

Abstract
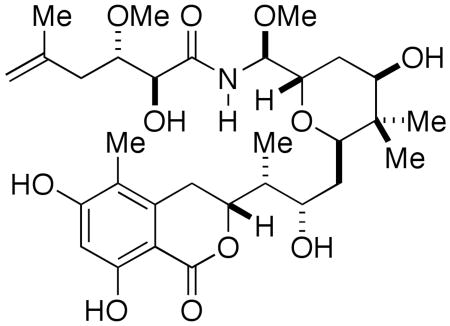
The total synthesis of (+)-iriciniastatin A (psymberin) is reported in 19 steps and 6% overall yield. Key reactions include a highly convergent enolsilane-oxocarbenium ion union to generate the C8-C25 fragment and a late stage coupling of a hemiaminal and acid chloride to complete the synthesis.
In 2004, Pettit and Crews independently reported the isolation of two remarkably potent and selective cancer cell growth inhibitors from the extracts of deep water sponges. Irciniastatin A (1) was obtained from Ircinia ramosa by Pettit,1 and psymberin from Psammocina sp. by Crews.2 The two secondary metabolites were shown to be structurally related based on spectroscopic studies, however, incomplete and conflicting stereochemical analyses precluded a definitive assignment of absolute configuration. Each displayed incredibly powerful and selective cancer cell growth inhibition. Irciniastatin A was reported to exhibit a greater than 104 differential in activity across related cell lines. The limited natural abundance and lack of an absolute stereochemical assignment, as well as interest in further exploration of the unique chemotherapeutic profile and mode of action, sparked immediate interest within the synthetic community.
Early reports shed light on the stereochemical ambiguities through fragment synthesis3 and natural product degradation.4 Ultimately, it was the seminal total synthesis by DeBrabander5 that confirmed the absolute configuration and demonstrated that irciniastatin A (1) and psymberin were identical. Subsequent reports, including fragment synthesis,6 a formal synthesis,7 two total syntheses,8,9 and two reports of analog syntheses,10,11 all offered creative and distinct approaches toward the unique structural elements of irciniastatin A.
Our interest in (+)-irciniastatin A (psymberin) focused on developing a highly diastereoselective and modular synthesis that might be suitable for analog preparation. Our efforts toward (+)-1, recently culminated in a completed total synthesis which is reported here.
Retrosynthetically (Scheme 1), (+)-irciniastatin A (1) was envisioned to arise from two key disconnections. A late stage attachment of the psymberic acid chain would be accomplished by coupling of acid chloride 2 with hemiaminal 3, the product of a Curtius rearrangement of the carboxylic acid derived from benzyl ether 4. This tactic would allow for a highly stereocontrolled entry to the C8 hemiaminal and efficient incorporation of the side chain. Tetrahydropyran 4 would arise from the stereoselective addition of enolsilane 6 to the oxocarbenium-ion derived from acetate 5. These two key disconnections segregate the three major subunits of irciniastatin A (1); each of similar size and complexity and accessible in a highly stereocontrolled fashion from standard aldol synthons.
Scheme 1.

Retrosynthetic Analysis of Irciniastatin A
All reported syntheses of the psymberate side chain have relied on functionalizing commercially available chiral pools or enzymatic resolutions. In addition, analog studies10,11 have shown the side chain to be required for high activity. Therefore, an enantioselective and highly tunable synthesis of the side chain would be ideal for further investigation of side chain function. We reasoned that an oxazolidinethione asymmetric glycolate aldol reaction12 would allow for enantioselective entry to the psymberate side chain as well as provide sufficient opportunities for derivitization and congener synthesis. The synthesis of acid chloride 2 (Scheme 2) began with the known13 anti-aldol12a reaction of glycolate 9 and 3-methyl-but-3-enal to deliver aldol adduct 10 that was converted to known TIPS ether 11.13 Methylation of the alcohol was followed by removal of the allyl protecting group under Kulinkovich conditions13,14 to give alcohol 12. Protection of alcohol 12 as the SEM ether was followed by selective removal of the primary TIPS ether. The resultant primary alcohol 13 was oxidized under Smith's conditions giving the corresponding acid over two steps. The acid (9 steps, 28% overall) was converted to acid chloride 2.
Scheme 2.

Synthesis of the Psymberic Acid Side Chain
The synthesis of the acetate 5 (Scheme 3) began from known p-methoxybenzylidine acetal 1415 available from 2-deoxy-D-ribose in two steps. Methylation of alcohol 14 was followed by a dihydroxylation-oxidative cleavage sequence to reveal aldehyde 16. A catalyst controlled Kiyooka16 aldol reaction of aldehyde 16 and enolsilane 1717 provided carbinol 18 in 84% yield and 9:1 dr. Protection of alcohol 18 to give TBS ether 19 was followed by acid-catalyzed cyclization to provide a 10:1 mixture of lactone 20 and the corresponding diol, which was converted to lactone 20 by exposure to CF3CO2H. Protection of the primary alcohol gave benzyl ether 21 and subsequent one-pot reductive acetylation18 afforded acetate 5 in quantitative yield (9 steps, 34% overall from 2-deoxy-D-ribose).
Scheme 3.

Synthesis of Acetate 5
The synthesis of enolsilane 6 (Scheme 4) began from known catechol 246,19 prepared by cycloaddition of allene 2320 and diene 22.21 Protection of catechol 24 as the bis-TIPS ether was followed by selective ester reduction to give aldehyde 25 in 78% yield over 2 steps. An asymmetric propionate aldol22 reaction of aldehyde 25 and propionyl thiazolidinethione 26 afforded the Evans-syn-aldol adduct 27 in 94% yield and >20:1 dr. Direct displacement of the chiral auxiliary with Weinreb's amine was followed by protection of the secondary alcohol as a TBS ether to deliver silyl ether 28. The methyl ketone prepared from Weinreb's amide 28, was then readily converted to desired enolsilane 6 (7 steps; 66% overall from catechol 24).
Scheme 4.

Synthesis of Enolsilane 6
Having devised highly stereocontrolled routes to all three key fragments, the union of enolsilane 6 and acetate 5, was investigated (Scheme 5). Rigorous experimentation revealed that BF3●OEt2 was the optimum Lewis acid and that TBS enolsilane 6 performed better than the corresponding TMS enolsilane for the assembly of 5 and 6. Most intriguing, however, was that addition of a solution of enolsilane 6 to a premixed solution of 5 and BF3●OEt2 at -40 °C was required for efficient coupling. The union proceeded with high diastereoselectivity, giving tetrahydropyran 4 as the only detectable diastereomer. The high diastereoselectivity can be rationalized by well precedented23 pseudoaxial addition of the nucleophile to the oxocarbenium conformer 29, proceeding through a favorable chair-like conformation. Conformer 29 would also be expected to be favored as a result of through-space stereoelectronic stabilization23 of the oxocarbenium ion by the axially positioned C11 ether.
Scheme 5.

Coupling of Acetate 5 with Enolsilane 6
With the C8-C25 carbon framework in place, conditions to set the C15 stereocenter were investigated (Scheme 6). Standard achiral reducing agents proved to be non-selective or completely selective for the undesired diasteromer,24 however, the use of the chiral (R)-CBS25 agent exclusively provided desired diastereomer 3026 in 82% yield. After protection of the secondary alcohol as its TBS ether and cleavage of the benzyl ether, carbinol 31 was oxidized over 2 steps to carboxylic acid 32. Initial studies showed the one-pot Curtius reaction with (O,O)-diphenylphosphoryl azide27 to be ineffective, providing significant quantities of inseparable carbamoyl azide impurity.28 Therefore, Weinstock's procedure9,29 was employed to generate the intermediate isocyanate, which under mild conditions using copper (I) chloride afforded Teoc-protected hemiaminal 33.30 Much to our dismay, a screen of reported conditions31 for acylation of stucturally related intermediates never yielded N-acyl hemiaminal 34. A survey of the previous syntheses of irciniastatin A finds that all feature a late stage aminal and side chain incorporation with a preformed lactone of the dihydroisocoumarin. Albeit unlikely that such remote functionality should have any effect on the hemiaminal coupling, the limited number of available options warranted pursuit of this lead.32
Scheme 6.

Completion of the Synthesis of (+)-Irciniastatin A
Strategically, after the CBS reduction, the lactone was formed concomitantly with removal of the TBS ether to give 35 adding only one synthetic step over the original plan (bottom of Scheme 6). An analogous sequence of TBS protection of the C15 carbinol and hydrogenolysis of the benzyl ether gave carbinol 36, which was subjected to a two step oxidation to acid 37. Acid 37 was subjected to the previously established Curtius conditions to give hemiaminal 3. With hemiaminal 3 featuring the lactonized dihydroisocoumarin, it was found that i-PrMgCl, a base not previously used for this transformation, successfully effected reaction with acid chloride 2 in a remarkable 87% yield. Finally, global deprotection with TASF in DMF at 50 °C provided (+)-irciniastatin A (1) in 94% yield.
In summary, the total synthesis of (+)-irciniastatin A (1) was completed in 19 steps with a 6% overall yield from 2-deoxy-D-ribose. The successful application of this strategy allowed for rapid assembly of the three key fragments with high diastereocontrol. Studies directed toward analog synthesis and further evaluation of the antitumor activity of irciniastatn A are underway in collaboration with the Lineberger Comprehensive Cancer Center.
Supplementary Material
Acknowledgments
Financial support from the National Institute of General Medical Sciences (GM60567) is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental details and spectral data for new compounds as well as psymberin. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pettit GR, Xu JP, Chapuis JC, Pettit RK, Tackett LP, Doubek DL, Hooper JNA, Schmidt JM. J Med Chem. 2004;47:1149. doi: 10.1021/jm030207d. [DOI] [PubMed] [Google Scholar]
- 2.Cichewicz RH, Valeriote FA, Crews P. Org Lett. 2004;6:1951. doi: 10.1021/ol049503q. [DOI] [PubMed] [Google Scholar]
- 3.Kiren S, Williams LJ. Org Lett. 2005;7:2905. doi: 10.1021/ol0508375. [DOI] [PubMed] [Google Scholar]
- 4.Green ME, Rech JC, Floreancig PE. Org Lett. 2005;7:4117. doi: 10.1021/ol051396s. [DOI] [PubMed] [Google Scholar]
- 5.Jiang X, Garcia-Fortanet J, DeBrabander JK. J Am Chem Soc. 2005;127:11254. doi: 10.1021/ja0537068. [DOI] [PubMed] [Google Scholar]
- 6.Rech JC, Floreancig PE. Org Lett. 2005;7:5175. doi: 10.1021/ol0520267.Henssen B, Kasparyan E, Marten G, Pietruszka J. Heterocycles. 2007;74:245.Pietruszka J, Simon R. Eur J Org Chem. 2009:3628. The following report appeared during the preparation of this manuscript:Brown LE, Landaverry YR, Davies JR, Milinkevich KA, Ast S, Carlson JS, Oliver AG, Konopelski JP. J Org Chem. 2009 doi: 10.1021/jo9009003. ASAP
- 7.Shangguan N, Kiren S, Williams LJ. Org Lett. 2007;9:1093. doi: 10.1021/ol063143k. [DOI] [PubMed] [Google Scholar]
- 8.Huang X, Shao N, Palani A, Aslanian R, Buevich A. Org Lett. 2007;9:2597. doi: 10.1021/ol071068n. [DOI] [PubMed] [Google Scholar]
- 9.Smith AB, III, Jurica JA, Walsh SP. Org Lett. 2008;10:5625. doi: 10.1021/ol802466t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Huang X, Shao N, Huryk R, Palani A, Aslanian R, Seidel-Dugan C. Org Lett. 2009;11:867. doi: 10.1021/ol802772s. [DOI] [PubMed] [Google Scholar]; (b) Huang X, Shao N, Palani A, Aslanian R, Buevich A, Seidel-Dugan C, Huryk R. Tetrahedron Lett. 2008;49:3592. [Google Scholar]
- 11.Jiang X, Williams N, De Brabander JK. Org Lett. 2007;9:227. doi: 10.1021/ol062656o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Crimmins MT, McDougall PJ. Org Lett. 2003;5:591. doi: 10.1021/ol034001i. [DOI] [PubMed] [Google Scholar]; (b) Crimmins MT, She J. Synlett. 2004:1371. [Google Scholar]
- 13.Crimmins MT, McDougall PJ, Emmitte KA. Org Lett. 2005;7:4033. doi: 10.1021/ol051543m. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Cha JK. Tetrahedron Lett. 1996;37:3663. [Google Scholar]
- 15.(a) Fürstner A, Schlede M. Adv Synth Catal. 2002;344:657. [Google Scholar]; (b) Uehara H, Oishi T, Inoue M, Shoji M, Nagumo Y, Kosaka M, Le Brazidec JY, Hirama M. Tetrahedron. 2002;58:6493. [Google Scholar]
- 16.Kiyooka SI, Kira H, Hena MA. Tetrahedron Lett. 1996;37:2597. [Google Scholar]
- 17.Juaristi E, Cruz-Sanchez S. J Org Chem. 1988;53:3334. [Google Scholar]
- 18.(a) Kopecky DJ, Rychnovsky SD. J Org Chem. 2000;65:191. doi: 10.1021/jo9914521. [DOI] [PubMed] [Google Scholar]; (b) Dahanukar VH, Rychnovsky SD. J Org Chem. 1996;61:8317. doi: 10.1021/jo961205m. [DOI] [PubMed] [Google Scholar]
- 19.Langer P, Kracke B. Tetrahedron Lett. 2000;41:4545. [Google Scholar]
- 20.(a) Node M, Fujiwara T, Ichihashi S, Nishide K. Tetrahedron Lett. 1998;39:6331. [Google Scholar]; (b) Isobe T, Ishikawa T. J Org Chem. 1999;64:6984. [Google Scholar]
- 21.Langer P, Schneider T, Stoll M. Chem-Eur J. 2000;6:3204. doi: 10.1002/1521-3765(20000901)6:17<3204::aid-chem3204>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 22.(a) Crimmins MT, King BW, Tabet EA. J Am Chem Soc. 1997;119:7883. [Google Scholar]; (b) Crimmins MT, Chaudhary K. Org Lett. 2000;2:775. doi: 10.1021/ol9913901. [DOI] [PubMed] [Google Scholar]; (c) Crimmins MT, King BW, Tabet EA, Chaudhary K. J Org Chem. 2001;65:894. doi: 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]; (d) Crimmins MT, Shamszad M. Org Lett. 2007;9:149. doi: 10.1021/ol062688b. [DOI] [PubMed] [Google Scholar]
- 23.Ayala L, Lucero CG, Romero JAC, Tabacco SA, Woerpel KA. J Am Chem Soc. 2003;125:15521. doi: 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]
- 24.DIBAL in toluene at -78 °C was completely selective for the undesired isomer.
- 25.Corey EJ, Bakshi RK, Shibata S, Chen CP, Singh VK. J Am Chem Soc. 1987;109:7925. For a review see: Corey EJ, Helal CJ. Angew Chem Int Ed Engl. 1998;37:1987. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z.
- 26.Rychnovsky SD, Rogers B, Yang G. J Org Chem. 1993;58:3511. [Google Scholar]; Also see supporting information.
- 27.Shioiri T, Yamada S, Ninomiya K. J Am Chem Soc. 1972;94:6203. doi: 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]
- 28.(a) Shioiri T, Yamada SI. Chem Pharm Bull. 1974;22:855. doi: 10.1248/cpb.22.1795. [DOI] [PubMed] [Google Scholar]; (b) Csuk R, Schabel MJ, von Scholz YV. Tetrahedron: Asymmetry. 1996;7:3505. [Google Scholar]; (c) Sibi MP, Lu J, Edwards J. J Org Chem. 1997;62:5864. [Google Scholar]
- 29.Weinstock J. J Org Chem. 1961;26:3511. Also see: Smith AB, III, Safonov IG, Corbett RM. J Am Chem Soc. 2002;124:11102. doi: 10.1021/ja020635t.
- 30.(a) Duggan ME, Imagire JS. Synthesis. 1989:131. [Google Scholar]; (b) Evans SD, Houghton RP. J Mol Catal. 2000;164:157. [Google Scholar]
- 31.(a) Jewett JC, Rawal VH. Angew Chem Int Ed Engl. 2007;46:6502. doi: 10.1002/anie.200701677. [DOI] [PubMed] [Google Scholar]; (b) Marron TG, Roush WR. Tetrahedron Lett. 1995;36:1581. [Google Scholar]; (c) Roush WR, Pfeiffer LA. Org Lett. 2000;2:859. doi: 10.1021/ol005629l. [DOI] [PubMed] [Google Scholar]; (d) Kagawa N, Ihara M, Toyota M. J Org Chem. 2006;71:6796. doi: 10.1021/jo060803q. [DOI] [PubMed] [Google Scholar]
- 32.NOE experiments on related compounds by DeBrabander indicate a close spatial relationship between these two regions of the molecule (see reference 11).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.