

Abstract
Free radical generation is one of the primary causes of myocardial ischemia/reperfusion (I/R) injury. Melatonin is an efficient free radical scavenger and induces the expression of antioxidant enzymes. We have previously shown that melatonin can prevent free radical-induced myocardial injury. To date, the underlying mechanism of melatonin’s cardioprotective effect is not clear. In this study, we assessed the ability of melatonin to protect against I/R injury in mice deficient in glutathione peroxidase 1 (Gpx1). Hearts were subjected to 40 min of global ischemia in vitro followed by 45 min reperfusion. Myocardial I/R injury (expressed as % of recovery of left ventricular developed pressure × heart rate) was exacerbated in mice deficient in Gpx-1 (51 ± 3% for Gpx1+/+ mice vs. 31± 6 % for Gpx1-/- mice, p<0.05). Administration of melatonin for 30 min protected against I/R injury in both Gpx1+/+ mice (72 ± 4.8%) and Gpx1-/- mice (63 ± 4.7%). This protection was accompanied by a significant improvement of left ventricular end-diastolic pressure and a two-fold decrease in lactate dehydrogenase (LDH) released from melatonin-treated hearts. In another set of experiments, mice were subjected to 50 min of ligation of the left descending anterior coronary artery in vivo followed by 4 h of reperfusion. The infarct sizes, expressed as the percentage of the area at risk, were significantly larger in Gpx1-/- mice than in Gpx1+/+ mice (75 ± 9% vs. 54 ± 6%, P<0.05) and were reduced significantly in melatonin-treated mice (31 ± 3.7% Gpx1-/- mice and 33 ± 6.0 % Gpx1+/+ mice). In hearts subjected to 30 min of coronary artery occlusion followed by 3 h of reperfusion, melatonin-treated hearts had significantly fewer in situ oligo ligation positive myocytes and less protein nitration. Our results demonstrate that the cardioprotective function of melatonin is independent of Gpx1.
Keywords: glutathione peroxidase, melatonin, free radical scavenger, apoptosis, myocardial reperfusion injury
1. Introduction
Myocardial ischemia/reperfusion injury is mediated at least in part by the generation of reactive oxygen species such as hydroxyl radicals, superoxide anions, and H2O2 [1,2]. In previous studies, we showed that transgenic mice overexpressing antioxidant enzymes such as mitochondrial MnSOD or cytosolic CuZnSOD are more resistant to myocardial I/R injury [3,4]. Other studies have shown that transgenic mice overexpressing glutathione peroxidase are more resistant to I/R injury [5], whereas transgenic mice deficient in glutathione peroxidase are more susceptible to I/R injury [6]. These studies clearly demonstrate that free radicals play an important role in I/R injury.
The downstream events of free radical-induced I/R injury include apoptosis, or programmed cell death. Indeed, Kajstura et al [7, 8] showed that apoptosis was the predominant mode of cardiac cell death induced by coronary artery occlusion. Furthermore, our studies have shown that overexpression of the anti-apoptotic proteins Bcl-2, IAP-2, and bifunctional apoptosis regulator (BAR) inhibits apoptosis and protects against myocardial I/R injury in transgenic mice [9-11].
Because there is strong evidence that free radicals contribute to post-ischemic injury and therefore apoptosis, an effective anti-free radical agent would be highly useful in the prevention of I/R injury. One potential candidate is melatonin, a pineal indoleamine that has been shown to act as an antioxidant and a free radical scavenger of hydroxyl radicals, peroxyl radicals, and superoxide anions [12,13]. Melatonin is 5- and 14-times more effective in scavenging hydroxyl radical than glutathione and mannitol, respectively [14]. It is twice as efficient as vitamin E in removing peroxyl radicals [15], and it has been shown to directly scavenge hydrogen peroxide [16]. In addition to melatonin, several of its metabolites are likewise effective free radical scavengers [17].
We and others have demonstrated that melatonin protects the heart against I/R injury in wild-type mice and rats [18-25]. The protective effect of melatonin against apoptosis has also been observed in the brain, kidney, pancreas, and liver [26-29]. The mechanism of this protection likely involves melatonin’s direct scavenging function, but it may also include the induction of antioxidant enzymes such as Gpx1 [30-32].
The following experiments were designed to assess whether melatonin’s cardioprotective effect is mediated by induction of Gpx1 expression.
2. Methods and methods
2.1. Materials
The CytoTox 96 assay kit was obtained from Promega Corp. (Madison, WI). The ApopTag in situ oligo ligation (ISOL) kit was obtained from Chemicon (Temecula, CA). All other reagents were of the highest grades commercially available.
2.2. Animal studies
The generation of Gpx1-/- mice was described previously [33]. Since Gpx1 is the predominant form of the Gpx isozyme in the heart, the Gpx activity in knockout mice was less than 10% of the activity of wild-type mice [33]. These mice were housed in filtered-hood, large plastic cages. They were provided with clean, sterilized bedding and were permitted free access to food and water. These animals were free of pathogens during the entire course of experiments. All the animal protocols were approved by East Tennessee State University’s Animal Care and Use Committee and performed according to Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
2.3. Global ischemia in vitro
Gpx1+/+ and Gpx1-/- mice were injected with sodium heparin (500 units/kg body weight i.p.) 30 min prior to anesthetization with sodium pentobarbital (120 mg/kg, i.p.). Melatonin (150 μg/kg, i.p.) was given 30 min before the hearts were removed for in vitro perfusion. Hearts were rapidly excised and perfused retrogradely at 60 mm Hg by Langendorff technique with Krebs-Henseleit bicarbonate buffer as described [3]. After 30 min of preliminary perfusion, a range of end-diastolic pressures was tested to construct a functional curve. After 40 min of ischemia, hearts were reperfused for 45 min. Coronary effluent was collected for measurement of lactate dehydrogenase (LDH) release. At the end of perfusion, another functional index was measured.
Cardiac functions, such as left ventricular developed pressures (LVDP), ±dP/dt, heart rates, and coronary flow rates, recorded before ischemia and 45 min after reperfusion, were used for comparison. Percentages of functional recovery were calculated by dividing each of the functional recovery values at the end of reperfusion by their corresponding pre-ischemic values. End diastolic pressures prior to ischemia and after 45 min of reperfusion were calculated from the functional curves. The perfusate flowing out of the heart was collected and measured. Coronary flow rate was determined by the amount of perfusate measured in a specific time period.
2.4. Lactate dehydrogenase release
Cardiac injury was also assessed by measuring LDH release in the perfusate, as described previously [3]. LDH released from the heart was determined by a CytoTox 96 assay and an automated microplate reader (EL312, Bio-Tek Instruments, Winooski, VT). The amount of LDH released was expressed as total mU/mg heart protein.
2.5. Regional ischemia in vivo
Gpx1+/+ and Gpx1-/- mice (4-5 months old) were anesthetized with tribromoethanol (275 mg/kg, i.p.). Melatonin (150 μg/kg, i.p.) was given 30 min before left anterior descending coronary artery (LAD) ligation was performed. After 50 min of LAD ligation, the heart was reperfused for 4 h. Macroscopic staining with triphenyltetrazolium chloride (TTC) was used to quantify the infarct sizes. The area at risk was expressed as the percentage of the left ventricle, and the area of infarct was expressed as the percentage of the area at risk [3].
2.6. Detection of apoptosis by in situ oligo ligation
Our data showed that mouse hearts subjected to 50 min of focal ischemia developed more than 50% infarction. The presence of mixed populations of live and dead cells hampers the interpretation of apoptosis results in the risk area. Thus, a period of 30 min of ischemia and 3 h of reperfusion in the absence of cell death based on TTC staining was chosen for apoptosis analysis. In situ staining of DNA strand breaks in the heart was detected by the ApopTag in situ oligo ligation (ISOL) kit using oligo A according to the manufacturer’s instructions with some modifications. The endogenous biotin was blocked with an Avidin/Biotin Blocking kit (BioGenex, San Ramon, CA). TACS Blue label was used as a peroxidase substrate, and nuclear fast red was used as a counterstain (Trevigen, Gaithersburg, MD). The percentage of positively stained immunolabeled nuclei of myocytes was determined by randomly counting 10 fields per section.
2.7. Immunohistochemistry
To detect protein nitration, mice were subjected to a period of 30 min of LAD ligation and 3 h of reperfusion. Nitrotyrosine immunostaining was performed using monoclonal anti-nitrotyrosine antibody at a dilution of 1:200 (Cayman, Ann Arbor, MI) at 4°C overnight. Antigen-antibody complexes were detected by the super-sensitive alkaline phosphatase kit (BioGenex, San Ramon, CA) using fast red as a chromogen and hematoxylin as a counterstain. A SPOT camera was used to capture images, which were transferred to digital image analysis software (Image Pro Plus, Media Cybernetics, Silver Spring, MD). LV immunoreactivity was determined by applying intensity thresholding analysis. The sum density of total LV image pixels in the 100-to-255 range was used as a semi-quantitative measure of relative immunoreactivity.
2.8. Statistical analysis
All data were expressed as mean ± SEM. Statistical difference was assessed by the ANOVA test (one-way analysis of variance) followed by the Student-Newman Keuls multiple comparison tests. Values of P<0.05 were considered statistically significant.
3. Results
At baseline, there was no difference in age, body weight, or heart protein content between the Gpx1-/- and Gpx1+/+ groups. All mice were healthy and showed no apparent phenotypic differences.
We compared the cardiac parameters of hearts from Gpx1-/- mice with those of their normal littermates after 30 min of equilibration perfusion (Table 1). Left ventricular developed pressures (LVDP) and maximum rates of pressure development during both contraction and relaxation (±dP/dt) were essentially the same in both groups. Heart rates (HR) and coronary flow rates (CFR) were similar in both groups.
Table 1.
Basal values of hearts from Gpx1+/+ and Gpx1-/- mice pretreated with or without melatonin
| Parameter | Gpx1+/+ | Gpx1+/+ +Mel | Gpx1-/- | Gpx1-/-+Mel |
|---|---|---|---|---|
| +dP/dt (mm Hg/sec) | 2304±170 | 2390±77 | 2536±154 | 2405±146 |
| -dP/dt (mm Hg/sec) | 1733±76 | 1775±53 | 1956±35 | 1940±103 |
| LVDP (mm Hg) | 105±8.2 | 111±4.7 | 103±5.9 | 105±12 |
| Heart Rate (beats/min) | 237±22 | 224±16 | 256±12 | 254±26 |
| LVDPxHR | 23,851±1297 | 24,697±1369 | 27,877±1765 | 26,797±2067 |
| Coronary flow (ml/min) | 0.58±0.17 | 1.39±0.07 | 1.74±0.07 | 1.73±0.15 |
| LVEDP (mm Hg) | 4.06±1.48 | 3.24±0.62 | 3.54±0.69 | 4.18±0.78 |
Mice were pretreated with melatonin (150 μg/kg, i.p.) for 15 min and hearts were removed for perfusion as Langendorff preparations. Measurements of ±dP/dt, LVDP, heart rate and coronary flow were made after 30 min of preliminary perfusion. All values are mean ± SEM of 6 hearts. There is no significant difference among all four groups. Cardiac functions are defined as follows: +dP/dt, maximum rate of pressure development during contraction; -dP/dt, maximum rate of pressure development during relaxation; LVDP, left ventricular developed pressure; HR, Heart rate; CFR, Coronary flow rate.
A comparison of the functional recovery of the Gpx1-/- and Gpx1+/+ hearts subjected to 40 min of ischemia and 45 min of reperfusion revealed significant dysfunction in the hearts of Gpx1-/- mice (Fig. 1). Functional recovery, expressed as LVDP × HR, was 32 ± 6% for the Gpx1-/- mice and 51 ± 3% for the Gpx1+/+ group (P<0.05). Percentages of recovery in Gpx1-/- vs. Gpx1+/+ mice were 48 ± 5% vs. 65 ± 5% for +dP/dt, 49 ± 6% vs. 63 ± 5% for -dP/dt, and 32 ± 6% vs. 55 ± 6% for LVDP.
Fig. 1.
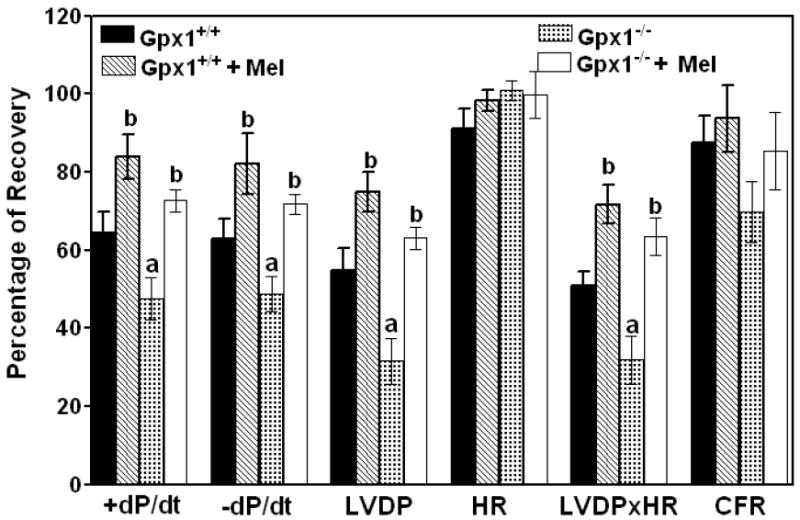
Effect of melatonin on the cardiac function and coronary flow rate of hearts from Gpx1-/- and wild type mice. Melatonin (150 μg/kg, i.p.) was given 30 min before the hearts were removed for in vitro perfusion. Results are expressed as the percentages of recovery of pre-ischemic values. Values are mean ± SEM of 6 hearts. a = P<0.05, Gpx1-/- vs. Gpx1+/+; b = P< 0.05, melatonin-treated group vs. nontreated group;
Melatonin treatment improved the cardiac function in both groups of mice. Percentages of recovery in melatonin-treated Gpx1-/- vs. Gpx1+/+ mice were 73 ± 3% vs. 84 ± 6% for +dP/dt, 72 ± 3% vs. 82 ± 8% for -dP/dt, 63 ± 3% vs. 75 ± 5% for LVDP and 63 ± 5% vs. 72 ± 5% for LVDP × HR. HR, CFR, and pre-ischemic end-diastolic pressures) were not significantly different (Figs. 1 and 2). End diastolic pressures increased to 39.6 ± 5.3 mm Hg for the Gpx1+/+ hearts and 65.9 ± 9.7 mm Hg for the Gpx1-/- hearts after 45 min of post-ischemic reperfusion. End-diastolic pressures decreased significantly in melatonin-treated hearts (16.7 ± 3.9 mm Hg for Gpx1+/+ vs. 26.9 ± 3.6 mm Hg for Gpx1-/-).
Fig. 2.
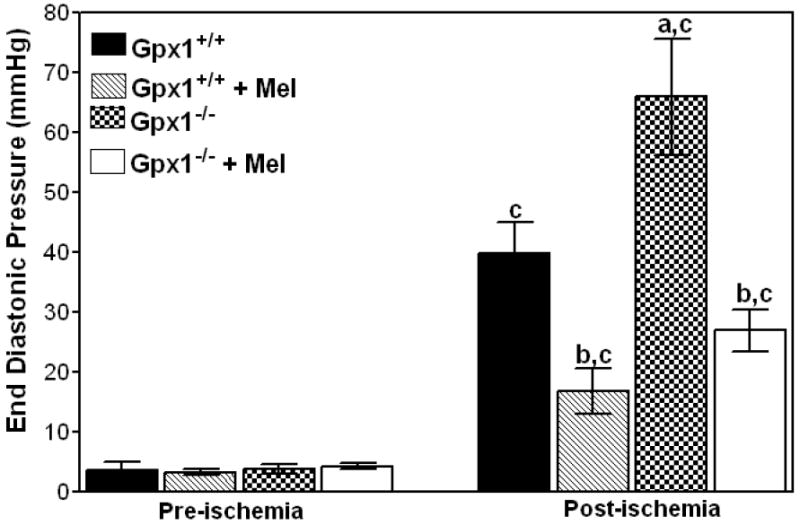
Melatonin improves left ventricular end-diastolic pressures of Gpx1+/+ and Gpx1-/- mouse hearts. Left ventricular end-diastolic pressure was measured after 40 min of global ischemia and 45 min of reperfusion in isolated hearts from Gpx1+/+ and Gpx1-/- mice with or without pretreatment with melatonin (150 μg/kg). Values are mean ± SEM of 6 hearts. a = P<0.05, Gpx1-/- vs. Gpx1+/+; b = P< 0.05, melatonin-treated group vs. nontreated group, c = P<0.05, post-ischemic values vs. respective pre-ischemic values.
Cardiac injury was also assessed by measuring the release of LDH in the perfusate. Total LDH release from the Gpx1-/- hearts was two times higher than LDH release from Gpx1+/+ hearts (Fig. 3). Specifically, the total release of LDH during 45 min of reperfusion after 40 min of global ischemia of the Gpx1+/+ hearts and Gpx1-/- hearts group was 1642 ± 113 mU/mg protein and 3692 ± 266 mU/mg protein, respectively. Melatonin treatment significantly reduced the release of LDH in both groups of mice (1022 ± 122 mU/mg protein, Gpx1+/+ vs. 1425 ± 136 mU/mg protein, Gpx1-/-).
Fig. 3.
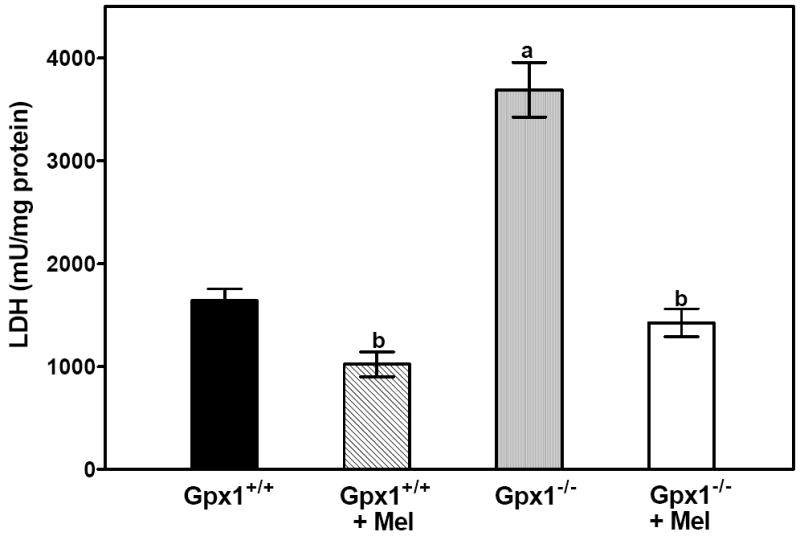
Melatonin reduces LDH release in vitro. The release of LDH during 45 min of reperfusion after 40 min of global ischemia was measured. Values were mean ± SEM of 6 hearts. a = P<0.05, Gpx1-/- vs. Gpx1+/+; b = P< 0.05, melatonin-treated group vs. nontreated group.
To study the effect of melatonin treatment on regional I/R injury in vivo, mice were subjected to 50 min of LAD ligation followed by 4 h of reperfusion (Fig. 4). The infarct size (expressed as a percentage of LV) of Gpx1+/+ and Gpx1-/- hearts was 15.3 ± 2.3% and 23.6 ± 2.5%, respectively. Melatonin treatment reduced the infarct sizes to 8.8 ± 1.5% for Gpx1+/+ hearts and 9.6 ± 0.9% for the Gpx1-/- hearts. Infarct sizes expressed as the percentage of the risk area for the Gpx1+/+ hearts and Gpx1-/- hearts were 53.7 ± 6.6% and 75.1 ± 9.4%, respectively. Similarly, melatonin treatment decreased the infarct sizes to 30.4 ± 4.9% for Gpx1+/+ hearts and 31.1 ± 3.8% for Gpx1-/- hearts. The risk area, expressed as the percentage of the left ventricle (LV), was comparable among the four groups. In this experiment, we ligated the LAD 3-4 mm from the tip of the left auricle, resulting in only 27-31% risk area/LV. As such, the mice survived even when the infarct size/risk area was as high as 75%. Our results indicate that melatonin treatment limits infarct size in an in vivo regional ischemia model in both groups of mice.
Fig. 4.
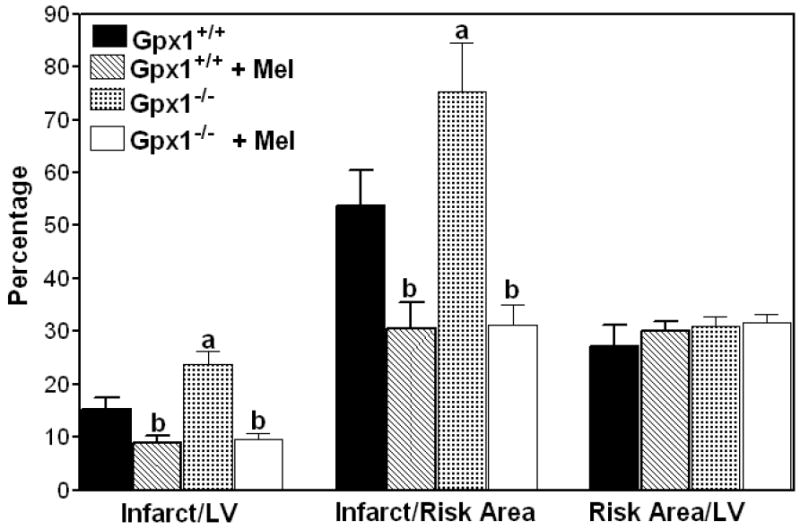
Protective effect of melatonin on myocardial infarction in Gpx1+/+ and Gpx1-/- mouse hearts. Mice were treated with 150 μg/kg melatonin for 30 min. They were subjected to 50 min of LAD ligation followed by 4 h of reperfusion. Values are mean ± SEM of 6 hearts. a = P<0.05, Gpx1-/- vs. Gpx1+/+; b = P<0.05, melatonin-treated group vs. nontreated group.
To examine whether the protection of melatonin is related to apoptosis, DNA fragmentation analyses were performed. Our results showed that mouse hearts subjected to 50 min of ischemia developed more than 50% infarction. The heterogeneity of mixed populations of live and dead cells will hamper the interpretation of apoptosis results. Therefore, a period of 30 min of ischemia and 3 h of reperfusion in the absence of cell death based on TTC staining was chosen for apoptosis analysis. We performed an in situ ligation assay (ISOL) that uses hairpin oligonucleotide probes to identify apoptotic nuclei. Whereas the labeling in the nuclei myocytes was 15.3 ± 0.55%, Gpx1-/- vs. 11.7 ± 0.84%, Gpx1+/+, ISOL-positive nuclei in melatonin-treated hearts from Gpx1-/- and Gpx1+/+ mice were 7.22 ± 0.14% and 7.18 ± 0.39%, respectively (Fig. 5).
Fig. 5.
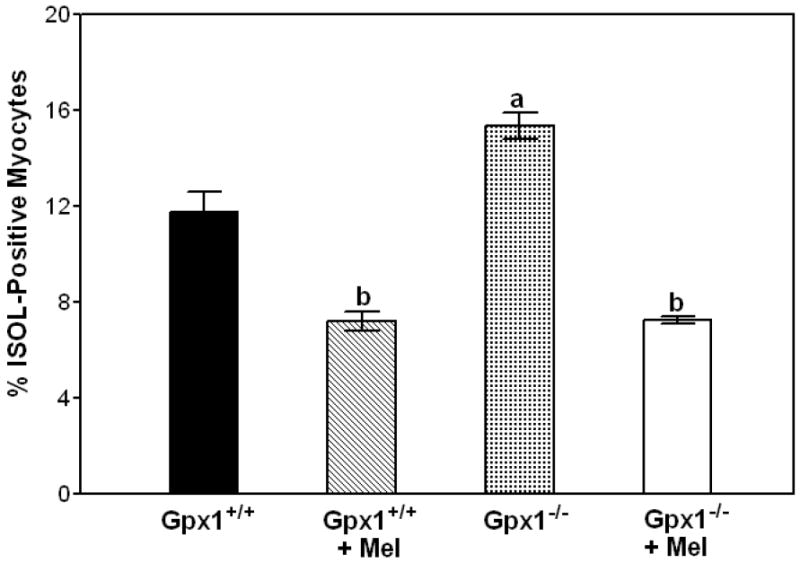
Melatonin attenuates apoptosis in Gpx1+/+ and Gpx1-/- mouse hearts. Mice were subjected to a period of 30 min of LAD ligation and 3 h of reperfusion. Paraffin-embedded sections of hearts were stained by ISOL procedure. Immunolabeled nuclei of myocytes were determined by random counting of 10 fields per section. Each bar represents mean ± SEM of six hearts. a = P<0.05, Gpx1-/- vs. Gpx1+/+; b = P< 0.05, melatonin-treated group vs. nontreated group.
One of the products of oxidative stress is the nitration of proteins on tyrosine residues leading to the formation of 3-nitro-L-tyrosine (3-NT). We next examined the protective effect of melatonin on protein nitration. Immunostaining of 3-NT was performed in heart sections using nitrotyrosine specific antibodies. Diffuse cytoplasmic staining was observed throughout the heart sections. Mouse hearts from Gpx1+/+ (A) and Gpx1-/- (B) mice showed increased immunostaining, and melatonin treatment lowered immunostaining density by two-fold in Gpx1+/+ (C) and Gpx1-/- hearts (D) (Fig. 6). In the absence of primary nitrotyrosine antibody, minimal immunostaining was observed (data not shown).
Fig. 6.
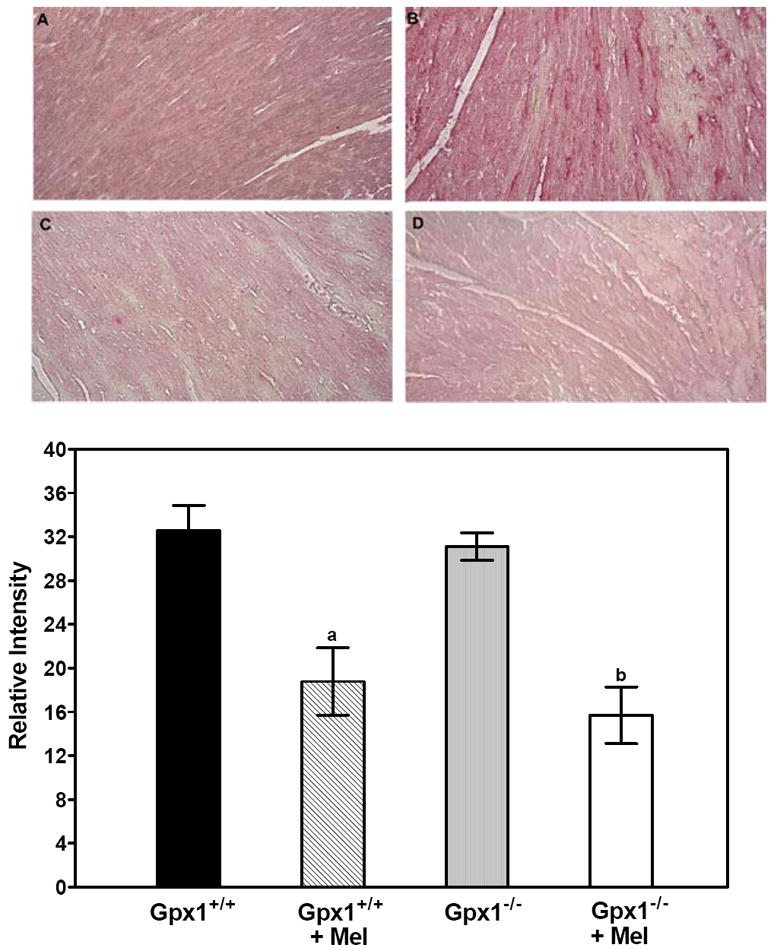
Melatonin reduces nitrotyrosine staining in Gpx1+/+ and Gpx1-/- mouse hearts. Mice were subjected to a period of 30 min of LAD ligation and 3 h of reperfusion. Immunohistochemical staining of mouse hearts was performed with anti-nitrotyrosine antibody. Sections from Gpx1+/+ (A), Gpx1+/+ + Mel (B), Gpx1-/- (C), and Gpx1-/- + Mel mouse hearts (D) were stained with anti-tyrosine antibody. Semi-quantitative analysis was performed using Image Pro Plus. Each bar represents mean ± SEM of 4 hearts. a = P < 0.05, Gpx1+/+ + Mel vs. Gpx1+/+; b = P<0.05, Gpx1-/- + Mel vs. Gpx1-/-.
4. Discussion
The present studies demonstrate three major findings regarding the effects of melatonin on apoptosis and myocardial I/R injury. First, hearts from Gpx1-deficient mice were more susceptible to I/R injury than hearts from Gpx1+/+ as indicated by three parameters: a reduction of functional recovery, a rise in LVEDP, and a significant increase of LDH release. Pretreatment of mice with melatonin (150 μg/kg, i.p.) for 30 min protected the hearts of both groups of mice against I/R injury (Figs. 1 and 2). This protection was evidenced by a reduction in the release of LDH from the hearts of animals pretreated with melatonin (Fig. 3). Second, when mouse hearts were subjected to 50 min of LAD ligation followed by reperfusion, melatonin pretreatment (150 μg/kg for 30 min) was able to reduce the infarct size/area at risk as well as the infarct size/LV in both groups of mice (Fig. 4). This result corroborates our previous finding that melatonin can protect the heart against I/R injury [18]. Finally, and most interestingly, there was significantly more apoptosis and protein nitration in hearts from Gpx1-/- mice than Gpx1+/+ mice, and melatonin attenuated apoptosis and protein nitration in both groups of mice (Figs. 5 and 6).
Recent studies have demonstrated that melatonin is a very effective cardioprotective agent. One important advantage of melatonin as a cardioprotective agent is that it has the ability to penetrate the membranes of all cells in the heart and thus reach a desired concentration in every subcellular compartment, where it can scavenge free radicals in situ. A second advantage of melatonin is that it can scavenge multiple partially-reduced species of oxygen, including superoxide anions, hydroxyl radicals, and hydrogen peroxide which are all generated intracellularly as byproducts of oxidative metabolism. However, since melatonin is also capable of inducing the expression of antioxidant enzymes, including Gpx1 [30-32], it is not clear whether the cardioprotective function of melatonin is mediated through its free radical scavenging activity or its ability to enhance the activity of antioxidant enzymes. As demonstrated in the present studies, melatonin is effective in protecting mouse hearts that are completely deficient in Gpx1, suggesting that the action of melatonin is independent of its effect on this antioxidant enzyme.
It has been shown previously that melatonin can reduce the extent of lipid peroxidation and increases the GSH levels in rats subjected to in vivo I/R injury [19]. In our study, we show that melatonin can inhibit protein nitration (Fig. 6). This study implies that the effect of melatonin is mediated at least in part by its antioxidant activities. Several issues remain unclear, however. First, it is unclear whether melatonin functions upstream of Gpx1 through removal of superoxide anions by preventing the formation of hydrogen peroxide, or acts downstream of Gpx1 by scavenging hydrogen peroxide. Second, some have suggested that melatonin’s cardioprotective effects involve mel1a receptors in the mouse heart, but our results using mice deficient for the MT1 melatonin receptor argue against this possibility [18]. In addition, we showed that 8-methoxy-2-propionamidotetralin, a melatonin receptor agonist with no antioxidant activity, offered no protection on myocardial I/R injury or doxorubicin-induced cardiotoxicity [18, 34]. On the other hand, Lochner et al [35] reported that the protective effect of melatonin on I/R injury in rats was blocked by luzindole, a relatively selective MT2 antagonist. The discrepancy could be attributed to different species or differences in the dose of melatonin and antagonist used. More studies need to be done to clarify this issue.
In the present study, mice deficient in Gpx1 experienced significantly more apoptosis than wild-type mice. Since Gpx1 is an important intracellular antioxidant enzyme, this result suggests that an increased level of hydrogen peroxide and/or lipid hydroperoxides due to a lack of Gpx1 activity, can lead to apoptosis. This notion is further supported by our observations that both Gpx1+/+ and Gpx1-/- mice treated with melatonin experienced significantly reduced apoptosis in response to cardiac I/R injury, as detected by ISOL analysis (Fig. 5). Although these results suggest that melatonin prevents I/R induced apoptosis by scavenging reactive oxygen species, there are likely many other mechanisms by which melatonin can modulate cellular apoptotic pathways. For instance, melatonin directly inhibits the mitochondrial permeability transition pore with an IC50 of 0.8 μM in mouse striatal neurons [36]. This is significant because mitochondria are known to play a critical role in apoptotic cell death by releasing cytochrome c and activating caspases. As another example, melatonin has been shown to inhibit the intrinsic pathway of apoptosis by impairing the activation/dimerization of Bax and re-localization of Bcl-2 to mitochondria in U937, a human tumor cell line [37]. The delineation of the exact mechanism of melatonin’s protective effects against I/R injury will require further research.
In conclusion, our studies demonstrate that mice deficient in Gpx1 exhibit increased apoptosis in response to I/R injury. Furthermore, we demonstrate that melatonin is an effective cardioprotective agent that can attenuate apoptosis independently of the endogenous Gpx1. Given that melatonin is a natural molecule with few side effects and that it has been shown to provide protection to critical organs, it is clear that melatonin could become a very powerful cardioprotective agent with great clinical potential.
Acknowledgments
This study was supported by NIH grant HL087271, a grant from the Department of Veterans Affairs Merit Review and a grant-in-aid from the American Heart Association Southeast Affiliate to B.H.L.C.; and by American Heart Association Midwest Affiliate grants 0455876Z and 0655631Z to Y.-S. H. The use of equipment in the Imagine and Cytometry Facility Core was supported by a Center grant P30 ES06639.
References
- 1.MURPHY E, STEENBERGEN C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DAS M, DAS DK. Molecular mechanism of preconditioning. IUBMB Life. 2008;60:199–203. doi: 10.1002/iub.31. [DOI] [PubMed] [Google Scholar]
- 3.CHEN Z, SIU B, HO YS, et al. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cel Cardiol. 1998;30:2281–2289. doi: 10.1006/jmcc.1998.0789. [DOI] [PubMed] [Google Scholar]
- 4.CHEN Z, HO YS, OBERLEY TD, et al. Overexpression of CuZnSOD in coronary vascular cells attenuates myocardial ischemia/reperfusion injury. Free Rad Biol Med. 2000;29:589–596. doi: 10.1016/s0891-5849(00)00363-4. [DOI] [PubMed] [Google Scholar]
- 5.YOSHIDA T, WATANABE M, ENGELMAN D, et al. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 1996;28:1759–1767. doi: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 6.YOSHIDA T, MAULIK N, ENGELMAN RM, et al. Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation. 1997;96:II-216–II-220. [PubMed] [Google Scholar]
- 7.KAJSTURA J, CHENG W, REISS K, et al. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 8.GOTTLIEB RA, BURELESON KO, KLONER RA. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CHEN Z, CHUA CC, HO YS, et al. Overexpression of bcl-2 attenuates apoptosis and protects the heart from ischemia/reperfusion injury. Am J Physiol. 2001;280:H2313–H2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 10.CHUA CC, GAO J, HO YS, et al. Overexpression of IAP-2 attenuates apoptosis and protects against myocardial ischemia/reperfusion injury in transgenic mice. Biochim Biophys Acta. 2007;1773:577–583. doi: 10.1016/j.bbamcr.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.CHUA CC, GAO J, HO YS, et al. Over-expression of a modified bifunctional apoptosis regulator protects against myocardial ischemia/reperfusion injury and doxorubicin-induced cardiotoxicity in transgenic mice. Cardiovas Res. 2008 Oct 14; doi: 10.1093/cvr/cvn257. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.REITER RJ, TANG L, GARCIA JJ. Pharmacological actions of melatonin in oxygen radical pathophysiology. Life Sci. 1997;60:2255–2271. doi: 10.1016/s0024-3205(97)00030-1. [DOI] [PubMed] [Google Scholar]
- 13.REITER RJ, MELCHIORRI D, SEWERYNEK E, et al. A review of the evidence supporting melatonin’s role as an antioxidant. J Pineal Res. 1995;18:1–11. doi: 10.1111/j.1600-079x.1995.tb00133.x. [DOI] [PubMed] [Google Scholar]
- 14.HARDELAND R, REITER RJ, POEGGELER B, et al. The significance of the metabolism of the neurohormone melatonin: Antioxidative protection and formation of bioactive substances. Neurosci Biobehav Rev. 1993;17:347–357. doi: 10.1016/s0149-7634(05)80016-8. [DOI] [PubMed] [Google Scholar]
- 15.PIERI C, MARRA M, MORONI F, et al. Melatonin: A peroxyl radical scavenger more effective than vitamin. E Life Sci. 1994;15:PL271–PL276. doi: 10.1016/0024-3205(94)00666-0. [DOI] [PubMed] [Google Scholar]
- 16.TAN DX, MANCHESTER LC, REITER RJ, et al. Melatonin directly scavenges hydrogen peroxide: a potentially new metabolic pathway of melatonin biotransformation. Free Rad Biol Med. 2000;29:1177–1185. doi: 10.1016/s0891-5849(00)00435-4. [DOI] [PubMed] [Google Scholar]
- 17.TAN DX, MANCHESTER LC, TERRON MP, et al. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42:28–42. doi: 10.1111/j.1600-079X.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 18.CHEN Z, CHUA CC, GAO J, et al. Protective effect of melatonin on myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;284:H1618–H1624. doi: 10.1152/ajpheart.00874.2002. [DOI] [PubMed] [Google Scholar]
- 19.SAHNA E, PARLAKPINAR H, TURKOZ Y, et al. Protective effects of melatonin on myocardial ischemia/reperfusion induced infarct size and oxidative changes. Physiol Res. 2005;54:491–495. [PubMed] [Google Scholar]
- 20.REITER RJ, TAN DX. Melatonin: a novel protective agent against oxidative injury of the ischemic/reperfused heart. Cardiovasc Res. 2003;58:10–19. doi: 10.1016/s0008-6363(02)00827-1. [DOI] [PubMed] [Google Scholar]
- 21.SAHNA E, ACET A, OZER MK, et al. Myocardial ischemia-reperfusion in rats: reduction of infarct size by either supplemental physiological or pharmacological doses of melatonin. J Pineal Res. 2002;33:234–238. doi: 10.1034/j.1600-079x.2002.02924.x. [DOI] [PubMed] [Google Scholar]
- 22.REITER RJ, TAN DX, SAINZ RM, et al. Melatonin protects the heart against both ischemia/reperfusion injury and chemotherapeutic drugs. Cardiovasc Drugs Ther. 2002;16:5–6. doi: 10.1023/a:1015376328431. [DOI] [PubMed] [Google Scholar]
- 23.SZARZOI O, ASEMU G, VANACEK J, et al. Effects of melatonin on ischemia and reperfusion injury of the rat heart. Cardiovasc Drugs Ther. 2001;15:251–257. doi: 10.1023/a:1011920407691. [DOI] [PubMed] [Google Scholar]
- 24.SALIE R, HARPER I, CILLIE C, et al. Melatonin protects against ischaemic-reperfusion myocardial damage. J Mol Cell Cardiol. 2001;33:343–357. doi: 10.1006/jmcc.2000.1306. [DOI] [PubMed] [Google Scholar]
- 25.LAGNEAUX C, JOYEUX M, DEMENGE P, et al. Protective effects of melatonin against ischemia-reperfusion injury in the isolated rat heart. Life Sci. 2000;66:503–509. doi: 10.1016/s0024-3205(99)00620-7. [DOI] [PubMed] [Google Scholar]
- 26.KOH PO. Melatonin attenuates the focal cerebral ischemic injury by inhibiting the dissociation of pBad from 14-3-3. J Pineal Res. 2008;44:101–106. doi: 10.1111/j.1600-079X.2007.00495.x. [DOI] [PubMed] [Google Scholar]
- 27.KUNDUZOVA OR, ESCOURROU G, SEGUELAS MH, et al. Prevention of apoptotic and necrotic cell death, caspase-3 activation, and renal dysfunction by melatonin after ischemia/reperfusion. FASEB J. 2003;17:872–874. doi: 10.1096/fj.02-0504fje. [DOI] [PubMed] [Google Scholar]
- 28.MUNOZ-CASARES FC, PADILLO FJ, BRICENO J, et al. Melatonin reduces apoptosis and necrosis induced by ischemia/reperfusion injury of the pancreas. J Pineal Res. 2006;40:195–203. doi: 10.1111/j.1600-079X.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 29.KIM SH, LEE SM. Cytoprotective effects of melatonin against necrosis and apoptosis induced by ischemia/reperfusion injury in rat liver. J Pineal Res. 2008;44:165–171. doi: 10.1111/j.1600-079X.2007.00504.x. [DOI] [PubMed] [Google Scholar]
- 30.RODRIGUEZ C, MAYO JC, SAINZ RM, et al. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res. 2004;36:1–9. doi: 10.1046/j.1600-079x.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- 31.DZIEGIEL P, MURAWKSA-CIALOWICZ E, JETHON Z, et al. Melatonin stimulates the activity of protective antioxidative enzymes in myocardial cells of rats in the course of doxorubicin intoxication. J Pineal Res. 2003;35:183–187. doi: 10.1034/j.1600-079x.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 32.PABLOS MI, AGAPITO MT, GUTIERREZ R, et al. Melatonin stimulates the activity of the detoxifying enzyme glutathione peroxidase in several tissues of chicks. J Pineal Res. 1995;19:111–115. doi: 10.1111/j.1600-079x.1995.tb00178.x. [DOI] [PubMed] [Google Scholar]
- 33.HO YS, MAGNENAT JL, BRONSON RT, et al. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 34.LIU X, CHEN Z, CHUA CC, et al. Melatonin as an effective protector against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. 2002;283:H254–263. doi: 10.1152/ajpheart.01023.2001. [DOI] [PubMed] [Google Scholar]
- 35.LOCHNER A, GENADE S, DAVIDS A, et al. Short- and long-term effects of melatonin on myocardial post-ischemic recovery. J Pineal Res. 2006;40:56–63. doi: 10.1111/j.1600-079X.2005.00280.x. [DOI] [PubMed] [Google Scholar]
- 36.ANDRABI SA, SAYEED I, SIEMEN D, et al. Direct inhibition of mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004;18:869–871. doi: 10.1096/fj.03-1031fje. [DOI] [PubMed] [Google Scholar]
- 37.RADOGNA F, CRISTOFANON S, PATERNOSTER L, et al. Melatonin antagonizes the intrinsic pathway of apoptosis via mitochondrial targeting of Bcl-2. J Pineal Res. 2008;44:316–325. doi: 10.1111/j.1600-079X.2007.00532.x. [DOI] [PubMed] [Google Scholar]