

Abstract
Joubert Syndrome Related Disorders (JSRDs) are autosomal recessive pleiotropic conditions sharing a peculiar cerebellar and brainstem malformation known as the “molar tooth sign” (MTS). Recently, mutations in a novel ciliary gene, RPGRIP1L, have been shown to cause both JSRDs and Meckel-Gruber syndrome. We searched for RPGRIP1L mutations in 120 patients with proven MTS and phenotypes representative of all JSRD clinical subgroups. Two homozygous mutations, the previously reported p.T615P in exon 15 and the novel c.2268_2269delA in exon 16, were detected in two out of 16 families with cerebello-renal presentation (~12%). Conversely, no pathogenic changes were found in patients with other JSRD phenotypes, suggesting that RPGRIP1L mutations are largely confined to the cerebello-renal subgroup, while they overall represent a rare cause of JSRD (<2%).
Keywords: RPGRIP1L, Joubert Syndrome Related Disorders, molar tooth sign, nephronophthisis
Introduction
Joubert Syndrome Related Disorders (JSRDs) are clinically and genetically heterogeneous autosomal recessive conditions with multi-organ involvement. Their pathognomonic feature is a complex midbrain-hindbrain malformation evident on brain magnetic resonance, that is characterized by hypo/dysplasia of the cerebellar vermis, elongated, thickened and mal-oriented superior cerebellar peduncles and a deep interpeduncular fossa (“Molar Tooth Sign”, MTS) (1). Besides the typical neurological features of Joubert syndrome (JS, MIM213300), the organs most commonly affected are the kidneys and the retina, with nephronophthisis (NPH) and retinal dystrophy of variable severity. Based on these organs’ involvement, four major JSRD subgroups are defined, namely pure JS, JS plus retinopathy (cerebello-retinal phenotype), JS plus renal involvement (cerebello-renal phenotype) and cerebello-oculo-renal phenotype (COR). Additional clinical features can be found in all subgroups, including other central nervous system (CNS) malformations, chorioretinal colobomas, hepatic fibrosis, polydactyly, oro-facial midline and laterality defects. Some of these features may co-occur in definite associations, such as COACH (JS plus colobomas and hepatic fibrosis, MIM216360) and Oro-Facio-Digital syndrome type VI (JS plus polydactyly and oro-facial midline defects, MIM277170) (2).
The phenotypic complexity of JSRDs mirrors their genetic heterogeneity. Two loci (JBTS1 and JBTS2) have been mapped to chromosomes 9 and 11, while mutations in AHI1, NPHP1, CEP290, MKS3 and, more recently, RPGRIP1L have been detected in JSRD patients with variable phenotypes. Adding more complexity, mutations in many JSRD genes have been found also to cause other ciliopathies, including isolated NPH (NPHP1), Senior-Loken syndrome (NPHP1, CEP290), Leber Congenital Amaurosis (CEP290) and Meckel-Gruber syndrome (MKS3, CEP290, RPGRIP1L) (2).
Molecular screens of large cohorts of JSRD patients representative of all subgroups have been performed only for some genes, allowing the identification of valuable genotype-phenotype correlates. Mutations in CEP290 have been shown nearly invariable to cause the COR phenotype (3,4), NPHP1 mutations have been detected in patients with JS always associated to renal involvement and occasionally with retinopathy (5–8), while AHI1 mutations cause either pure JS or JS plus retinopathy, with kidneys nearly always spared (9–10).
The RPGRIP1L gene was recently found mutated in about 8–10% of patients with cerebello-renal phenotype (11–13) but its mutation frequency in the full spectrum of JSRDs has not been evaluated. Here we report the mutation analysis of this gene in a large cohort of clinically well-characterised JSRD patients and discuss genotype-phenotype correlates.
Materials and methods
Patients
Mutation analysis was performed in a cohort of 120 JSRD patients recruited worldwide. In all cases, the clinical diagnosis was confirmed by neuroradiological demonstration of the MTS. Detailed clinical data were obtained by referring clinicians through a standardised questionnaire assessing the possible involvement of all organs, including CNS, retina, kidneys, liver and the occurrence of associated malformations. Data were collected at the CSS-Mendel Institute of Rome and at the University of California San Diego and samples were obtained through referring physicians or the JS BioBank (http://www.joubertsyndrome.org/BioBank.asp). Patients were divided in six different cohorts as follows: i) 44 with pure JS; ii) 24 with JS plus retinal involvement; iii) 16 with JS plus renal involvement; iv) 19 with COR phenotype; v) 11 with COACH syndrome; vi) 6 with OFDVI syndrome.
Informed consent was obtained from all families and the study was approved by the local ethic committees. Patients previously found to harbour mutations in either NPHP1, AHI1, CEP290 or MKS3 were excluded from the screening.
Molecular analysis
Genomic DNA was extracted from peripheral blood lymphocytes following standard methods or obtained directly from referral centres. The whole RPGRIP1L (GeneBank NM_015272) coding region (exons 2–27) was tested for mutations. Each exon including exon-intron junctions was PCR-amplified and then analysed by means of denaturing high-performance liquid chromatography (DHPLC) using a Wave DNA Fragment Analysis System (Transgenomics) at column temperatures recommended by Navigator Software v. 1.5.4 (Transgenomics). Primers, PCR and DHPLC conditions are listed in Table 1.
Table 1.
Primers, PCR and DHPLC conditions for RPGRIP1L analysis.
| Exons | Primer sequence (5′ to 3′) |
Amplico n size (bp) |
PCR annealing temp. °C |
Cycles | DHPLC oven temp.°C |
Start % Buffer B |
|---|---|---|---|---|---|---|
| 2 | TTGGTTCATTCCATTGCATAGGGACTATATAGTTACTTAAATGG | 275 | 56 | 40 | 58.8 | 51.3 |
| 3 | CTCAGATGAGTGCATCAGAGTACCCTCCAAATTTTACTGAGGATTTTATGCTGACTTCTCAC | 294 | 56 | 40 | 53.6/57.6/59.8 | 56.9/52.9/50.7 |
| 4 | GTGTTAAACAATCTAATAAGACCTTATTATAGCAAATCAAAGATCC | 406 | 56 | 40 | 56.1/57.8 | 59.5/57.8 |
| 5 | AGACATTATCAATAACCAAAGTTCTGCTTGCTCTTTTTCATGAAG | 229 | 56 | 40 | 53.5/56 | 54.2/51.7 |
| 6 | GTATTTTAAACTAATTTTGACCCAAAAACCTCATTGACTGATG | 295 | 56 | 40 | 53.5/55.3/57.2 | 58.4/56.6/54.7 |
| 7 | CTTGAATACTACTTTTGAATCCATCTGTATAAGGTGAACTTCG | 313 | 56 | 40 | 55.6 | 55.1 |
| 8 | GTTACCAAGAAGTTGTAATGCCATGTTAACCTTCTCTCATATAG | 325 | 56 | 40 | 55.9/56.9 | 54.8/53.8 |
| 9 | ATATAAAAGAGCTCAAACAAGACGAAGCTGATTTCCAAGCTTTC | 228 | 56 | 40 | 52.9/55 | 53.8/51.7 |
| 10 | GTACTGACTGATTCAATGAAGCAAGACTGGTGTGGAACAG | 256 | 56 | 40 | 56.6/60.2 | 55.3/51.7 |
| 11 | GTAGTATAGTGCTTTCTCTATGGGAATGTCTCTTGTATTTTCC | 283 | 56 | 40 | 52.7/55.2 | 54.7/57.2 |
| 12 | CAGATAAGGGTCATCATTATGTATATCAGGACTTCCTTTGTC | 185 | 56 | 40 | 56/57 | 51.9/50.9 |
| 13 | GATAACAGGTGATAACATACAGCTTCTGATTTGTGAATATTGCC | 416 | 56 | 40 | 53.1/58 | 58.3/50.8 |
| 14 | CCATTCCTCTAGTTACTGTGCCAGTTTTCATTTTCTCATGTC | 346 | 56 | 40 | 54.7/56.8 | 59.5/56 |
| 15 | CAAATTAGTAATTCAAACACCCCTGCTATCTATTTTAATGTATG | 559 | 56 | 40 | 55.1/56.3 | 61.5/60.3 |
| 16 | TTTTAGTTTAAAGCACGACACGCTGTTCATCTTTTAACTGTG | 292 | 56 | 40 | 54.6/57.6 | 55.1/52.1 |
| 17 | GAATCATATCCATAACACTAAGGAGTAATAAAACTTCACTCTAGC | 510 | 56 | 40 | 51.1/56.4/60.4 | 62.2/60.1/55.2 |
| 18–19 | CAGGTAGGGAATAATATGCCGATAGAATGATTTGGGATTTAG | 575 | 56 | 40 | 51/55.5/56.4 | 64.2/59.7/60.2 |
| 20 | CTATGACTTCCTGAGTCATGGCTATAAAATGTGATCTAAGC | 263 | 56 | 40 | 54.2/56.3 | 55.5/52.4 |
| 21 | CAGATAAAACAAAGCACAGGCAGGGTATAAGTAGAATTGG | 274 | 56 | 40 | 54.8/57.7 | 56.4/53.5 |
| 22 | GTACTGTGAATGAAAGGCAGGCATTTAATGTGCTTCTTAATC | 195 | 56 | 40 | 54.8/57.8 | 52.9/49.9 |
| 23 | ACTAGATTTTGTATAAAGATCTGGTGGAAAGTAATAGTGGATC | 354 | 56 | 40 | 56/60.4/62.2 | 60.1/57.7/55.9 |
| 24 | GAGAAATAAACTTTCTCTCTGGTAGCCTATTCCTGAGTGAC | 350 | 56 | 40 | 53.4/56.5/59.5 | 58/54.9/51.9 |
| 25 | GTCCTGCTACAGAGATCTACGTGATTTCTTCTCACTTGGTG | 200 | 56 | 40 | 56.2 | 52.7 |
| 26 | GAGTTTGGAGTTCAGCAATTGTACTGTTTCTGTCTGCGAGC | 241 | 56 | 40 | 59.6/63.3 | 54.7/51 |
| 27 | ACTTTCATGTGAGCATTTACTG | 245 | 56 | 40 | 57.9/60.3 | 54.8/52.4 |
In order to preserve DNA from affected children and to overcome DHPLC limits in identifying homozygous nucleotide changes, DNA from both parents was analysed in the screening. All exons showing abnormal elution profiles underwent bidirectional sequencing after purification of the PCR product (Millipore) by using BigDye chemistry and an ABI 3100 Capillary Array Sequencer (Applied Biosystems). Segregation of mutations detected in healthy parents was evaluated by direct sequencing of the proband and other available family members.
Results
Two families in the cerebello-renal subgroup harboured mutations in RPGRIP1L, while no pathogenic changes were found in patients with other JSRD phenotypes. Single nucleotide polymorphisms and not-segregating variants of unknown significance are listed in Table 2.
Table 2.
Polymorphisms and variants observed in the RPGRIP1L gene.
| DNA alteration | Reference SNP | Protein alteration | Exon/Intron | Allele frequency (n=96 chromosomes) |
|---|---|---|---|---|
| IVS4-29G>A | - | - | Intron 4 | n.s. |
| 685G>A | - | A229T | Exon 6 | common |
| IVS7-30delAAT | - | - | Intron 7 | n.s. |
| 2231G>A | rs2302677 | R744Q | Exon 16 | common |
| 2304G>A | - | S768S | Exon 16 | common |
| IVS19-32G>A | rs7203525 | Intron 19 | common | |
| 3073G>A | rs2111119 | G1025S | Exon 21 | common |
| 3428C>G | - | T1143S | Exon 23 | common |
| IVS23+37 C>T | - | - | Intron 23 | n.s. |
| IVS23+67G>A | - | - | Intron 23 | common |
| 3790G>A | rs3213758 | D1264N | Exon 26 | common |
| 3936C>T | rs4784320 | D1312D | Exon 27 | common |
common: allele frequency greater than 2%; n.s.: variation found in the parent but not segregating in affected offspring
Family A consisted of two affected siblings born from apparently non-consanguineous healthy parents of Swiss origin. Both siblings were homozygous and both parents were heterozygous for the c.1843A>C mutation in exon 15, resulting in the p.T615P missense change. While this work was in progress, the two sibs have been reported as part of a cohort of patients with NPH and JS (Family F138 in 13), and we provide here a more detailed description of their phenotype.
The 22-year-old male proband and his younger sister presented developmental delay, growth and mental retardation, NPH and severe scoliosis. Visual acuity and fundus examination, as well as liver function, repeatedly tested normal in both patients. Several features presented marked intrafamilial variability, being detected either in the sister (post-axial polydactyly of hands, bilateral ptosis, abnormal neonatal breathing) or in the proband (oculomotor apraxia). The degree of growth and psychomotor delay also differed widely between the two sibs, being much more severe in the sister. Indeed, she showed a dramatic failure to thrive, with body weight and height that have always been well below the third centile. She never achieved the abilities to stand unaided and to produce expressive language, was always fully dependent for daily life activities and died at the age of 17,5 years from end stage renal failure. Conversely, the proband reached independent walking at 30 months and attended a special school where he acquired many skills of daily life, becoming nearly self-sufficient. He underwent kidney transplant at age 11 years, that is still functioning well, and surgical treatment for severe scoliosis at age 18 years. His growth parameters are within the normal limits.
The second mutated family (B) consisted of a 4-year-old affected female born from first-cousin healthy parents of Moroccan origin. She was homozygous for a c.2268_2269delA mutation in exon 16 resulting in frameshift and premature protein truncation (p.I756fsX769). At birth, she presented occipital meningoencephalocele which was surgically removed, bilateral post-axial polydactyly of hands and feet, club foot and right-sided inguinal hernia. Several episodes of hyperpnea followed by periods of apnea were recorded and her milestones were severely delayed along with a marked failure to thrive. Renal dysfunction became manifested at age 1 year, when she was hospitalized for an episode of acute increase of plasma creatinine levels and reduced glomerular filtration. Kidney ultrasounds repeatedly showed small kidneys with increased echogenicity, loss of corticomedullary differentiation and multiple cysts compatible with NPH. Ocular examination showed horizontal nystagmus and alternating internal strabismus while other investigations including fundoscopy were negative. Now, at age 4 years, the patient presents chronic renal failure, marked growth retardation (body weight, height and head circumference well below the third centile) and severe psychomotor delay, with lack of head control and inability to speak any meaningful word.
Affected members from both families presented the typical MTS (figure).
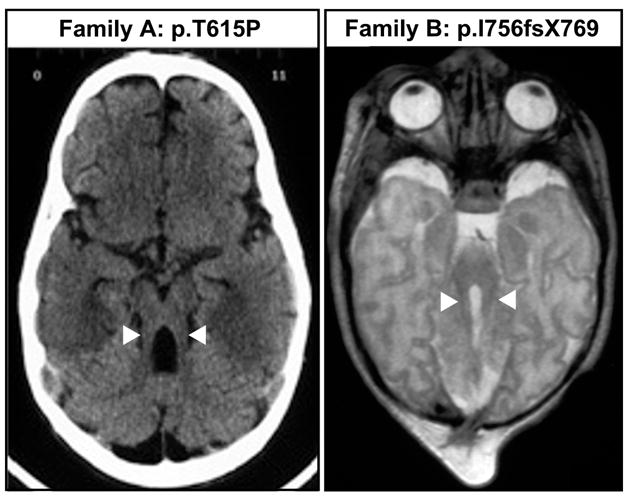
Axial magnetic resonance imaging showing the MTS in probands from families A and B. Note the thickening and malorientation of superior cerebellar peduncles in both cases (arrowheads).
Discussion
In this study, we performed mutation analysis of the RPGRIP1L gene in a cohort of 120 patients with proven MTS, comprehensive of the whole JSRD clinical spectrum. We identified pathogenic mutations only in two out of 16 families with cerebello-renal phenotype, leading to a ~12% frequency in this specific clinical subgroup, in line with previous data (13). Conversely, pathogenic mutations were not detected in patients presenting with pure JS, JS plus retinopathy, COR, COACH and OFDVI syndromes, thus making RPGRIP1L a rare genetic cause of JSRDs (<2%). Overall, these data support the existence of a correlate between this gene and the cerebello-renal presentation. Indeed, such phenotype has been found in 13 out of the 18 RPGRIP1L-mutated JSRD patients so far described (11–13, this study). In the remaining five cases, three had pure JS, yet they were all young (1, 3 and 5 years) and still at risk to develop renal disease later in life; the other two patients presented atypical phenotypes with possible retinal involvement but no proven MTS (12,13).
The other major gene causative of JS plus renal involvement is NPHP1, with mutation frequencies also in the range of 5–14% (5–8). Nevertheless, some differences can be observed when comparing NPHP1- and RPGRIP1L- associated phenotypes. Firstly, the MTS aspect in NPHP1-deleted patients appears to be consistently “mild”, with superior cerebellar peduncles that are elongated but not thickened, at difference with RPGRIP1L patients presenting a classical MTS (figure). Secondly, NPHP1 is nearly invariably associated with juvenile NPH, while RPGRIP1L mutations have been shown to cause both the infantile and juvenile forms of NPH. Thirdly, liver involvement, encephalocele and severe scoliosis have been reported in a number of patients with RPGRIP1L mutations but never in association with NPHP1.
Finally, retinal involvement is detected in about one third of NPHP1 patients, while it has been reported only in two out of 18 RPGRIP1L cases. In the first patient (F491-1 in 12), only one heterozygous missense change could be identified, and it is possible that unidentified mutations in other genes could contribute to his phenotypic diversity. The other patient (A166, II-3 in 13), homozygous for the p.T615P mutation, has been reported with unspecified “vision problems”, but it is currently unclear whether these could result from an underlying retinal degeneration.
The rare association of RPGRIP1L mutations with retinal defects is worthy of note, since mutations in the closely related RPGRIP1 gene account for about 6% of isolated Leber Congenital Amaurosis, one of the commonest genetic causes of visual impairment at birth (14–15). A plausible hypothesis is that the two homologue genes may have complementary and partially redundant functions in specific tissues, i.e. the kidneys and the retina respectively.
So far, 16 different RPGRIP1L mutations have been described. About half of them cluster in exon 15, including p.T615P, that is the only mutation recurring in five unrelated families. Thus, exon 15 likely represents a mutational hot spot and deserves molecular investigation in patients with cerebello-renal JSRD. The second mutation reported here is novel and is the first so far described in RPGRIP1L exon 16. This homozygous 1-bp deletion is predicted to cause premature protein termination before the second C2 domain, and is the second homozygous truncating mutation so far reported in JSRD patients (11). Hence, such mutations are associated not only with Meckel-Gruber syndrome, but also with JSRD phenotypes comparable to those caused by missense changes. This finding argues against the proposed correlation between complete loss of function of the protein and more severe clinical syndromes (12), and underlies the existence of still unknown genetic modifiers.
The presence of possible epistatic effects is further highlighted by the phenotypic variability observed within some RPGRIP1L mutated families. For instance, patient A166, II-3 reported by Wolf et al (13) presented with mental retardation, NPH and impaired vision in the absence of cerebellar vermis hypoplasia and MTS, while this malformation was detected in her 5-year-old brother with developmental delay and NPH. Similarly, intrafamilial variability was observed in our family A regarding several clinical features such as polydactyly, oculomotor apraxia, growth and psychomotor delay.
In conclusion, significant correlates are emerging between JSRD causative genes and specific clinical phenotypes, despite the wide genetic heterogeneity. The exclusion of RPGRIP1L mutations in patients within most JSRD subgroups strengthens the correlation between this gene and the cerebello-renal presentation.
Acknowledgments
This work was supported by grants from the Italian Ministry of Health (Ricerca Corrente 2008; Ricerca Finalizzata 2006 ex. articolo 56), the U.S. National Institute of Neurological Disease and Stroke, the March of Dimes, the Burroughs Wellcome Fund Award in Translational Research and the NIH. We thank Remi Salomon and Sophie Saunier (INSERM U-574, Hopital Necker-Enfants Malades and Universite Paris Descartes) for early sharing of RPGRIP1L data.
Appendix
Other members of the International JSRD Study Group are:
Richard Leventer (Parkville, Australia); Padraic Grattan-Smith (Sydney, Australia); Andreas Janecke (Innsbruck, Austria); Marc D’Hooghe (Brugge, Belgium); Rudy Van Coster (Ghent, Belgium); Karin Dias, Carla Moco, Ana Moreira (Porto Alegre, Brazil); Chong Ae Kim (Sao Paulo, Brazil); Gustavo Maegawa (Toronto, Canada); Ghada M.H. Abdel-Salam, Alice Abdel-Aleem, Maha S. Zaki (Cairo, Egypt); Itxaso Marti, Susana Quijano-Roy (Garches, France); Pascale de Lonlay, Stephane Romano, Alain Verloes (Paris, France); Renaud Touraine (St. Etienne, France); Michel Koenig, Clotilde Lagier-Tourenne, Jean Messer (Strasbourg, France); Heike Philippi (Mainz, Germany); Sofia Kitsiou Tzeli (Athens, Greece); Saevar Halldorsson, Jonina Johannsdottir, Peter Ludvigsson (Reykjavik, Iceland); Shubha R. Phadke (Lucknow, India); Bernard Stuart (Dublin, Ireland); Alex Magee (Belfast, Northern Ireland); Dorit Lev, Marina Michelson (Holon, Israel); Bruria Ben-Zeev (Ramat-Gan, Israel); Rita Fischetto, Mattia Gentile (Bari, Italy); Silvia Battaglia, Lucio Giordano, Lorenzo Pinelli (Brescia, Italy); Loredana Boccone (Cagliari, Italy); Martino Ruggieri (Catania, Italy); Stefania Bigoni, Alessandra Ferlini (Ferrara, Italy); Maria Alice Donati, Elena Procopio (Florence, Italy); Gianluca Caridi, Francesca Faravelli, Gianmarco Ghiggeri (Genoa, Italy); Silvana Briuglia, Carmelo D. Salpietro, Gaetano Tortorella (Messina, Italy); Stefano D’Arrigo, Chiara Pantaleoni, Daria Riva, Graziella Uziel (Milan, Italy); Anna Maria Laverda, Alberto Permunian (Padova, Italy); Stefania Bova (Pavia, Italy); Roberta Battini (Pisa, Italy); Maria Roberta Cilio, Marilu Di Sabato, Vincenzo Leuzzi, Pasquale Parisi (Rome, Italy); Alessandro Simonati (Verona, Italy); Asma A. Al-Tawari, Laila Bastaki, Ahmad (Kuwait City, Kuwait), Mirjam M. de Jong (Groningen, The Netherlands); Roshan Koul, Anna Rajab (Muscat, Oman); Matloob Azam (Islamabad, Pakistan); Clara Barbot (Oporto, Portugal); Berta Rodriguez (La Coruna, Spain); Ignacio Pascual-Castroviejo (Madrid, Spain); Hulya Kayserili, Sinan Comu (Istanbul, Turkey); Mustafa Akcakus (Kayseri, Turkey); Lihadh Al Gazali, Laszlo Sztriha (Al Ain, UAE); David Nicholl (Birmingham, UK); C. Geoffrey Woods (Cambridge, UK); Christopher Bennett, Jane Hurst (Leeds, UK); Raoul Hennekam, Melissa Lees (London, UK); Saunder Bernes (Mesa, Arizona, US); Henry Sanchez (Fremont, California, US); Aldon E. Clark (Laguna Niguel, California, US); Elysa DeMarco, Clement Donahue, Elliot Sherr (San Francisco, California, US); Jin Hahn, Terence D. Sanger (Stanford California, US); Tomas E. Gallager (Manoa, Hawaii, US); William B. Dobyns (Chicago, Illinois, US); Cynthia Daugherty (Bangor, Maine, US); Kalpathy S. Krishnamoorthy, Dean Sarco, Christopher A. Walsh (Boston, Massachusetts, US); Trudy McKanna (Grand Rapids, Michigan, US); Joanne Milisa (Albuquerque, New Mexico, US); Wendy K. Chung, Darryl C. De Vivo, Hillary Raynes, Romaine Schubert (New York, New York, US); Alison Seward (Columbus, Ohio, US); David G. Brooks (Philadephia, Pennsylvania, US); Amy Goldstein (Pittsburg, Pennsylvania, US); James Caldwell, Eco Finsecke (Tulsa, Oklahoma, US); Bernard L. Maria (Charleston, South Carolina, US), Kenton Holden (Mt. Pleasant, South Carolina, US); Robert P. Cruse (Houston, Texas, US); Kathryn J. Swoboda, Dave Viskochil (Salt Lake City, Utah, US).
Footnotes
Competing Interests
The authors declare that they have no competing financial interests.
References
- 1.Maria BL, Quisling RG, Rosainz LC, et al. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14 (6):368–376. doi: 10.1177/088307389901400605. [DOI] [PubMed] [Google Scholar]
- 2.Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008;51 (1):1–23. doi: 10.1016/j.ejmg.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Brancati F, Barrano G, Silhavy JL, et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2007;81 (1):104–113. doi: 10.1086/519026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helou J, Otto EA, Attanasio M, et al. Mutation analysis of NPHP6/CEP290 in patients with Joubert-Syndrome and Senior-Loken-Syndrome. J Med Genet. 2007;44 (10):657–663. doi: 10.1136/jmg.2007.052027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parisi MA, Bennett CL, Eckert ML, et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75 (1):82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castori M, Valente EM, Donati MA, et al. NPHP1 gene deletion is a rare cause of Joubert syndrome related disorders. J Med Genet. 2005;42 (2):e9. doi: 10.1136/jmg.2004.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caridi G, Dagnino M, Rossi A, et al. Nephronophthisis type 1 deletion syndrome with neurological symptoms: prevalence and significance of the association. Kidney Int. 2006;70 (7):1342–1347. doi: 10.1038/sj.ki.5001768. [DOI] [PubMed] [Google Scholar]
- 8.Tory K, Lacoste T, Burglen L, et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18 (5):1566–1575. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- 9.Valente EM, Brancati F, Silhavy JL, et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol. 2006;59 (3):527–534. doi: 10.1002/ana.20749. [DOI] [PubMed] [Google Scholar]
- 10.Parisi MA, Doherty D, Eckert ML, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43 (4):334–339. doi: 10.1136/jmg.2005.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arts HH, Doherty D, van Beersum SE, et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39 (7):882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 12.Delous M, Baala L, Salomon R, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39 (7):875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 13.Wolf MTF, Saunier S, O’Toole JF, et al. Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int. 2007;72:1520–1526. doi: 10.1038/sj.ki.5002630. [DOI] [PubMed] [Google Scholar]
- 14.Dryja TP, Adams SM, Grimsby JL, et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68 (5):1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerber S, Perrault I, Hanein S, et al. Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur J Hum Genet. 2001;9 (8):561–571. doi: 10.1038/sj.ejhg.5200689. [DOI] [PubMed] [Google Scholar]