRF1, have been recently shown to influence mitochondrial membrane dynamics in mammalian cells (Karbowski et al., 2007; Li et al., 2008; Nakamura et al., 2006; Neutzner et al., 2008; Yonashiro et al., 2006). Mitochondrial fission and fusion factors, such as the fission factors Drp1 and hFis1, have been shown to be directly ubiquitinated by some of these E3 ligases (Nakamura et al., 2006; Yonashiro et al., 2006), and there is evidence that mitochondrial fission/fusion proteins might be degraded by the ubiquitin proteasomal pathway (Neutzner et al., 2008). However, E3 ligase-related activity does not always result in changes in levels of fission/fusion factors, suggesting that ubiquitination as a cell-signaling, rather than degradation, mechanism could be occurring (Karbowski et al., 2007). This suggests the possibility that parkin could induce similar regulation of mitochondrial fission and fusion factors after translocation to mitochondria, or potentially, even in the cytosol.
Parkin, mitochondrial dynamics, and mitophagy?
The role of parkin in mitochondrial dynamics may go beyond regulating fission and fusion. A recent elegant study suggests an entirely new role for parkin, targeting dysfunctional mitochondria for autophagic degradation (Narendra et al., 2008). The authors showed that mitochondrial depolarization with the protonophore CCCP resulted in recruitment of parkin to mitochondria in several mammalian cell types, and that subsequently, these mitochondria undergo fission and mitophagy. This is particularly interesting in light of the evidence suggesting that regulated fusion/fission can target mitochondria for degradation (Twig et al., 2008). Although one previous study showed parkin being released nonspecifically from mitochondria after exposure to CCCP in proliferating dopaminergic SH-SY5Y cells (Kuroda et al., 2006a), these new findings suggest the possibility that one of the functions of parkin in mitochondrial dynamics could be to function as a regulator of mitochondrial maintenance and homeostasis, surveying the cell and responding to specific mitochondrial stressors by targeting damaged mitochondria for degradation (Figure 1C). Considering the proposed role for a PINK1/parkin pathway in regulation fission and fusion, it is tantalizing to speculate that the two pathways are linked, and that PINK1 and parkin work in tandem to initiate fission of depolarized mitochondria, and subsequently target any resulting dysfunctional mitochondria for mitophagy. This relationship, however, remains to be explored.
Parkin and PINK1 affecting mitochondrial transport?
Parkin may even influence mitochondrial transport and distribution within the neuron. Parkin was shown to bind to and stabilize microtubules (Yang et al., 2005). Since mitochondrial transport is dependent on the microtubule system in neuronal processes (Hollenbeck and Saxton, 2005), one could hypothesize that parkin, or mutations in parkin, could affect mitochondrial transport throughout the neuron.
New evidence suggests that PINK1 may also have a role in mitochondrial transport. Weihofen et al. (2009) recently reported that PINK1 forms a complex with mitochondrial proteins Miro and Milton, both key in mitochondrial transport. Interestingly, abnormal mitochondrial morphology associated with a loss of functional PINK1 was ameliorated by Milton and Miro overexpression. Thus, dysregulation of proper mitochondrial transport may play a role in the effects of PINK1 mutations on altered mitochondrial morphology and PD-associated pathogenesis.
Other potential protein interactions?
Conflicting evidence examines whether PINK1 participates in a pathway with another putative PD-related protein Omi/HtrA2 (Plun-Favreau et al., 2008). Omi/HtrA2 is a mitochondrial serine protease, and mutations affecting protease activity have been linked to increased risk of PD (PARK13) (Bogaerts et al., 2008; Strauss et al., 2005), though recent evidence has questioned this finding (Ross et al., 2008; Simon-Sanchez and Singleton, 2008). Omi/HtrA2 knockout mice and mice expressing a protease-inactive mutant form Omi/HtrA2, found that both demonstrated premature death associated with parkinsonian-like neurodegeneration (Jones et al., 2003; Martins et al., 2004). In one study, PINK1 was identified as a binding partner for Omi/HtrA2, and it was found that Omi/HtrA2 phosphorylation was decreased in brain tissue from PD patients who had PINK1 mutations (Plun-Favreau et al., 2007). Phosphorylation appeared to increase serine protease activity of Omi/HtrA2 and was necessary for Omi/HtrA2-mediated protection of mitochondria when cells were exposed to various toxins (Plun-Favreau et al., 2007). One study in Drosophila has reported that Omi/HtrA2 genetically interacts with, and downstream of, PINK1 in a pathway independent from parkin (Whitworth et al., 2008). In that study, overexpression of PINK1 alone or PINK1 and Omi/HtrA2 together in the fly eye resulted in a disorganized eye phenotype, and the downregulation of Omi/HtrA2 protected against PINK1 overexpression. However, some of the results of that study have been directly contradicted by a genetic loss-of-function study recently reported, which found no strong evidence for in vivo interaction between Omi/HtrA2 and PINK1 (Yun et al., 2008). Given this conflicting evidence, it is unclear whether this mitochondrial protease will prove to be involved in the PINK1 pathway of mitochondrial dynamics.
Another studied mitochondrial target is the intermembrane protease Rhomboid-7, which has not been previously linked to PD pathogenesis. Rhomboid-7 was demonstrated to be important for normal mitochondrial fusion in Drosophila (McQuibban et al., 2006). Overexpression of Rhomboid-7 has also been shown to enhance the PINK1 overexpression phenotypes in Drosophila, and has been suggested to function upstream of PINK1 and parkin (Whitworth et al., 2008). The relationship of these findings to PD pathogenesis, if any, remain to be studied, but this work does provide a step toward further defining the pathway by which PINK1 and parkin affect mitochondrial dynamics.
Conclusions
Accumulating evidence supports the possibility that changes in mitochondrial dynamics could critically influence PD-related cellular degeneration. One could hypothesize that these are the types of changes that could cause damage first in distal portions of neurons, where mitochondrial distribution, fusion, and fission play an important role and where early pathogenesis is thought to occur (Figure 2).
Figure 2. Potential susceptibility of neurons to impaired mitochondrial dynamics.
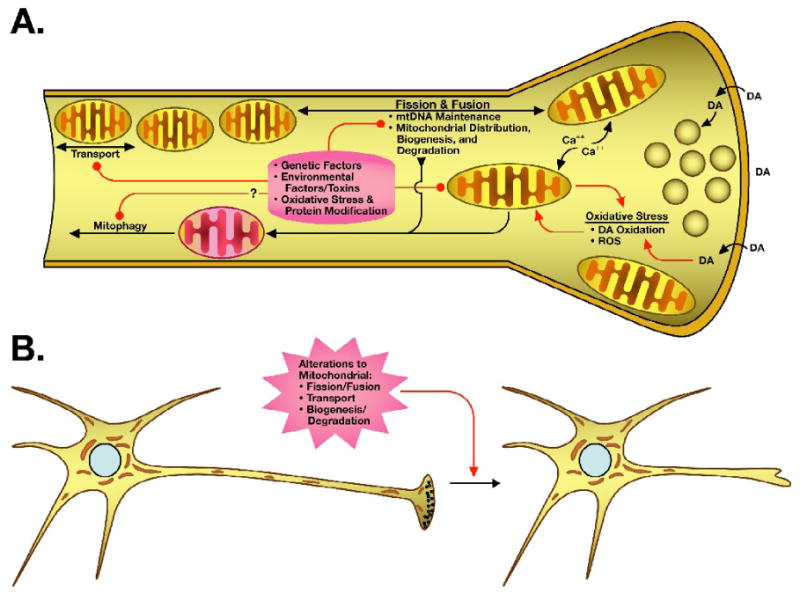
The unique energy demands of axons and nerve terminals, particularly those of the dopaminergic nigrostriatal pathway, are dependent on proper mitochondrial distribution and function. At the terminus, mitochondria are critical for calcium buffering and energy for maintaining the vesicular pool. Environmental and genetic factors may contribute to increased mitochondrial and oxidative stress, impacting mitochondrial function, fission and fusion, transport, and perhaps even mitochondrial maintenance systems (red connectors). The dopaminergic neurons of the substantia nigra are known to have increased oxidative stress, due in part to the oxidative nature of the neurotransmitter dopamine (DA). Mitochondrial dysfunction also has the potential to set up a vicious cycle of increased oxidative stress, through production of reactive oxygen species (ROS) and decreased energy availability (red arrows). B. Altered or disrupted mitochondrial dynamics, which may result in an accumulation of damaged mitochondria or decreased distribution of healthy mitochondria at the nerve terminal and within the axon, may ultimately lead to stress and terminal loss, propagating back to the cell body.
When considering the role of mitochondrial dynamics in neurodegeneration, it is important to take into account that mitochondrial fission and fusion are not isolated events in the cell, but surely are interrelated with other mitochondrial maintenance functions such as biogenesis and degradation (e.g. Berman et al., 2009). Thus, influencing one feature of this system will likely influence the entire balance of dynamic mitochondrial maintenance. It is also important to consider that the specific pathways by which PINK1 and parkin are mediating mitochondrial dynamics are not fully characterized. Both proteins, particularly parkin, have identified functions beyond mitochondrial dynamics, and thus may further affect mitochondrial function via other pathways. Additionally, the specific regulatory roles these proteins play in fission and fusion may differ between differing cell types or cell-specific situations. This may explain the apparent contradictions between studies as to the effect of PINK1 and parkin on mitochondrial morphology.
In familial forms of PD in which mutations in PINK1 or parkin occur, the evidence thus far suggests that mutations could lead to problems in the interaction of these proteins, and potentially the recruitment of parkin to the mitochondria. This could lead to dysregulation of normal mitochondrial dynamics, disrupting pathways necessary for the mitochondria to maintain health. In addition, dysfunctional parkin, or the inability to recruit parkin, could also prevent proper targeting of irreparably-damaged mitochondria for mitophagy, leading to a buildup of toxic, dysfunctional mitochondria, ultimately leading to death in more susceptible neurons.
But what about sporadic PD? The above scenario is harder to fit to an otherwise normally-functioning system. It is possible that other factors, such as toxic exposure, increased oxidative stress, or contributing genetic factors, could limit the availability of parkin and other proteins for proper mitochondrial maintenance. It has been shown that parkin is a target for covalent modification and inactivation by dopamine quinone, which may contribute to the increased susceptibility of the dopaminergic neurons (LaVoie et al., 2005). Alternatively, these toxic, metabolic, or genetic factors may directly or indirectly influence mitochondrial dynamics themselves, leading to degeneration through similar mechanisms. As a result, cells such as the dopaminergic neurons of the substantia nigra, which have a lower mitochondrial mass and increased stress in general, may not be able to maintain a distribution of healthy mitochondrial and eventually succumb to degeneration, contributing to PD pathogenesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alam M, Schmidt WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136:317–324. doi: 10.1016/s0166-4328(02)00180-8. [DOI] [PubMed] [Google Scholar]
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Starkov AA, Beal MF, Thomas B. Mitochondrial dysfunction in the limelight of Parkinson's disease pathogenesis. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbadis.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbas NR. Cognitive, affective, and psychiatric features of Parkinson's disease. Clin Geriatr Med. 2006;22:773–796. v–vi. doi: 10.1016/j.cger.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Barroso N, Campos Y, Huertas R, Esteban J, Molina JA, Alonso A, Gutierrez-Rivas E, Arenas J. Respiratory chain enzyme activities in lymphocytes from untreated patients with Parkinson disease. Clin Chem. 1993;39:667–669. [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. Embo J. 2006 doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria and neurodegeneration. Novartis Found Symp. 2007;287:183–192. doi: 10.1002/9780470725207.ch13. discussion 192-186. [DOI] [PubMed] [Google Scholar]
- Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- Berman SB, Chen Y, McCaffery JM, Rucker EB, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Blandini F, Nappi G, Greenamyre JT. Quantitative study of mitochondrial complex I in platelets of parkinsonian patients. Mov Disord. 1998;13:11–15. doi: 10.1002/mds.870130106. [DOI] [PubMed] [Google Scholar]
- Bogaerts V, Nuytemans K, Reumers J, Pals P, Engelborghs S, Pickut B, Corsmit E, Peeters K, Schymkowitz J, De Deyn PP, Cras P, Rousseau F, Theuns J, Van Broeckhoven C. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum Mutat. 2008;29:832–840. doi: 10.1002/humu.20713. [DOI] [PubMed] [Google Scholar]
- Borland MK, Trimmer PA, Rubinstein JD, Keeney PM, Mohanakumar KP, Liu L, Bennett JP., Jr Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson's disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol Neurodegener. 2008;3:21. doi: 10.1186/1750-1326-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Brockmann K, Dreha-Kulaczewski S, Dechent P, Bonnemann C, Helms G, Kyllerman M, Bruck W, Frahm J, Huehne K, Gartner J, Rautenstrauss B. Cerebral involvement in axonal Charcot-Marie-Tooth neuropathy caused by mitofusin2 mutations. J Neurol. 2008;255:1049–1058. doi: 10.1007/s00415-008-0847-1. [DOI] [PubMed] [Google Scholar]
- Cerveny KL, Tamura Y, Zhang Z, Jensen RE, Sesaki H. Regulation of mitochondrial fusion and division. Trends Cell Biol. 2007;17:563–569. doi: 10.1016/j.tcb.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- Dagda R, Kulich SM, Cherra SJ, III, Tandon A, Chu CT. PINK1 modulates mitochondrial morphology and autophagy in 6-OHDA-injured SH-SY5Y cells. Washington, DC: Society for Neuroscience Neuroscience Meeting Planner; 2008. Program No. 410.10. [Google Scholar]
- Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Kruger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk HA, Yaffe MP. A role for ubiquitination in mitochondrial inheritance in Saccharomyces cerevisiae. J Cell Biol. 1999;145:1199–1208. doi: 10.1083/jcb.145.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson's disease. Ann N Y Acad Sci. 2003;991:111–119. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Zhu C, Fernagut PO, Mehta A, DiCarlo CD, Seaman RL, Chesselet MF. Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Exp Neurol. 2004;187:418–429. doi: 10.1016/j.expneurol.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Muqit MM, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, Latchman DS, Holton JL, Wood NW, Revesz T. PINK1 protein in normal human brain and Parkinson's disease. Brain. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, Shults CW. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol. 1995;37:714–722. doi: 10.1002/ana.410370604. [DOI] [PubMed] [Google Scholar]
- Haque ME, Thomas KJ, D'souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A. 2008;105:1716–1721. doi: 10.1073/pnas.0705363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The Machines that Divide and Fuse Mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003;53 3:S26–36. doi: 10.1002/ana.10483. discussion S36-28. [DOI] [PubMed] [Google Scholar]
- Jin J, Hulette C, Wang Y, Zhang T, Pan C, Wadhwa R, Zhang J. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol Cell Proteomics. 2006;5:1193–1204. doi: 10.1074/mcp.M500382-MCP200. [DOI] [PubMed] [Google Scholar]
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnoczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–727. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Krige D, Carroll MT, Cooper JM, Marsden CD, Schapira AH. Platelet mitochondrial function in Parkinson's disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann Neurol. 1992;32:782–788. doi: 10.1002/ana.410320612. [DOI] [PubMed] [Google Scholar]
- Kuroda Y, Mitsui T, Kunishige M, Matsumoto T. Parkin affects mitochondrial function and apoptosis in neuronal and myogenic cells. Biochem Biophys Res Commun. 2006a;348:787–793. doi: 10.1016/j.bbrc.2006.06.201. [DOI] [PubMed] [Google Scholar]
- Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006b;15:883–895. doi: 10.1093/hmg/ddl006. [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Beal MF, Manfredi G. The role of mitochondria in inherited neurodegenerative diseases. J Neurochem. 2006;97:1659–1675. doi: 10.1111/j.1471-4159.2006.03990.x. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Liu F. Genetic factors involved in the pathogenesis of Parkinson's disease. Brain Res Rev. 2008;58:354–364. doi: 10.1016/j.brainresrev.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS ONE. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Liang CL, Wang TT, Luby-Phelps K, German DC. Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson's disease. Exp Neurol. 2007;203:370–380. doi: 10.1016/j.expneurol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, Schapira AH. Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann Neurol. 1994;36:876–881. doi: 10.1002/ana.410360612. [DOI] [PubMed] [Google Scholar]
- Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuibban GA, Lee JR, Zheng L, Juusola M, Freeman M. Normal mitochondrial dynamics requires rhomboid-7 and affects Drosophila lifespan and neuronal function. Curr Biol. 2006;16:982–989. doi: 10.1016/j.cub.2006.03.062. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Matuda S, Yoshino H, Mori H, Hattori N, Ikebe S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson's disease. Ann Neurol. 1994;35:204–210. doi: 10.1002/ana.410350212. [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, Wood NW, Willems PH, Smeitink JA, Cookson MR, Bandmann O. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol. 2008;64:555–565. doi: 10.1002/ana.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, Elibol B, Dalmizrak O, Ercan A, Kulaksiz G, Ogus H, Dalkara T, Ozer N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord. 2004;19:544–548. doi: 10.1002/mds.10695. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, Healy DG, Gilks WP, Lees AJ, Holton J, Revesz T, Parker PJ, Harvey RJ, Wood NW, Latchman DS. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98:156–169. doi: 10.1111/j.1471-4159.2006.03845.x. [DOI] [PubMed] [Google Scholar]
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006 doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzner A, Benard G, Youle RJ, Karbowski M. Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann N Y Acad Sci. 2008;1147:242–253. doi: 10.1196/annals.1427.012. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. 2001;28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- Orth M, Schapira AH. Mitochondrial involvement in Parkinson's disease. Neurochem Int. 2002;40:533–541. doi: 10.1016/s0197-0186(01)00124-3. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Pallone JA. Introduction to Parkinson's disease. Dis Mon. 2007;53:195–199. doi: 10.1016/j.disamonth.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. 2009;378:518–523. doi: 10.1016/j.bbrc.2008.11.086. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Parks JK. Mitochondrial ND5 mutations in idiopathic Parkinson's disease. Biochem Biophys Res Commun. 2005;326:667–669. doi: 10.1016/j.bbrc.2004.11.093. [DOI] [PubMed] [Google Scholar]
- Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, Barja F, Martinou JC. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE. 2008;3:e3257. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn AM, Roberts T, Hodder J, Allen PS, Zhu G, Martin WR. Generalized mitochondrial dysfunction in Parkinson's disease detected by magnetic resonance spectroscopy of muscle. Neurology. 1995;45:2097–2099. doi: 10.1212/wnl.45.11.2097. [DOI] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Pham NA, Richardson T, Cameron J, Chue B, Robinson BH. Altered mitochondrial structure and motion dynamics in living cells with energy metabolism defects revealed by real time microscope imaging. Microsc Microanal. 2004;10:247–260. doi: 10.1017/S143192760404005X. [DOI] [PubMed] [Google Scholar]
- Plun-Favreau H, Gandhi S, Wood-Kaczmar A, Deas E, Yao Z, Wood NW. What have PINK1 and HtrA2 genes told us about the role of mitochondria in Parkinson's disease? Ann N Y Acad Sci. 2008;1147:30–36. doi: 10.1196/annals.1427.032. [DOI] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Raimondi A, Mangolini A, Rizzardini M, Tartari S, Massari S, Bendotti C, Francolini M, Borgese N, Cantoni L, Pietrini G. Cell culture models to investigate the selective vulnerability of motoneuronal mitochondria to familial ALS-linked G93ASOD1. Eur J Neurosci. 2006;24:387–399. doi: 10.1111/j.1460-9568.2006.04922.x. [DOI] [PubMed] [Google Scholar]
- Rapaport D, Brunner M, Neupert W, Westermann B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J Biol Chem. 1998;273:20150–20155. doi: 10.1074/jbc.273.32.20150. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23:7881–7888. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riparbelli MG, Callaini G. The Drosophila parkin homologue is required for normal mitochondrial dynamics during spermiogenesis. Dev Biol. 2007;303:108–120. doi: 10.1016/j.ydbio.2006.10.038. [DOI] [PubMed] [Google Scholar]
- Ross OA, Soto AI, Vilarino-Guell C, Heckman MG, Diehl NN, Hulihan MM, Aasly JO, Sando S, Gibson JM, Lynch T, Krygowska-Wajs A, Opala G, Barcikowska M, Czyzewski K, Uitti RJ, Wszolek ZK, Farrer MJ. Genetic variation of Omi/HtrA2 and Parkinson's disease. Parkinsonism Relat Disord. 2008;14:539–543. doi: 10.1016/j.parkreldis.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samii A, Nutt JG, Ransom BR. Parkinson's disease. Lancet. 2004;363:1783–1793. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Sheehan JP, Swerdlow RH, Parker WD, Miller SW, Davis RE, Tuttle JB. Altered calcium homeostasis in cells transformed by mitochondria from individuals with Parkinson's disease. J Neurochem. 1997;68:1221–1233. doi: 10.1046/j.1471-4159.1997.68031221.x. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y. Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol. 1999;45:668–672. doi: 10.1002/1531-8249(199905)45:5<668::aid-ana19>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Singleton AB. Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum Mol Genet. 2008;17:1988–1993. doi: 10.1093/hmg/ddn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H. Mono- and double-mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet. 2007;16:2377–2393. doi: 10.1093/hmg/ddm083. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Muller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Kruger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Jr, Davis RE, Parker WD., Jr Origin and functional consequences of the complex I defect in Parkinson's disease. Ann Neurol. 1996;40:663–671. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- Takatori S, Ito G, Iwatsubo T. Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett. 2008;430:13–17. doi: 10.1016/j.neulet.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Krige D, Barnes PR, Kemp GJ, Carroll MT, Mann VM, Cooper JM, Marsden CD, Schapira AH. A 31P magnetic resonance spectroscopy study of mitochondrial function in skeletal muscle of patients with Parkinson's disease. J Neurol Sci. 1994;125:77–81. doi: 10.1016/0022-510x(94)90245-3. [DOI] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16(Spec No 2):R183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- Trimmer PA, Borland MK, Keeney PM, Bennett JP, Jr, Parker WD., Jr Parkinson's disease transgenic mitochondrial cybrids generate Lewy inclusion bodies. J Neurochem. 2004;88:800–812. doi: 10.1046/j.1471-4159.2003.02168.x. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Wang C, Lu R, Ouyang X, Ho MW, Chia W, Yu F, Lim KL. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci. 2007;27:8563–8570. doi: 10.1523/JNEUROSCI.0218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lim PJ, Karbowski M, Monteiro MJ. Effects of overexpression of Huntingtin proteins on mitochondrial integrity. Hum Mol Genet. 2008a doi: 10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008b;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet. 2008;17:602–616. doi: 10.1093/hmg/ddm334. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 Forms a Multiprotein Complex with Miro and Milton, Linking Pink1 Function to Mitochondrial Trafficking (dagger) Biochemistry. 2009 doi: 10.1021/bi8019178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub D, Comella CL, Horn S. Parkinson's disease--Part 1: Pathophysiology, symptoms, burden, diagnosis, and assessment. Am J Manag Care. 2008;14:S40–48. [PubMed] [Google Scholar]
- Weintraub D, Stern MB. Psychiatric complications in Parkinson disease. Am J Geriatr Psychiatry. 2005;13:844–851. doi: 10.1176/appi.ajgp.13.10.844. [DOI] [PubMed] [Google Scholar]
- Westermann B. Merging mitochondria matters: cellular role and molecular machinery of mitochondrial fusion. EMBO Rep. 2002;3:527–531. doi: 10.1093/embo-reports/kvf113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003;10:260–261. doi: 10.1038/sj.cdd.4401147. [DOI] [PubMed] [Google Scholar]
- Whitworth AJ, Lee JR, Ho VM, Flick R, Chowdhury R, McQuibban GA. Rhomboid-7 and HtrA2/Omi act in a common pathway with the Parkinson's disease factors Pink1 and Parkin. Dis Model Mech. 2008;1:168–174. doi: 10.1242/dmm.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler-Stuck K, Kirches E, Mawrin C, Dietzmann K, Lins H, Wallesch CW, Kunz WS, Wiedemann FR. Re-evaluation of the dysfunction of mitochondrial respiratory chain in skeletal muscle of patients with Parkinson's disease. J Neural Transm. 2005;112:499–518. doi: 10.1007/s00702-004-0195-y. [DOI] [PubMed] [Google Scholar]
- Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AS, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, Taylor J, Heales S, Duchen MR, Latchman D, Tabrizi SJ, Wood NW. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Jiang Q, Zhao J, Ren Y, Sutton MD, Feng J. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J Biol Chem. 2005;280:17154–17162. doi: 10.1074/jbc.M500843200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura-Hoshino M, Sada K, Hotta H, Yamamura H, Inatome R, Yanagi S. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. Embo J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino H, Nakagawa-Hattori Y, Kondo T, Mizuno Y. Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson's disease. J Neural Transm Park Dis Dement Sect. 1992;4:27–34. doi: 10.1007/BF02257619. [DOI] [PubMed] [Google Scholar]
- Yun J, Cao JH, Dodson MW, Clark IE, Kapahi P, Chowdhury RB, Guo M. Loss-of-function analysis suggests that Omi/HtrA2 is not an essential component of the PINK1/PARKIN pathway in vivo. J Neurosci. 2008;28:14500–14510. doi: 10.1523/JNEUROSCI.5141-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM, Battologlu E. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]