

Abstract
Adult exposure to NMDA receptor antagonists, such as ketamine, produces psychosis in humans, and exacerbates symptoms in schizophrenic patients. We recently showed that ketamine activates the innate immune enzyme NADPH-oxidase in brain, and that the superoxide produced leads to dysfunction of a subset of fast-spiking inhibitory interneurons expressing the calcium-binding protein parvalbumin (PV). Here we show that neuronal production of interleukin-6 (IL-6) is necessary and sufficient for ketamine-mediated activation of NADPH-oxidase in brain. Removal of IL-6 in neuronal cultures by anti-IL-6 blocking antibodies, or in vivo by use of IL-6-deficient mice, prevented the increase in superoxide by ketamine and rescued the interneurons. Accumulating evidence suggests that schizophrenia patients suffer from diminished antioxidant defenses, and a recent clinical trial showed that enhancing these defenses may ameliorate symptoms of the disease. Our results showing that ketamine-induced IL-6 is responsible for the activation of NADPH-oxidase in brain suggest that reducing brain levels of this cytokine may protect the GABAergic phenotype of fast-spiking PV-interneurons and thus attenuate the propsychotic effects of ketamine.
Keywords: schizophrenia, interleukin-6, parvalbumin, interneurons, NADPH-oxidase, prelimbic
Introduction
Acute exposure in adulthood to subanesthetic doses of NMDA receptor (NMDA-R) antagonists such as phencyclidine (PCP) and ketamine reproduce both negative and positive symptoms of schizophrenia, and has therefore been used to model its pathophysiological features (Javitt, 2007). The acute propsychotic effects of these antagonists are believed to arise from specific effects on inhibitory circuits that lead to disinhibition of excitatory systems (Olney et al., 1999; Tomitaka et al., 2000; Holcomb et al., 2005; Homayoun and Moghaddam, 2007).
Although acute exposures to NMDA-R antagonists produce increased metabolism in frontal regions, these effects are transient. Subchronic exposures, however, produce more persistent neurochemical changes that include depression in brain metabolic activity in the prefrontal cortex (PFC), as well as within structures of the auditory system and the reticular nucleus of the thalamus, and resemble more closely the alterations observed in schizophrenia patients (Jentsch and Roth, 1999; Cochran et al., 2003). These subchronic exposures lead to reduced expression of the calcium-binding protein parvalbumin (PV) in GABAergic interneurons in rodents and nonhuman primates (Cochran et al., 2003; Keilhoff et al., 2004; Rujescu et al., 2006; Morrow et al., 2007), a deficiency that has been consistently observed in postmortem brain samples of schizophrenic patients. Initial studies showing decreased expression of GAD67, the main isoform synthesizing GABA (Akbarian et al., 1995), were subsequently supported by studies showing that the expression of PV in a subset of fast-spiking inhibitory interneurons was also reduced in postmortem samples of schizophrenic patients (Beasley and Reynolds, 1997; Hashimoto et al., 2003). This loss of GABAergic phenotype in PV-interneurons led to the suggestion that dysfunction of these fast-spiking inhibitory interneurons may be a core feature of the disease (Lewis et al., 2005). We have recently confirmed that prolonged exposure to ketamine and other NMDA-R antagonists decreases GAD67 and parvalbumin in the same neurons in vitro (Kinney et al., 2006), and that exposure to subanesthetic doses on 2 consecutive days is sufficient to induce the loss of phenotype of these interneurons in mouse PFC (Behrens et al., 2007).
Accumulating evidence suggests that schizophrenia patients suffer from diminished antioxidant defenses, and a recent clinical trial showed that increasing these defenses may ameliorate symptoms of the disease (Berk et al., 2008). We have recently shown that the initial disinhibition caused by exposure to ketamine at subanesthetic doses induced the activation of the superoxide-producing enzyme NADPH oxidase (Nox) in vitro and in vivo, and that the superoxide produced is responsible for the loss of phenotype of PV-interneurons (Behrens et al., 2007). Furthermore, we showed that preventing the activation of Nox ameliorates the effects of ketamine on these interneurons. However, the mechanism(s) by which ketamine induces the activation of Nox were unknown. Here we present data showing that neuronal production of interleukin-6 (IL-6) is necessary and sufficient to induce and activate Nox on ketamine exposure in vitro and in vivo.
Materials and Methods
Materials.
Antibodies used were as follows: anti-parvalbumin, anti-calbindin, and anti-calretinin rabbit polyclonals from Swant, anti-GAD67 monoclonal from Millipore Bioscience Research Reagents, anti-Nox2 from Santa Cruz Biotechnology; anti-IL-6-blocking antibodies were goat anti-recombinant mouse IL-6 total IgG fraction, and rat anti-recombinant mouse IL-6 (IgG1) from R&D Systems. Unless otherwise stated, all reagents were from Sigma and tissue culture media from Invitrogen. The spin traps 5-(diisopropoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DIPPMPO) and 5-(diethylphosphoryl)-5-methyl-1-pyrroline-N-oxide (DEPMPO) were from Alexis Biochemicals.
Maintenance of mice and administration of ketamine and dihydroethidium.
Male C57BL/6, gp91phox−/−, and IL-6−/− mice were obtained from The Jackson Laboratory at 8 weeks of age and housed in our facility until 12–15 weeks, when they were used for experiments. Ketamine (30 mg/kg) was applied intraperitoneally singly or on 2 consecutive days at ∼4:00 P.M. Dihydroethidium (DHE) was applied 30 min after the last ketamine injection as described previously (Quick et al., 2008). Briefly, two serial intraperitoneal injections of freshly prepared dihydroethidium (27 mg/kg) were given at 30 min intervals. Eighteen hours later, mice were anesthetized with inhaled halothane and perfused intracardially with cold saline, followed by 4% paraformaldehyde in PBS for immunohistochemistry experiments, or their brains were dissected and their forebrains processed for synaptosomal preparations (see below). Experimental groups for immunohistochemistry were as follows: 1 d ketamine (30 mg/kg) treatment, 4 saline and 5 ketamine C57BL/6 mice; 2 d ketamine (30 mg/kg) and recovery, 5 ketamine (day 1 recovery), 5 ketamine (day 3 recovery), and 5 ketamine (day 10 recovery), 15 saline (5 of each killed at the same time as the ketamine groups); ketamine effects on gp91phox−/−, 5 C57BL/6 and 7 gp91phox−/− for saline, 4 C57BL/6 and 5 gp91phox−/− for ketamine (30 mg/kg) treatment; ketamine effects on IL-6−/−, 4 C57BL/6 and 4 IL-6−/− for saline, and 4 C57BL/6 and 4 IL-6−/− for ketamine (30 mg/kg) treatment. All animal studies were approved by the Animal Care Program at the University of California, San Diego, and are in accordance with the US Public Health Service Guide for the Care and Use of Laboratory Animals, US Department of Agriculture regulations, and the American Veterinary Medical Association Panel on Euthanasia.
Cortical neuronal cultures.
Cultures were prepared form cortices of Swiss Webster E15 mouse embryos and grown on poly-lysine-coated coverslips for 21 d as described previously (Kinney et al., 2006; Behrens et al., 2007).
Synaptosomal preparations.
Synaptosomes were obtained from adult mouse forebrains as described previously (Behrens et al., 2007).
RT-PCR.
RNA was extracted from primary cultures or forebrain using TRIzol. After reverse transcription of 1 μg of mRNA, the cDNA obtained was analyzed for IL-6, IL-1β, and tumor necrosis factor α (TNFα) expression using GAPDH as internal control. PCR conditions were as follows: 45 s at 95°C, 15 s at 45°C, and 1 min at 72°C for 35 cycles. IL-6 primers: forward, 5′-atggatgctaccaaactggat-3′ and reverse, 5′-tgaaggactctggctttgtct-3′. IL-1β primers: 5′-caaccaacaagtgatattctccatg-3′ and reverse: 5′-gatccacactctccagctgca-3′. TNFα primers: 5′-catcttctcaaaattcgagtgacaa-3′ and reverse: 5′-tgggagtagacaaggtacaaccc-3′. GAPDH primers: forward: 5′-gaacatcatccctgcctctactgg-3′ and reverse: 5′-tccaccaccctgttgctgta-3′.
Determination of superoxide production in live neuronal cultures by electron paramagnetic resonance spectroscopy.
Effects of ketamine treatment on the production of superoxide in neuronal cell cultures were determined by an electron paramagnetic resonance (EPR) spin trapping method using relatively lipophilic spin trap DIPPMPO (final concentration 30 mm) to allow rapid cellular delivery. Neurons were grown for 21 d, as described (Kinney et al., 2006), on “footed” plastic coverslips cut to the dimensions of the EPR tissue culture chamber. The cultures were treated with ketamine (0.5 μm) for the indicated times before transferring the coverslip to the EPR tissue culture chamber containing the spin trap in 40 μl of the growing media. The EPR cell was then tightly covered to avoid media leakage and inserted in the spectrometer to record EPR spectra using settings similar to those used in synaptosomes except that each spectrum was the average of 10 scans.
Superoxide detection in synaptosomes by EPR spectroscopy.
Detection of NADPH-dependent superoxide production in synaptosomal preparations was performed using spin-trapping EPR spectroscopy as described (Behrens et al., 2007). Immediately after mixing synaptosomes (∼0.1–0.2 mg of synaptosomal protein) with 70 mm DEPMPO and appropriate combinations of the substrates/inhibitors, the mixture was loaded into 50 μl glass capillary and introduced into the EPR cavity of a MiniScope MS200 Benchtop spectrometer (Magnettech) maintained at 37°C. We confirmed that the detected EPR signals are substrate specific, and not due to redox cycling in the studied mixtures, by lack of signals when DEPMPO was mixed with combinations of substrates and inhibitors in the absence of synaptosomes. EPR signals accumulated during the first 6 min after mixing with NADPH were quantified. To record quantifiable signals, the SOD inhibitor diethyldithiocarbamic acid (DETC; 5 mm) was included in the assay mixture. When analyzing the acute effect of IL-6 on superoxide production in synaptosomes, IL-6 (100 ng/ml final concentration) was applied for 5 min at room temperature before oxidase activity was triggered by the addition of NADPH (5 mm final concentration). EPR conditions were: microwave power, 5 mW; modulation amplitude, 2 G; modulation frequency, 100 kHz; sweep width, 150 G centered at 3349.0 G; scan rate, 7.5 G s−1, and each spectrum was the average of 5 scans.
Immunocytochemistry.
Fixation of neurons in culture was performed as described (Kinney et al., 2006). For double immunostaining, the coverslips were incubated in 2% normal goat serum containing the following primary antibodies (Ab): mAb against GAD67 (1:1000), Nox2 (1:200), a rabbit polyclonal Ab against Parvalbumin (1:4000), and incubated for 2 h at 37°C. Specific binding was detected by incubation for 45 min at room temperature with a 1:1000 dilution of secondary Abs conjugated to AlexaFluor dyes (568: red, 488: green).
Immunohistochemistry.
Brains were sliced using a vibratome into 50 μm coronal sections. Six sequential slices, encompassing the prefrontal cortex region (from Bregma 2.0–1.3), were processed for floating-section fluorescence immunohistochemistry for the detection of the calcium binding proteins parvalbumin, calbindin or calretinin, the last two costained for GAD67. Immunostaining for GAD67 cannot be performed in slices of animals that had been treated with DHE. Double-fluorescence immunohistochemistry for the calcium binding proteins and GAD67 was performed as described (Behrens et al., 2007). Briefly, slices were incubated in 1% sodium-borohydride for 15 min, followed by blocking in 10% normal goat serum (NGS). Primary antibodies were diluted in 2% NGS at the following dilutions: mouse anti-GAD67, 1:2000; rabbit anti-PV, 1:3000, rabbit anti-Calbindin, 1:5000; rabbit anti-calretinin 1:2000.
Confocal microscopy and image analysis.
Mounted slices or coverslips were evaluated for fluorescence under settings for 488 and 568 excitation on a LSM510 Meta multiphoton laser confocal microscope using a 10× (slices) and 40× (slices and coverslips) water-immersion objective. Oxidized DHE fluorescence was obtained using Ex λ 543 nm, Em λ > 590 nm. For slice imaging, each slice was imaged across the prelimbic region between bregmas 1.3 and 2.0 (two images per slice). Six slices were imaged per animal. For image quantification a z-stack of 8 images was obtained (corresponding to 1.4 μm on the z-axis). All PV-neurons in the images were analyzed for their parvalbumin and DHE content which corresponds to a mean of 300 ± 20 neurons per animal.
Image analysis of the neuronal population in primary cultures was essentially as described (Kinney et al., 2006). Briefly, coverslips are scanned to obtain 200–400 neurons (∼26–30 images captured per coverslip per condition using a 40× water-immersion objective). Each image analyzed consists of a stack of 16 0.2 μm Z-stage images taken from the base of the neurons and across 3.2 μm depth. When analyzing PV-interneurons in particular, the coverslips are scanned to obtain images as before but all PV-interneurons on the coverslip are imaged. Experiments were performed in duplicate coverslips of neurons prepared from the same dissection across at least three different dissections.
Confocal microscope settings were maintained constant for each series of experiments so that the resulting images could be analyzed by densitometry and the treatment-dependent changes in fluorescence compared and expressed as percentage of control (for primary cultures) or saline (for in vivo) conditions. For each experiment, both in vitro and in vivo, controls (saline) were processed in parallel such that variability due to antibody batch or laser power could be avoided. Neurons in the images were then analyzed for their somatic median green and red fluorescence intensity per cell using MetaMorph (scale of 0–255 arbitrary units).
Statistical analysis.
All intensity values were normalized by the mean obtained for the control (primary cultures) or saline (in vivo) conditions for each experiment, processed in parallel with the experimental group, and expressed as a percentage of this mean. To obtain the mean fluorescence/cell/animal, percentage values were averaged across the six slices of the same animal (or experiment in the case of primary cultures in coverslips), and the mean fluorescence intensity/cell/animal (or per experiment in the case of primary cultures) was used to calculate the mean and SD per group. These were then used for statistical analysis using SigmaStat software. Values obtained per experiment were analyzed by one- or two-way ANOVA followed by Tukey's post hoc test for multiple comparisons as indicated in each figure. ANOVA results were considered significant when p < 0.05.
Results
We have recently shown that exposure to subanesthetic levels of ketamine on 2 consecutive days induces a pronounced increase in brain superoxide through activation of NADPH-oxidase, and that this leads to the loss of phenotype of PV-interneurons in prefrontal cortex (Behrens et al., 2007). The effects of ketamine on PV-interneurons in the prefrontal region were observed only after exposure on 2 consecutive days, and not present 24 h following a single exposure (Fig. 1) (main effect of exposure time: F(1,16) = 22.766, p < 0.001), as previously reported for rat (Cochran et al., 2002). Furthermore, as previously shown in microdialysis studies in rats 24 h after exposure to a single injection of ketamine (Zuo et al., 2007) we did not observe increase in DHE oxidation in the prelimbic region of mice 24 h after a single injection of ketamine (data not shown). These results support the idea that repeated exposure to NMDA-R antagonists is required to produce persistent changes in PV-interneuron phenotype and function (Cochran et al., 2003; Keilhoff et al., 2004; Rujescu et al., 2006; Behrens et al., 2007; Morrow et al., 2007). To test for the enduring effects of the 2 d ketamine treatment on the loss of phenotype of PV-interneurons, adult male C57BL/6 mice were treated with saline or ketamine (30 mg/kg) on 2 consecutive days and the PV-interneuronal population in the prelimbic region was analyzed on days 1, 3, and 10 after the last ketamine injection. As previously described (Behrens et al., 2007), a pronounced decrease in the expression of PV and GAD67 in PV-interneurons was observed the day after the two ketamine injections (main effect of ketamine: F(1,32) = 67.299, p < 0.001 for PV, and F(1,32) = 55.411, p < 0.001 for GAD67) (Fig. 1). This effect waned slowly after drug withdrawal, but values remained significantly below saline controls 10 d after the last ketamine injection (treatment × time interaction: F(3,32) = 14.318, p < 0.001 for PV, and F(3,32) = 13.435, p < 0.001 for GAD67) (Fig. 1). The decrease was specific for the PV-interneuronal population, as demonstrated by the lack of effects of the 2 d ketamine treatment on prelimbic inhibitory interneurons expressing the calcium binding proteins calbindin (CB) or calretinin (CR). After two injections of ketamine the levels of CB and GAD67 in calbindin cells were (mean intensity ± SD n = 5 animals per condition): CB saline = 215.6 ± 32.1, CB ketamine = 231.6 ± 25.6 (F(1,8) = 0.558, n.s.); GAD67 saline: 39.1 ± 14.3, GAD67 ketamine = 44.2 ± 14.5 (F(1,8) = 0.319, n.s.), and those of CR and GAD67 in calretinin-expressing cells were: CR saline = 133.6 ± 35.6, CR ketamine = 139.4 ± 28.7 (F(1,8) = 0.079, n.s.); GAD67 saline: 30.1 ± 14.3, GAD67 ketamine = 44.2 ± 14.5 (F(1,8) = 0.583, n.s.).
Figure 1.
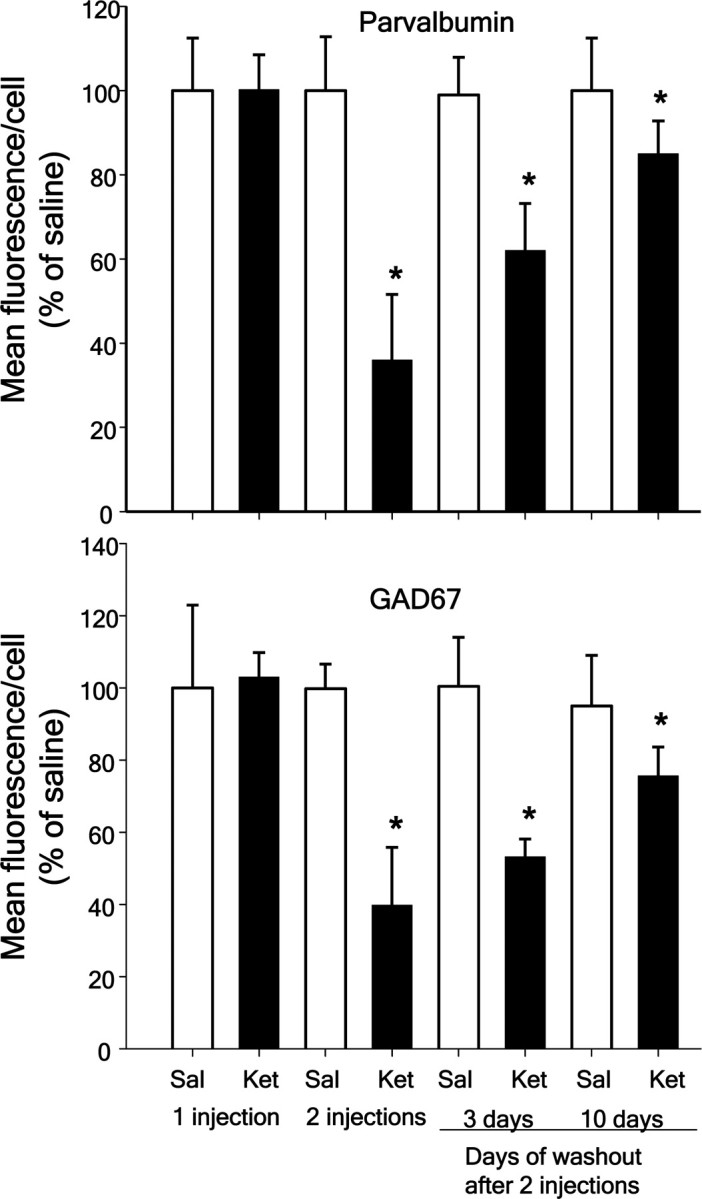
Slow reversal of ketamine effects on PV-interneurons in vivo. C57BL/6 mice (3 month-old males) were treated with ketamine (30 mg/kg i.p.) on 1 or 2 consecutive days as described previously (Behrens et al., 2007). Animals were killed either 24 h after a single injection (1 injection), 24 h after the second injection (2 injections), or 3 and 10 d after withdrawal of the second ketamine injection. Coronal brain sections comprising the prelimbic region were analyzed by fluorescence immunohistochemistry for PV and GAD67, and expressed as percentage of the 1 d saline-treated controls. A slow increase in fluorescence intensity for both proteins is observed at 3 d after the second ketamine injection. *p < 0.05 by two-way ANOVA followed by Tukey's post hoc multiple comparisons. Each time point consisted of five animals treated with saline and five animals treated with ketamine. Data are means ± SD. Mean fluorescence intensity for 1 d of saline: PV = 160.6 ± 13.3; GAD67 = 110.4 ± 8.6.
To confirm the role of Nox2-dependent NADPH oxidase (Nox2) in the superoxide mediated loss of phenotype of PV-interneurons, we exposed adult Nox2-deficient (gp91phox−/−) male mice to ketamine (30 mg/kg) on 2 consecutive days, and injected dihydroethidium (DHE) 30 min after the last ketamine treatment to measure superoxide production as described previously (Behrens et al., 2007). Analysis of the prelimbic region showed that deletion of Nox2 prevented the increase in superoxide induced by ketamine (Fig. 2A), and protected the phenotype of PV-interneurons (Fig. 2B) (genotype × treatment: F(1,17) = 13.480, p < 0.01 for PV, and F(1,17) = 55.472, p < 0.001 for oxDHE). These results confirm the specific role of Nox2-dependent superoxide production in the loss of phenotype of PV-interneurons caused by ketamine exposure. Increased basal level of superoxide production in gp91phox−/− animals were previously observed, and attributed to developmental compensatory mechanisms that lead to increased expression of other Nox subunits (Byrne et al., 2003; Liu et al., 2007). We also observed an increased basal level of DHE oxidation in brains of Nox2-deficient animals (Fig. 2A) (main effect of genotype F(1,17) = 5.614, p < 0.05). However, this level of superoxide production was not sufficient to affect PV-interneurons (Fig. 2B). These results give strong support to a specific role of Nox2-dependent activation in the effects of NMDA-R antagonists on PV-interneurons.
Figure 2.
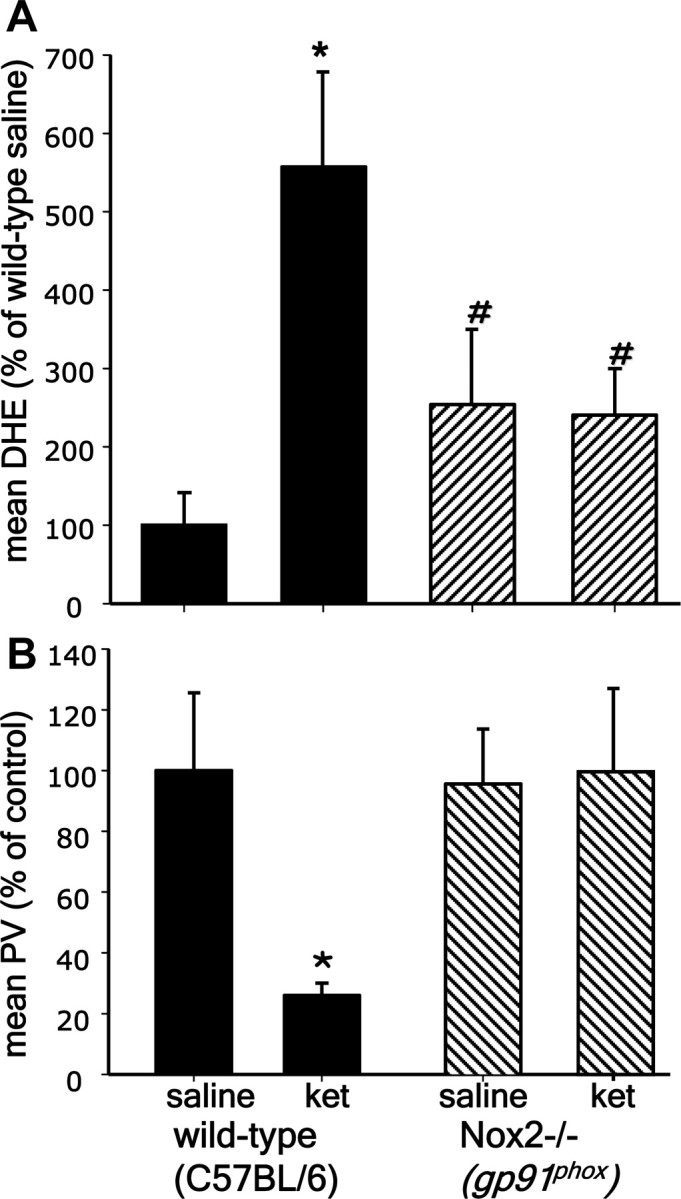
Absence of ketamine effects in the PFC of Nox2 knock-out mice. Three-month-old wild-type (C57BL/6) or gp91phox−/− mice were treated with ketamine (30 mg/kg i.p. on 2 consecutive days) followed by DHE injections. Coronal sections comprising the prelimbic and infralimbic regions were analyzed for (A) oxidized DHE, and (B) PV immunofluorescence. Fluorescence intensity is expressed as percentage of saline-treated C57BL/6 animals. *p < 0.001 and #p < 0.05. Data are means ± SD; n = 4–5 for wild type and 5–7 animals for gp91phox. Mean baseline fluorescence intensity for saline C57BL/6 control: ox-DHE = 9.8 ± 1.5; PV = 147.2 ± 23.3.
IL-6 reproduces ketamine effects on PV-interneurons
The important role played by Nox2-dependent NADPH oxidase in inflammatory processes outside the central nervous system led us to consider the possible involvement of proinflammatory molecules in the effects of ketamine. We focused on the proinflammatory cytokine IL-6 in the selective vulnerability of the PV-interneuronal population because of results showing that mice overexpressing IL-6 in astrocytes have a reduced number of PV-interneurons in adult brain (Samland et al., 2003). When primary neuronal cultures were exposed to IL-6 (10 ng/ml for 24 h), we observed a decrease in parvalbumin and GAD67 in PV-interneurons (Fig. 3), suggesting that IL-6 is able to fully reproduce the ketamine effects we previously showed in cultured neurons (Kinney et al., 2006; Behrens et al., 2007). IL-6 effects on PV-interneurons were prevented by coexposure to the NADPH oxidase inhibitor apocynin (4-hydroxy-3-methoxyacetophenone). One-way ANOVA analysis yielded a significant effect of IL-6 (PV: F(2,9) = 7.796, p < 0.001, and GAD67: F(2,9) = 30.004, p < 0.001), indicating that, similar to ketamine, the interleukin effects were mediated by activation of Nox2-dependent NADPH oxidase superoxide production (Fig. 3). To confirm that IL-6 mediates the increase in Nox2, we analyzed Nox2 expression by immunocytochemistry and its activity by determination of oxidized dihydroethidium (oxDHE). Primary cortical neurons exposed to IL-6 for 24 h showed a pronounced increase in the expression of Nox2 (F(1,8) = 13.337, p < 0.05), as well as an increase in superoxide production (F(1,6) = 17.224, p < 0.05) (Fig. 4). The superoxide production was eliminated when apocynin was added along with IL-6, whereas Nox2 induction by IL-6 was not affected by the oxidase inhibitor (main effect of IL-6 treatment oxDHE: F(2,12) = 11.238, p < 0.01; Nox2: F(2,12): 9.054, p < 0.001). These results suggest that IL-6 is the downstream mediator of ketamine in the induction of Nox2.
Figure 3.

IL-6 exposure leads to the loss of phenotype of PV-interneurons in primary neuronal cultures. Neuronal cultures were treated with IL-6 (10 ng/ml) in the absence (control) or presence of the Nox2 inhibitor apocynin (0.5 mm) for 24 h. Fluorescence confocal images of representative fields depicting the expression of PV and GAD67 in PV-interneurons. Bar graph represents the quantification of fluorescence expressed as percentage of control. (*p < 0.01, #p < 0.001. n = 4 experiments per condition.) Data are means ± SEM. Baseline intensities: PV = 135 ± 32; GAD67 = 114 ± 26.
Figure 4.
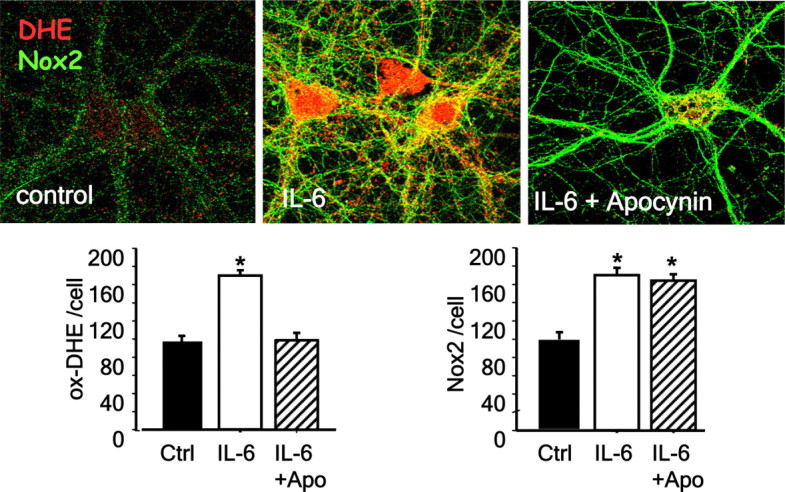
IL-6 increases superoxide production and Nox2 expression in neurons. Neuronal cultures were treated with IL-6 (10 ng/ml) in the absence (control) or presence of the Nox2 inhibitor apocynin (0.5 mm) for 24 h. DHE (1 μg/ml) was added during the last hour of treatment. Images show the increase in Nox2 immunoreactivity and oxidized DHE on treatment with IL-6. Bar graphs show the results of quantification of oxidized DHE and Nox2 fluorescence expressed as percentage of control. (*p < 0.05. n = 5 experiments per condition.) Data are means ± SD. Baseline intensities: DHE = 21 ± 4.7; Nox2 = 18.7 ± 2.6.
Ketamine exposure induces IL-6 expression
To directly examine whether ketamine exposure induced the expression of the cytokine in neurons, we exposed primary cortical cultures to ketamine and analyzed IL-6, IL-1β and TNFα mRNA at different time points during the 24 h exposure. PCR amplification of reverse transcribed mRNA showed that ketamine exposure induced a sustained increase in IL-6 transcript (F(3,6) = 28.274, p = 0.001) (Fig. 5, supplemental Fig. S1, available at www.jneurosci.org as supplemental material), without affecting the levels of other proinflammatory cytokines (supplemental Fig S1, available at www.jneurosci.org as supplemental material). A slight decrease in TNFα message was observed during ketamine exposure in vitro, in agreement with results observed after a 2 h exposure to PCP in vivo (Paterson et al., 2006). The level of IL-6 mRNA remained significantly elevated with respect to control conditions 24 h after ketamine (180 ± 18.1%, p < 0.01).
Figure 5.

Ketamine increases IL-6 mRNA expression. Primary neuronal cultures were exposed to ketamine (0.5 μm) for the times indicated and the abundance of IL-6 mRNA was determined by PCR using specific primers after reverse-transcription of mRNA obtained from the cultures. Values for IL-6 mRNA abundance were obtained after normalization by the expression of GAPDH mRNA in the samples. (*p < 0.05 with respect to control. n = 3 experiments per time-point.)
Sharp increases in IL-6 production have been shown under inflammatory conditions in brain, where the main sources of the cytokine are reactive astrocytes and microglia (Bedard and Krause, 2007). We have not observed signs of neurodegeneration or glial cell activation after ketamine exposures in primary cultures (Kinney et al., 2006), which suggests the absence of an overt inflammatory process under these conditions. Nevertheless, to test whether glial cells were responsible for the increase in IL-6 on ketamine exposure, we applied the NMDA-R antagonist to neurons in the absence of the astrocytic layer, and analyzed the PV-interneuronal population 24 h later. Ketamine produced a similar increase in DHE oxidation and loss of phenotype of PV-interneurons in the presence or absence of the astrocytic layer (Fig. 6) (PV: F(3.14) = 7.569, p = 0.003; GAD67: F(3,14) = 10.103, p < 0.001; oxDHE: F(3,14) = 94.583, p < 0.001), suggesting that if IL-6 mediates these effects, it must be of neuronal origin. To confirm this hypothesis, we applied IL-6 blocking antibodies (Smith et al., 2007) during the 24 h exposure of primary neurons to ketamine in the absence of the astrocytic layer. Blocking IL-6 completely prevented ketamine effects on PV-interneurons (Fig. 7A, supplemental Fig. S2, available at www.jneurosci.org as supplemental material) (PV: F(4,15) = 4.564, p < 0.05, GAD67: F(4,15) = 27.512, p < 0.001) and also the increase in superoxide (Fig. 7B) (oxDHE: F(4,10) = 46.415, p < 0.001), indicating that IL-6 is the downstream mediator of ketamine effects on Nox2 induction and activation. For these experiments we used varying concentrations of two different blocking antibodies [goat anti-IL-6 total IgG (Fig. 7; supplemental Fig. S3, available at www.jneurosci.org as supplemental material) and rat anti-IL-6 IgG1 (supplemental Figs. S2, S3, available at www.jneurosci.org as supplemental material), both made against mouse IL-6). The blocking capacity of these two antibodies differ by a factor of 10 (as described by manufacturer), and we have observed a similar difference when blocking ketamine effects (Fig. 7; supplemental Fig. S2, available at www.jneurosci.org as supplemental material, compare concentration dependence). To control for nonspecific effects of the blocking antibodies, these were heat inactivated and added to the cultures at 1 μg/ml (goat anti-rmIL-6) or 0.1 μg/ml (rat anti-IL-6). As a further control we, also used another antibody produced in rat (anti-GFAP, IgG2A) at a similar concentration as the rat anti-IL-6 blocking antibody (0.1 μg/ml). Under these conditions, ketamine effects on PV-interneurons were prevented only by the active IL-6-blocking antibodies (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) (F(6,26) = 33.726, p < 0.001).
Figure 6.

Ketamine effects on PV-interneurons do not require the presence of astrocytes. Primary neuronal cultures were grown on glass coverslips with “feet” as described (Kinney et al., 2006). After 21 d of development in vitro, the cultures were treated with ketamine (0.5 μm for 24 h) in the presence or absence of the astrocytic layer. For this, the coverslips containing neurons were separated from the astrocytic layer by transfer of the coverslip together with its media into an empty well. DHE was added for the last hour of treatment as described (Behrens et al., 2007). After treatment, neurons were fixed and processed for immunofluorescence for detection of either PV or GAD67 or for oxidized DHE. *,#Statistical significant difference compared with control conditions at p < 0.001. n = 4–5 experiments per condition. Data are means ± SEM. Baseline intensities: PV = 210 ± 32; GAD67 = 195 ± 26.
Figure 7.

Blocking IL-6 with antibodies prevents ketamine effects on PV-interneurons. Primary neuronal cultures were exposed to ketamine in the absence of the astrocytic monolayer and in the presence of an anti-mouse IL-6 blocking antibody produced in goat (α-mIL-6). A, Increasing concentrations of α-mIL-6 prevented the decrease in PV and GAD67 after 24 h of ketamine exposure. Bar graph show results for fluorescence quantification of both antigens in PV-interneurons expressed as % of control. *,**p < 0.05; #,## p < 0.001. n = 4 experiments per condition. Baseline intensities: PV = 165 ± 30; GAD67 = 127 ± 28. B, Neuronal cultures were treated as in A, and DHE was added for the last hour of treatment. After fixation, the coverslips were processed for immunocytochemistry for parvalbumin (PV, green). Bar graphs show results for oxidized DHE fluorescence (red) intensity analysis in all neurons including PV-interneurons. (*p < 0.001 compared with control and **p < 0.001 compared with ketamine. n = 3 experiments per condition.) Baseline intensities: DHE = 25.4 ± 5.4.
Ketamine does not lead to loss of GABAergic phenotype of PV-interneurons in IL-6−/− mice
To assess whether IL-6 and other inflammatory cytokines were induced in brain after ketamine exposure we analyzed the levels of mRNA for IL-6, IL-1β and TNFα, as previously shown for cultured neurons. Exposure to ketamine on 2 consecutive days only increased the levels of IL-6 mRNA (Fig. 8A), without affecting mRNA levels of IL-1β or TNFα (main effect of ketamine: IL-6: F(1,6) = 12.775, p = 0.012; TNFα: F(1,6) = 0.637, n.s; IL-1β: F(1,6) = 0.411, n.s). To further assess the role of IL-6 in ketamine effects in vivo, we exposed IL-6-deficient mice to ketamine on 2 consecutive days, and analyzed the PV-interneuronal population in the prefrontal region, as well as the activity of Nox2-dependent superoxide production by DHE oxidation. Lack of in vivo production of IL-6 prevented ketamine activation of NADPH oxidase, as determined by the diminished DHE oxidation in the IL-6-deficient mice (Fig. 8B,C). A two-way ANOVA (treatment × genotype) yielded significant effects of treatment in wild-type animals (oxDHE: F(1,11) = 31.432. p < 0.001; PV: F(1,11) = 31.690, p < 0.001). Moreover, the phenotype of PV-interneurons in the prefrontal region was preserved in the IL-6-deficient animals (Fig. 8, bar graphs). These results strongly suggest that CNS production of IL-6 is necessary and sufficient for the increase in Nox2-dependent NADPH oxidase activity that leads to the loss of phenotype of PV-interneurons observed after ketamine exposure.
Figure 8.
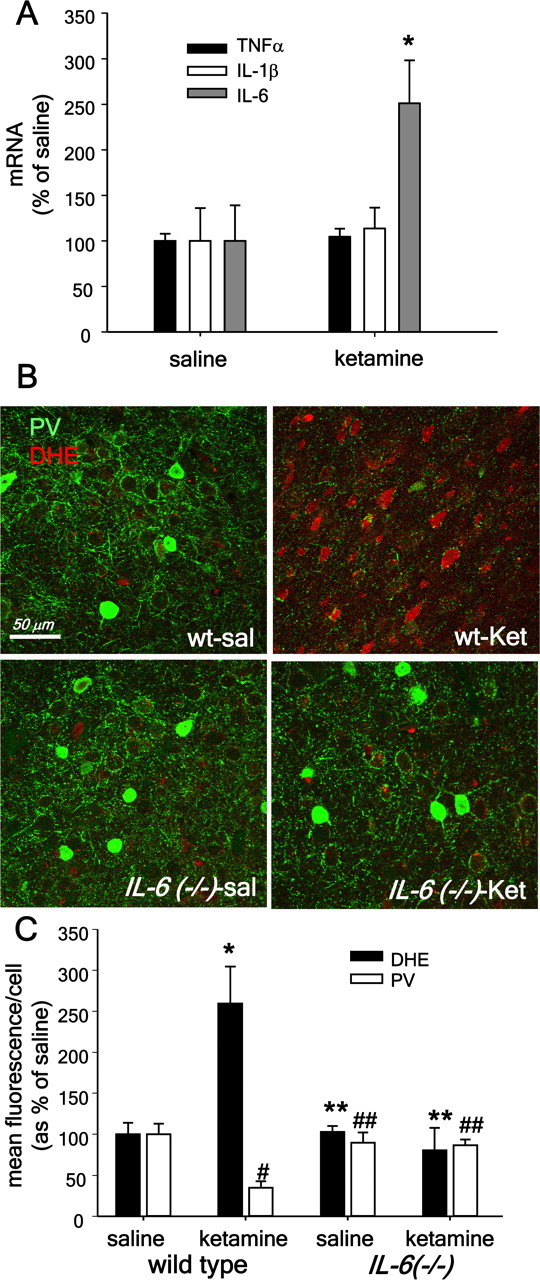
CNS production of IL-6 mediates ketamine effects on Nox and PV-interneurons in vivo. A, Animals were treated with saline or ketamine (30 mg/kg) on two consecutive days and the brains extracted for mRNA preparation 24 h after the last ketamine injection. The abundance of IL-6, IL-1β, and TNFα mRNA was determined by PCR using specific primers after reverse-transcription of mRNA obtained from forebrains. Values for mRNA abundance were obtained after normalization by the expression of GAPDH mRNA in the samples. (*Statistically significant differences compared with control conditions. n = 4 animals per condition.) B, C, Three-month-old C57BL/6 (wt) or IL-6-deficient (IL-6−/−) male mice were treated with ketamine (30 mg/kg) on 2 consecutive days, followed by DHE, as described previously (Behrens et al., 2007). Coronal sections comprising the prelimbic and infralimbic regions were analyzed by immunohistochemistry for PV and oxidized DHE fluorescence. Ketamine produced a substantial increase in oxidized DHE in wild-type mice but not in IL-6−/− animals (*p < 0.001 wt-sal vs wt-ket; **p = 0.001 wt-ket vs IL-6−/− sal or ket). The loss of parvalbumin expression induced by ketamine was prevented in the IL-6−/− animals. (#p < 0.001 wt-sal vs wt-ketamine; ##p < 0.001, wt-ketamine vs IL-6. n = 4 animals per condition). Data are means ± SD. Mean fluorescence intensity for saline: wild type, PV = 111.6 ± 9.3; ox-DHE = 10.1 ± 2.5; IL-6−/−, PV = 101.7 ± 10.2; ox-DHE = 11.7 ± 3.2.
IL-6 directly activates NADPH oxidase
Superoxide production by live neurons, as analyzed by EPR, increased rapidly after ketamine exposure (Fig. 9A). To confirm that this effect of ketamine was mediated by IL-6, we applied a blocking antibody against IL-6 during the exposure to ketamine and analyzed the activity of Nox by EPR in live cultures as before. Blocking IL-6 action with the antibody prevented the activation of Nox by ketamine (Fig. 9A) (F(2,24) = 7.786, p < 0.01). Moreover, to further test whether IL-6 triggers the signaling cascades that activate the oxidase, we exposed synaptosomal preparations to IL-6 (100 ng/ml) and assayed superoxide production by EPR. IL-6 produced a small but significant increase in superoxide that was completely blocked by coexposure to apocynin (F(3,22) = 12.056, p < 0.001), demonstrating that it was produced by Nox2-dependent NADPH oxidase (Fig. 9B).
Figure 9.

Ketamine-induced IL-6 release directly activates Nox. A, EPR assessment of superoxide production in live cultures on treatment with ketamine (0.5 μm). Primary cultures were exposed to ketamine for the times indicated in the absence or presence of an anti-mouse IL-6 blocking antibody produced in rat (αIL-6, 0.1 μg/ml). At the indicated times, the coverslips were transferred to a quartz chamber and superoxide production was followed by EPR spectroscopy using the spin-trap DIPPMPO. Ketamine induced a rapid increase in superoxide signals that were significantly reduced by the blocking antibody (*Statistically significant difference compared with control, p < 0.05 and p < 0.001 for 1 and 3 h, respectively. #Statistically significant difference compared with ketamine, p < 0.05. n = 3–6 experiments per condition.) B, IL-6 (100 ng/ml) increased basal NADPH oxidase activity in synaptosomes isolated from 3-month-old C57BL/6 male forebrains. IL-6 was preincubated with synaptosomal preparations for 5 min before triggering oxidase activity by addition of substrate, NADPH. Apocynin (0.4 mm) was applied 5 min before IL-6. Accumulation of superoxide during the first 6 min was analyzed using the spin trap DEPMPO. Data are means ± SEM (*p < 0.01. n = 6–7 experiments per condition.) Data are means ± SEM.
Discussion
Mild developmental derangements caused either by a genetic predisposition or by immune activation early during development are believed to contribute to the appearance of schizophrenic symptoms in early adulthood. Maternal viral infection has received special attention due to several reports showing a strong correlation between infections in middle-gestation and the incidence of schizophrenia in the offspring (for review, see Brown, 2006, 2008). Recent studies have shown that IL-6 is involved in the subsequent schizophrenia-like behavior observed in the offspring in the rodent maternal-infection model of the disease (Smith et al., 2007), and a specific vulnerability of PV-interneurons in adulthood was also observed in this model (Meyer et al., 2008a). Further confirmation for a role of IL-6 in the vulnerability of these interneurons derives from data showing that mice overexpressing IL-6 in astrocytes show selective loss of PV-interneurons (Samland et al., 2003). Our present data, showing that neuronal production of IL-6 during pharmacological inhibition of NMDA-R in vivo and in vitro is responsible for the induction of Nox2-dependent superoxide production and subsequent loss of phenotype of PV-interneurons, suggest that IL-6-mediated Nox activation may be responsible for the neurodevelopmental derangements occurring in the maternal infection and, possibly, in the neonatal NMDA-R antagonist models of the disease (Smith et al., 2007; Wang et al., 2008). In support of this idea, recent results show that decreased antioxidant capacity during this early postnatal period induce cognitive derangements that are relevant to schizophrenia (Cabungcal et al., 2007).
Decreased levels of glutathione (GSH), which is responsible for detoxification of reactive oxygen and other radical species, have been shown in CSF of drug-naive schizophrenia patients (Do et al., 2000), as well as in postmortem studies (Yao et al., 2006). Polymorphisms in genes coding for enzymes that participate in GSH synthesis have been linked to schizophrenia risk, and decreasing GSH levels during development leads to hypofunctional NMDA-Rs in adulthood (Tosic et al., 2006; Gysin et al., 2007). These data, together with our results showing that NMDA-R antagonist exposures lead to the activation of Nox (Behrens et al., 2007) and that this occurs through an IL-6-mediated mechanism (present results) strongly suggest that strategies that prevent increased oxidative mechanisms in brain may represent a valid therapeutic intervention to prevent the dysfunction of inhibitory circuits in schizophrenia. Indeed, a recent double-blind placebo controlled clinical trial has shown that augmenting production of GSH with N-acetyl cysteine treatment may be of clinical benefit in the treatment of schizophrenia (Berk et al., 2008).
The role of IL-6 in brain development and CNS disease is not well understood. Studies indicate that a correct balance of proinflammatory (such as IL-6) and anti-inflammatory cytokines (such as IL-10) is required for normal brain development (Meyer et al., 2008b). Several studies also suggest that IL-6 may have a neuroprotective role in conditions of overt neuronal damage (Gadient and Otten, 1997). However, it seems the levels and cell-types producing the interleukin may determine its role in neuroprotection or neurodegeneration (Jonakait, 2007). IL-6 and its receptor are expressed by neurons, astrocytes and microglia, and expression in the last two cell types increases substantially under inflammatory conditions (Gadient and Otten, 1997). However, a specific role for IL-6 production in neurons is suggested by studies showing that depolarization increases neuronal production of the cytokine (Sallmann et al., 2000), and by results showing that exposure to this cytokine inhibits neurotransmitter release and the spread of excitation in cerebral cortex (D'Arcangelo et al., 2000). Furthermore, production of IL-6 in the CNS was shown to be directly involved in the disruption of working memory caused by peripheral inflammation (Sparkman et al., 2006), as well as in the amount of stored information during in vivo LTP (Balschun et al., 2004).
The enduring effects of ketamine and IL-6 on inhibitory circuits described in our study agree with recent results in rat prelimbic cortex and dorsal horn neurons showing that ketamine (Zhang et al., 2008) or IL-6 (Kawasaki et al., 2008) can produce disinhibition of excitatory circuits by inducing a suppression of inhibitory transmission. Our results further show that neuronal IL-6 secretion is responsible for the activation of redox mechanisms, specifically Nox, after ketamine exposure. It is expected then that this increased pro-oxidant condition will affect glutamatergic transmission. Such a condition would occur during the disinhibition created by acute NMDA-R antagonist exposure. The sustained increase in glutamate release caused by lack of inhibitory control will lead to synthesis and release of IL-6, which then acts on neuronal targets to increase Nox activity and thus production of free radicals. Because the glutamatergic synapse is highly sensitive to oxidizing conditions (Kohr et al., 1994; Volterra et al., 1994; Pinteaux et al., 1996; Lipton et al., 2002; Herin and Aizenman, 2004; Mustafa et al., 2007), increased activity of Nox would lead to the oxidation of key synaptic proteins resulting in diminished release of, and response to, glutamate. Such a mechanism would be in essence a physiological neuroprotective strategy that does not produce changes in brain circuitry, as observed 24 h after exposure to NMDA-R antagonists (present results and Cochran et al., 2002; Zuo et al., 2007). However, this first exposure primes the brain to a sustained increase in IL-6 and to a pronounced activation of Nox on re-challenge (present results and Behrens et al., 2007), leading to the dysfunction of the subclass of fast-spiking interneurons critical to cognitive processes. In the adult brain, these effects would wane slowly on drug withdrawal, as shown here for mice. However, if such a mechanism occurs during the critical periods of PV-interneuron maturation, a deficiency in inhibitory circuits would be expected. NMDA-Rs in PV-interneurons show a different subunit composition than surrounding principal neurons (Kinney et al., 2006; Xi et al., 2008). This NMDA-R subunit composition, during the time-frame when active perisomatic innervation occurs (Chattopadhyaya et al., 2007), should make these cells highly sensitive to changes in oxidative conditions. Thus, prolonged inactivation of NMDA-Rs in PV-interneurons by Nox-dependent oxidation would trigger the intracellular processes resulting in reduced expression of its GABAergic markers, leading to the loss of their inhibitory capacity, and to the development of a chronically decreased inhibitory tone in cortex, as observed in schizophrenia.
Footnotes
This work was supported by The Larry Hillblom Foundation and The Hartford Foundation (L.L.D.). We thank Drs. Mary B. Kennedy, Athina Markou, and William F. Loomis for their insightful comments, and J. Lucero and G. Shekhtman for technical assistance.
References
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics.[comment] Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- Balschun D, Wetzel W, Del Rey A, Pitossi F, Schneider H, Zuschratter W, Besedovsky HO. Interleukin-6: a cytokine to forget. FASEB J. 2004;18:1788–1790. doi: 10.1096/fj.04-1625fje. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Reynolds GP. Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr Res. 1997;24:349–355. doi: 10.1016/s0920-9964(96)00122-3. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Judd F, Katz F, Katz P, Ording-Jespersen S, Little J, Conus P, Cuenod M, Do KQ, Bush AI. N-Acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64:361–368. doi: 10.1016/j.biopsych.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS. The risk for schizophrenia from childhood and adult infections. Am J Psychiatry. 2008;165:7–10. doi: 10.1176/appi.ajp.2007.07101637. [DOI] [PubMed] [Google Scholar]
- Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802–805. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- Cabungcal JH, Preissmann D, Delseth C, Cuénod M, Do KQ, Schenk F. Transitory glutathione deficit during brain development induces cognitive impairment in juvenile and adult rats: relevance to schizophrenia. Neurobiol Dis. 2007;26:634–645. doi: 10.1016/j.nbd.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 2007;54:889–903. doi: 10.1016/j.neuron.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran SM, Fujimura M, Morris BJ, Pratt JA. Acute and delayed effects of phencyclidine during mRNA levels of markers of glutamatergic and GABAergic neurotransmitter function in the rat brain. Synapse. 2002;46:206–214. doi: 10.1002/syn.10126. [DOI] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Tancredi V, Onofri F, D'Antuono M, Giovedi S, Benfenati F. Interleukin-6 inhibits neurotransmitter release and the spread of excitation in the rat cerebral cortex. Eur J Neurosci. 2000;12:1241–1252. doi: 10.1046/j.1460-9568.2000.00011.x. [DOI] [PubMed] [Google Scholar]
- Do KQ, Trabesinger AH, Kirsten-Krüger M, Lauer CJ, Dydak U, Hell D, Holsboer F, Boesiger P, Cuénod M. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci. 2000;12:3721–3728. doi: 10.1046/j.1460-9568.2000.00229.x. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten UH. Interleukin-6 (IL-6)–a molecule with both beneficial and destructive potentials. Prog Neurobiol. 1997;52:379–390. doi: 10.1016/s0301-0082(97)00021-x. [DOI] [PubMed] [Google Scholar]
- Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P, Deppen P, Preisig M, Ruiz V, Steullet P, Tosic M, Werge T, Cuénod M, Do KQ. Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci U S A. 2007;104:16621–16626. doi: 10.1073/pnas.0706778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herin GA, Aizenman E. Amino terminal domain regulation of NMDA receptor function. Eur J Pharmacol. 2004;500:101–111. doi: 10.1016/j.ejphar.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Holcomb HH, Lahti AC, Medoff DR, Cullen T, Tamminga CA. Effects of noncompetitive NMDA receptor blockade on anterior cingulate cerebral blood flow in volunteers with schizophrenia. Neuropsychopharmacology. 2005;30:2275–2282. doi: 10.1038/sj.npp.1300824. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. Glutamate and schizophrenia: phencyclidine, N-Methyl-d-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jonakait GM. The effects of maternal inflammation on neuronal development: possible mechanisms. Int J Dev Neurosci. 2007;25:415–425. doi: 10.1016/j.ijdevneu.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilhoff G, Becker A, Grecksch G, Wolf G, Bernstein HG. Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience. 2004;126:591–598. doi: 10.1016/j.neuroscience.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G, Eckardt S, Lüddens H, Monyer H, Seeburg PH. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron. 1994;12:1031–1040. doi: 10.1016/0896-6273(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- Liu S, Tang J, Ostrowski RP, Titova E, Monroe C, Chen W, Lo W, Martin R, Zhang JH. Oxidative stress after subarachnoid hemorrhage in gp91phox knockout mice. Can J Neurol Sci. 2007;34:356–361. doi: 10.1017/s031716710000682x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008a;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Yee BK. A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull. 2008b doi: 10.1093/schbul/sbn022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacology (Berl) 2007;192:283–290. doi: 10.1007/s00213-007-0708-0. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Kumar M, Selvakumar B, Ho GP, Ehmsen JT, Barrow RK, Amzel LM, Snyder SH. Nitric oxide S-nitrosylates serine racemase, mediating feedback inhibition of D-serine formation. Proc Natl Acad Sci U S A. 2007;104:2950–2955. doi: 10.1073/pnas.0611620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Paterson GJ, Ohashi Y, Reynolds GP, Pratt JA, Morris BJ. Selective increases in the cytokine, TNFalpha, in the prefrontal cortex of PCP-treated rats and human schizophrenic subjects: influence of antipsychotic drugs. J Psychopharmacol. 2006;20:636–642. doi: 10.1177/0269881106062025. [DOI] [PubMed] [Google Scholar]
- Pinteaux E, Copin JC, Ledig M, Tholey G. Modulation of oxygen-radical-scavenging enzymes by oxidative stress in primary cultures of rat astroglial cells. Dev Neurosci. 1996;18:397–404. doi: 10.1159/000111433. [DOI] [PubMed] [Google Scholar]
- Quick KL, Ali SS, Arch R, Xiong C, Wozniak D, Dugan LL. A carboxyfullerene SOD mimetic improves cognition and extends the lifespan of mice. Neurobiol Aging. 2008;29:117–128. doi: 10.1016/j.neurobiolaging.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, Giegling I, Genius J, McCarley RW, Möller HJ, Grunze H. A pharmacological model for psychosis based on N-methyl-D-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry. 2006;59:721–729. doi: 10.1016/j.biopsych.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Sallmann S, Juttler E, Prinz S, Petersen N, Knopf U, Weiser T, Schwaninger M. Induction of interleukin-6 by depolarization of neurons. J Neurosci. 2000;20:8637–8642. doi: 10.1523/JNEUROSCI.20-23-08637.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samland H, Huitron-Resendiz S, Masliah E, Criado J, Henriksen SJ, Campbell IL. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J Neurosci Res. 2003;73:176–187. doi: 10.1002/jnr.10635. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J Neurosci. 2006;26:10709–10716. doi: 10.1523/JNEUROSCI.3376-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomitaka S, Tomitaka M, Tolliver BK, Sharp FR. Bilateral blockade of NMDA receptors in anterior thalamus by dizocilpine (MK-801) injures pyramidal neurons in rat retrosplenial cortex. Eur J Neurosci. 2000;12:1420–1430. doi: 10.1046/j.1460-9568.2000.00018.x. [DOI] [PubMed] [Google Scholar]
- Tosic M, Ott J, Barral S, Bovet P, Deppen P, Gheorghita F, Matthey ML, Parnas J, Preisig M, Saraga M, Solida A, Timm S, Wang AG, Werge T, Cuénod M, Do KQ. Schizophrenia and oxidative stress: glutamate cysteine ligase modifier as a susceptibility gene. Am J Hum Genet. 2006;79:586–592. doi: 10.1086/507566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Floridi S, Racagni G. Reactive oxygen species inhibit high-affinity glutamate uptake: molecular mechanism and neuropathological implications. Ann N Y Acad Sci. 1994;738:153–162. doi: 10.1111/j.1749-6632.1994.tb21800.x. [DOI] [PubMed] [Google Scholar]
- Wang CZ, Yang SF, Xia Y, Johnson KM. Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology. 2008;33:2442–2455. doi: 10.1038/sj.npp.1301647. [DOI] [PubMed] [Google Scholar]
- Xi D, Keeler B, Zhang W, Houle JD, Gao WJ. NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J Neurosci Methods. 2008 doi: 10.1016/j.jneumeth.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Dis Markers. 2006;22:83–93. doi: 10.1155/2006/248387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Behrens MM, Lisman JE. Prolonged exposure to NMDAR antagonist suppresses inhibitory synaptic transmission in prefrontal cortex. J Neurophysiol. 2008;100:959–965. doi: 10.1152/jn.00079.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo DY, Wu YL, Yao WX, Cao Y, Wu CF, Tanaka M. Effect of MK-801 and ketamine on hydroxyl radical generation in the posterior cingulate and retrosplenial cortex of free-moving mice, as determined by in vivo microdialysis. Pharmacol Biochem Behav. 2007;86:1–7. doi: 10.1016/j.pbb.2006.05.010. [DOI] [PubMed] [Google Scholar]