

Abstract
Quinoid inhibitors of Cdc25B were designed based on the Linear Combination of Atomic Potentials (LCAP) methodology. In contrast to a published hypothesis, the biological activities and hydrogen peroxide generation in reducing media of three synthetic models did not correlate with the quinone half-wave potential, E1/2.
Introduction
Cyclin-dependent kinases (Cdk) are central regulators of the eukaryotic cell cycle that phosphorylate proteins responsible for the activation of structural and regulatory genes in G1, S, G2 and M cell phase transitions. Cdc25A, B, and C are members of the dual specificity protein phosphatase family, regulating Cdk by removing two inhibitory phosphate groups on adjacent Thr and Tyr residues near the amino terminus. The Cdc25 phosphatases control cell entry into all phases of the cell cycle,1 transform cells in culture, and harbor oncogenic potential.2 Many of the more potent Cdc25 phosphatase inhibitors reported to date are quinones,1,3–5 which can regulate phosphatase activity through a redox mechanism of reactive oxygen species (ROS) generation and irreversible oxidation of the catalytic cysteine of Cdc25.6–8
Ham, Carr and coworkers have postulated that the biological activity of quinone-derived inhibitors of Cdc25 and their ability to generate ROS can be modeled based on their reduction potential, and that the half-wave potential, E1/2, for the first reduction step of quinones is correlated with the energy of the lowest unoccupied molecular orbital (ELUMO), as calculated by AM1 semiempirical methods:5,9,10
| (1) |
Related correlations have been applied to other systems; for example, quinoxaline derivatives inhibit the growth of Trypanosoma cruzi in vitro, and this biological effect can be correlated with the LUMO energy of the agent.11 The LUMO energy of bispyridinium compounds also appears to correlate to their inhibition of choline kinase.12
The quinone pharmacophore is well represented in the clinically validated anticancer pharmacopoeia, with mitomycin C, daunorubicin, doxorubicin, idarubicin, epirubicin, geldanamycin, valrubicin and mitoxantrone providing representative examples. However, the in vivo use of quinones poses a major challenge since they can also cause acute toxicity.13,14 Off-target mechanisms include glutathione (GSH) depletion due to nucleophilic Michael additions of GSH and other protein thiols; as well as depletion of ATP due to redox cycling. Quinone radicals can also damage DNA and mitochondria through the formation of H2O2 and reactive oxygen/nitrogen species (ROS/RNS).15,16 A strategy for overcoming the intrinsic toxicity of quinones might be to utilize derivatives that are more stable in their reduced states, and thus are less likely to nucleate radicals and indiscriminately damage cells. Accordingly, we were interested in testing the hypothesis of Ham and Carr,5,9,10 by designing quinone based inhibitors of Cdc25 with half-wave potentials E1/2~105 mV – above the range necessary for spontaneous generation of superoxide radical anion, but not high enough to lead to fast, irreversible reduction by common organic and biological building blocks. While the highly fluorinated naphthoquinone reported by Ham and Carr was thought to represent a pure arylator of cysteine-containing proteins, without generating ROS, alternative explanations than an E1/2 effect could be responsible for this property, such as preferential protein binding affinities.
The systematic identification of diverse quinone-based inhibitors with specific half-wave potentials requires scanning large portions of chemical space.17 Some of us recently developed a linear combination of atomic potentials (LCAP) approach that transforms a molecular optimization into a continuous optimization problem,18–21 which enables an efficient survey of chemical space. Others have applied related ideas to drug design22 and protein folding.23 Herein we describe a modified version of the LCAP approach, where the gradient (which directs the next step of the optimization) is approximated by finite differences. It is a general algorithmic method for finding optimal solutions of various optimization problems, by keeping the best intermediate solution found. This method is a limiting case of the LCAP approach that resembles the “branch and bound” algorithm,24,25 and is especially useful in discrete and combinatorial optimizations.
A medium-sized quinone-based virtual library (1000 compounds) was designed for the computational search with the LCAP method. The goal of the search was to find diverse quinones with E1/2 close to ~105 mV. We also report here the synthesis and enzyme inhibition measurements of three new quinone-based inhibitors for Cdc25B, whose analysis of half-wave potentials provides a valid test case for the underlying hypothesis that E1/2 correlates with quinone toxicity (as measured by H2O2 generation and cytotoxicity analyses).
Methods
Computational studies
Fig. 1 shows the framework of the molecular library to be searched: A quinone scaffold, with four variable positions (X1, X2, Y and Z). The Y group determines the ring size as either five-or six-membered. The amine moiety serves to attach solubilizing substituents and to decrease the electrophile-accepting properties of the quinones. The total size of the library is 8 × 5 × 5 × 5 = 1000 possible targets (Fig. 2 and 3).
Fig. 1.
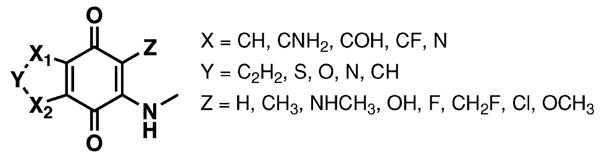
Molecular library used in designing inhibitors for Cdc25B.
Fig. 2.

Two typical optimization paths were found in the optimization, one long (Path 1), and one short (Path 2).
Fig. 3.

Results of enumeration of all 1000 library molecules, including (for each molecule) AM1 geometry optimization, calculating ELUMO, and evaluating E1/2 and the penalty function t, according to eqn (1). The molecule with largest t (Q1) is shown: E1/2 = 107.1 mV, t = 0.9995.
All geometries of sample molecules were optimized with the AM1 semiempirical method,26 as implemented in the DYNAMO27 code. For each optimized geometry, ELUMO was computed, and E1/2 was calculated using eqn (1). In order to locate molecules with E1/2 ~105 mV in the library, a penalty function was constructed, and the optimization target was changed to identify molecules with the highest t value:
| (2) |
In eqn (2), the denominator of the exponent changes the magnitude of the penalty, and the numerator determines where the maximum is located.
The optimization algorithm is:
Begin with a molecule A (by random selection of chemical groups for each of the j = 1. . .4 positions)
-
Calculate property PA of molecule A
-
Determine for each position which chemical group will give more desirable properties. There are j = 1. . .4 positions, and nRi possible groups for each Rj position. For each position Rj, freeze all other R, and alternate all possible groups for this position:
Build molecule B by changing only group nji at position Rj, keeping all other groups as in A
Calculate property PB of molecule B
Do AM1 geometry optimization for molecule A, calculate ELUMO in the new geometry, use eqn (1) to calculate E1/2 and use eqn (2) to calculate t
Find the group nji that has the largest t for each position Rj
Build the next molecule Anew, containing all nji groups (one per Rj position) that have the largest property
Test to see if the new molecule, Anew, was previously visited. If no – go to step 2 for another cycle. If yes – end optimization.
Synthesis
Construction of quinones WDP1079 and WDP1149 began from commercially available 2,5-dimethoxyaniline 1, which, upon treatment with Meldrum’s acid in the presence of trimethyl ortho-formate, afforded the known arylamino-methylene derivative 2 (Scheme 1).28 Bromination of the arene followed by thermal cyclization generated the desired quinolone 3.29 Exposure of 3 to POC1330 provided the 4-chloroquinoline derivative 4,29 which served as the common intermediate for the synthesis of quinone target molecules. Oxidative demethylation of 4 using cerium(IV) ammonium nitrate provided access to the quinone that was subsequently exposed to 4-(2-aminoethyl)morpholine to afford the functionalized quinone 5.31 The desired product, WDP1079, was obtained by treatment of 5 with N-chlorosuccinimide (NCS).32
Scheme 1.

Synthesis of the quinoline-5,8-dione WDP1079.
Starting from 4-chloroquinoline 4, treatment with NaOMe in MeOH resulted in formation of the trimethoxyquinoline 6 (Scheme 2). Analogous to the synthetic sequence shown in Scheme 1, substituted quinone 7 was readily obtained. Installation of a fluorine substituent was conducted utilizing Selectfluor® to afford WDP1149.33
Scheme 2.

Extension of the synthetic approach to WDP1149.
The synthesis of WDP1263 started from commercially available 2-amino-3-nitrophenol 8 (Scheme 3). Hydrogenation of the nitro group afforded the diamine derivative 9,34 which was then condensed with a glyoxal equivalent to access quinoxaline 10 in 82% yield over the two steps.35 Oxidation of the arene with hypervalent iodine reagent36 generated the known quinone 11.37 This step was followed by oxidative addition of 4-(2-aminoethyl)morpholine to the quinone scaffold to afford 12. Chlorination of the vinylogous amide with NCS completed the synthesis of WDP1263.
Scheme 3.

Synthesis of the quinoline-5,8-dione WDP1263.
Biological assays
The generic tyrosine phosphatase substrate O-methylfluorescein phosphate (OMFP) was used with the catalytic domain of Cdc25B to determine the inhibitory properties of inverse designed quinoids. The in vitro Cdc25B assay has been previously described.38 Briefly, recombinant human Cdc25B catalytic domain was incubated with test compounds and OMFP for 60 min, and the change in fluorescence intensity was measured (485 nm excitation/525 nm emission) using a Spectromax M5 microtiter plate reader (Molecular Devices). Percent inhibition was calculated relative to maximum and minimum controls and IC50 values were determined from a 10-point concentration curve from 25 μM to 0.2 μM fit to a four-parameter non-linear logistic model (also called the sigmoidal dose-response model) using GraphPad Prism 4.0 in two independent experiments performed in triplicate. Growth inhibition assays were conducted as previously described40 with minor modifications using human A549 lung cancer cells cultured in the presence of compounds for 48 h and CellTiter blue as described by the manufacturer (Promega, Madison, WI).
Hydrogen peroxide generation was quantified as previously described.39
Results and discussion
For the LCAP approach, 25 optimization runs were performed (each beginning with a different, random seed structure) with an average of 4.6 steps per optimization, and during each step the properties of 20 molecules were calculated. In 24 of these runs, Q1 (E1/2= 107.1 mV) was found among the optimization targets. Two other interesting molecules were also identified in the same optimizations, in the penultimate step, Q2 (E1/2= 118.1 mV) and the step before it (in only 12 runs), Q3 (E1/2= 89.8 mV). One optimization found a different quinone product from Q1 as the optimization target (E1/2 = 125.8 mV). Fig. 2 shows two typical optimization paths.
To validate that the optimization strategy used here indeed found the optimal compounds in the library (the molecules with highest t), we calculated the t value of all 1000 molecules in the library using direct enumeration. Table 1 shows the top 8 molecules (out of 1000) in the library ranked according to t values, and Q1 (the compound found in the optimization), was indeed the quinone with highest t, thus confirming the validity of the optimization algorithm. Fig. 3 shows the values of all calculated molecules versus t.
Table 1.
The eight highest calculated t values from a library of 1000 quinones
| Z | X1 | X2 | Y | ELUMO/eV | E1/2 (mV) | t | Name |
|---|---|---|---|---|---|---|---|
| OH | N | N | C2H2 | −1.770 | 107.1 | 0.9995 | Q1 |
| H | N | N | O | −1.771 | 108.5 | 0.9988 | |
| F | N | N | C2H2 | −1.782 | 118.1 | 0.9830 | Q2 |
| F | N | COH | C2H2 | −1.752 | 89.8 | 0.9772 | Q3 |
| F | N | CF | O | −1.790 | 125.9 | 0.9574 | |
| F | CF | COH | C2H2 | −1.741 | 79.9 | 0.9391 | |
| F | CF | N | O | −1.795 | 130.8 | 0.9356 | |
| F | CF | COH | C2H2 | −1.741 | 78.5 | 0.9324 |
The inverse design optimization found three molecules (Q1, Q2 and Q3) as candidates based on the computed redox properties. Before embarking on a large scale application of this methodology, three structurally related, synthetically readily accessible molecules (WDP1263, WDP1079 and WDP1149) were prepared. The measured Cdc25 inhibitory activities as well as the calculated AM1 ELUMO and half-wave potentials of these molecules are shown in Table 2. The IC50 values of WDP1263, WDP1079 and WDP1149 against the Cdc25B catalytic domain were 0.5 ± 0.1 lM, 1.1 ± 0.1 lM and 5.3 ± 0.6 lM (± SEM; N = 6), respectively. Subsequent cytotoxicity assays in the Cdc25B-expressing lung cancer cell line A549 positioned WDP1079 as the most active derivative, with an IC50 value of 2.7 lM (Table 3). WDP1149 and WDP1263 ranked closely behind with IC50 values of 9.5 and 22.3 μM. The cytotoxicity of the control quinones DA3003-1 and NSC95397 was in agreement with earlier measurements.40 This seemed to indicate that an optimal half-wave potential of E1/2 ~105 mV indeed provided some advantages at the cellular level, even though arguably both the enzyme inhibition data as well as the cellular activities of all three test compounds were within a relatively small single order of magnitude. A larger difference had been expected since their E1/2 covered a significant range of 270 mV from negative to positive values.
Table 2.
IC50 values for Cdc25B inhibition of quinone-based inhibitors, compared to their respective calculated properties
| Compound | IC50 ± SEM/lM (N = 6) | ELUMO/eV, calculated | E1/2/mV, calculated |
|---|---|---|---|
| WDP1149 | 5.3 ± 0.6 | −1.57 | −84 |
| WDP1079 | 1.1 ± 0.1 | −1.75 | 86 |
| WDP1263 | 0.5 ± 0.1 | −1.86 | 186 |
Table 3.
A549 cytotoxicity assays
| Compound | IC50 ± SEM/μM (N = 6) |
|---|---|
| WDP1149 | 9.52 ± 0.33 |
| WDP1079 | 2.69 ± 0.08 |
| WDP1263 | 22.28 ± 0.62 |
| DA3003-1 | 1.48 ± 0.04 |
| NSC95397 | 7.59 ± 0.44 |
Furthermore, while WDP1263, for which the lowest ELUMO and the highest half-wave potential were calculated, was the most potent Cdc25B inhibitor with an IC50 of 500 nM, this compound also produced very significant levels of H2O2 at 25 and 50 μM when incubated in the presence of 0.8 mM DTT (EC50 = 1.4 μM, Table 4). The E1/2 = 186 mV of WDP1263 should have prevented redox cycling through its reduced state. WDP1079 was also quite active in this assay with an EC50 value of 14 μM. Remarkably, WDP1149 in contrast was significantly less potent and only produced detectable levels of H2O2 at 50 μM (EC50 = 180 μM). Accordingly, no straightforward correlation can be drawn between a low E1/2 and the ability of quinones to engage in redox cycling and generate hydrogen peroxide in the presence of DTT. While the E1/2 remains likely to influence the overall reactivity of the quinone scaffold and determine relative rates in the reaction with charged species and nucleophiles, enzyme inhibition, redox cycling, and overall cytotoxicity are apparently more strongly influenced by other, less readily tractable structural features.
Table 4.
H2O2 generation as a measure for redox cycling potential
| Quinone | EC50/μM (N = 2) |
|---|---|
| WDP1149 | 180 ± 15 |
| WDP1079 | 14.0 ± 0.1 |
| WDP1263 | 1.4 ± 0.02 |
| NSC95397 | 5.6 ± 0.9 |
| DA3003-1 | 1.7 ± 0.04 |
It is possible that the relative inability of WDP1149 to generate hydrogen peroxide is due to the high level of captodative stabilization41 of the quinone radical anion. Classical resonance structures show the unpaired electron positioned a to the donor methoxy group as well as the strong fluoride acceptor substituent (Fig. 4).
Fig. 4.
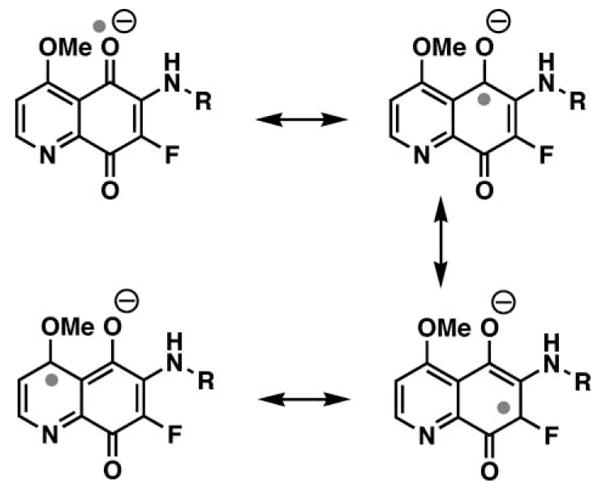
Resonance structures illustrating the captodative stabilization of the radical anion of WDP1149 (right).
Conclusions
Redox cycling and ROS formation by quinones are potentially unselective pathways for enzyme inhibition and can mediate a high level of undesirable toxicity. Computational methods are capable of fine-tuning the electronic properties of the quinone scaffold. The inverse design approach was applied to expedite quinone modification and structure–property correlations. Three probe molecules were synthesized to test the correlation between half-wave potential E1/2, Cdc25 inhibition, cellular toxicities, and redox cycling. Attractive Cdc25B inhibitory values and antitumor cell activities were accomplished. However, in contrast to a previously reported hypothesis, a correlation between redox cycling and E1/2 was not established. More likely, captodative stabilization of the quinone radical anion is responsible for a significant decrease in hydrogen peroxide formation in a DTT coupled redox cycling assay.
Experimental
General
All reactions were performed under a nitrogen or argon atmosphere unless otherwise noted. All reagents and solvents were used as received. Analytical thin layer chromatography (TLC) was performed on SiO2 60 F-254 plates available from Merck. Visualization was accomplished by UV irradiation at 254 nm and/or by staining with para-anisaldehyde (7.5 mL of para-anisaldehyde, 25 mL of conc. H2SO4, and 7.5 mL of acetic acid in 675 mL of 95% ethanol. Flash column chromatography was performed using SiO2 60 (particle size 0.040–0.055 mm, 230–400 mesh).
Melting points were obtained on a Meltemp capillary melting point apparatus fitted with a Fluke 51 II digital thermometer. Infrared spectral data were obtained from a Perkin Elmer Spectrum 100 FT-IR spectrometer using the Universal ATR Sampling Accessory for both oil and solid compounds. Proton and carbon NMR spectra were recorded at 300 MHz/75 MHz (1H NMR/13C NMR) and 600 MHz/150 MHz (1H NMR/13C NMR) in CDCl3 unless otherwise noted. Chemical shifts are reported as δ values in parts per million (ppm) as referenced to residual solvent. 1H NMR spectra are reported as follows: chemical shift, multiplicity (s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, ddd = doublet of doublet of doublets, m = multiplet), number of protons, and coupling constant(s). Mass spectra were obtained at the University of Pittsburgh Mass Spectrometry facility.

5-[(2,5-Dimethoxyphenylamino)methylene]-2,2-dimethyl-[1,3]dioxane-4,6-dione (2)
This compound was prepared according to the literature procedure by Valderrama.42 Mp 164–165 °C; IR (neat, cm−1) 3245.7, 2995.4, 2836.9, 1726.4, 1674.8, 1636.4, 1593.6, 1451.1, 1275.5; 1H NMR (CDCl3, 300 MHz) δ 11.56 (d, 1H, J = 14.4 Hz), 8.64 (d, 1H, J = 14.7 Hz), 6.93–6.88 (m, 2H), 6.75 (dd, 1H, J = 2.7, 9.0 Hz), 3.92 (s, 3H), 3.82 (s, 3H), 1.76 (s, 6H); 13C NMR (CDCl3, 150 MHz) δ 165.2, 163.9, 154.3, 150.8, 143.6, 127.5, 112.6, 111.7, 105.0, 101.7, 87.4, 56.5, 55.97, 27.05; HRMS (ESI) m/z calc for C15H17NO6Na (M + Na) 330.0954, found 330.0959.

5-[(4-Bromo-2,5-dimethoxyphenylamino)methylene]-2,2-dimethyl-[1,3]dioxane-4,6-dione
This compound was prepared according to the literature procedure by Echavarren.43 1H NMR (CDCl3, 300 MHz) δ 11.56 (d, 1H, J = 14.7 Hz), 8.63 (d, 1H, J = 14.4 Hz), 7.20 (s, 1H), 6.87 (s, 1H), 3.93 (s, 3H), 3.92 (s, 3H), 1.77 (s, 6H); HRMS (ESI) m/z calc for C15H16BrNO6Na (M + Na) 408.0059, found 408.0072.

6-Bromo-5,8-dimethoxy-1H-quinolin-4-one (3)
This compound was prepared according to the literature procedure by Echavarren.43 Mp 244–245 °C; 1 H NMR (DMSO-d6, 300 MHz) δ 11.3 (bs, 1H), 7.68–7.64 (m, 1H), 7.40 (s, 1H), 6.01 (d, 1H, J = 7.2 Hz), 3.97 (s, 3H), 3.70 (s, 3H); HRMS (EI) m/z calc for C11H10BrNO3 282.9844, found 282.9845.

6-Bromo-4-chloro-5,8-dimethoxyquinoline (4)43
Compound 3 (5.3 g, 18.7 mmol) was dissolved in POCl3 (70 mL) and heated at reflux for 30 min. After the reaction mixture was cooled to room temperature, it was carefully added to an Erlenmeyer flask containing ice–water (100 mL). Note: Large scale production of 4 required extreme caution in the addition to the ice water due to the highly exothermic nature of the process. The acidic aqueous solution was then neutralized with 5 N NaOH and extracted with CH2Cl2 (3 × 150 mL). The combined organic extracts were washed with H2O (100 mL), dried (MgSO4), and concentrated. The crude residue was purified by chromatography on SiO2 (50% EtOAc–hexanes → 100% EtOAc) to yield 4 (5.37 g, 17.7 mmol, 95%) as a pale yellow solid: Mp 91–92 °C; IR (neat, cm−1) 2938.1, 2838.5, 1574.6, 1490.7, 1231.7; 1H NMR (CDCl3, 300 MHz) δ 8.70 (d, 1H, J = 4.8 Hz), 7.50 (d, 1H, J = 4.8 Hz), 7.18 (s, 1H), 4.03 (s, 3H), 3.85 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 152.2, 148.4, 145.4, 141.6, 139.6, 125.1, 122.5, 116.5, 112.6, 62.1, 56.4; HRMS (EI) m/z calc for C11H9BrClNO2 300.9505, found 300.9507.

4-Chloro-6-(2-morpholin-4-yl-ethylamino)quinoline-5,8-dione (5)
Compound 4 (217 mg, 0.72 mmol) was dissolved in CH3CN (10 mL) and cooled in an ice bath. Cerium(IV) ammonium nitrate (CAN) (1.6 g, 2.9 mmol) in H2O (5 mL) was then added and the reaction mixture was allowed to stir for 4 hours with slow warming to room temperature. It was diluted with H2O (15 mL) and the solution was extracted with CHCl3 (3 × 50 mL). The combined organic extracts were dried (MgSO4) and concentrated. The crude material was taken onto the next step without further purification.
The crude quinone was dissolved in EtOH (12 mL), and CeCl3·7H2O (294 mg, 0.79 mmol) followed by 4-(2-aminoethyl)morpholine (0.103 mL 0.79 mmol) were added at room temperature. The resulting dark red reaction mixture was allowed to stir overnight at room temperature, concentrated and diluted with CH2Cl2 (20 mL). The CH2Cl2 solution was washed with H2O (10 mL), dried (MgSO4), filtered and concentrated to yield a red residue. The crude material was purified by chromatography on SiO2 (50% EtOAc–hexanes → 100% EtOAc → 100% CHCl3 → 10% MeOH–CHCl3) to yield 5 (103.2 mg, 0.32 mmol, 44% (2 steps)) as a red solid: Mp 179–181 °C; IR (neat, cm−1) 3325.6, 2960.2, 2835.7, 1672.3, 1610.9, 1548.3, 1344.5, 1108.5; 1H NMR (CDCl3, 300 MHz) δ 8.84 (d, 1H, J = 5.1 Hz), 7.59 (d, 1H, J = 5.1 Hz), 6.71 (bs, 1H), 5.92 (s, 1H), 3.76 (dd, 4H, J = 4.5, 4.8 Hz), 3.25 (ddd, 2H, J = 5.4, 5.4, 5.7 Hz), 2.71 (dd, 2H, J = 5.7, 6.0 Hz), 2.51 (dd, 4H, J = 4.5, 4.5 Hz); 13C NMR (CDCl3, 75 MHz) δ 179.7, 179.3, 153.9, 151.5, 148.0, 144.7, 129.2, 124.1, 101.1, 66.9, 55.4, 53.2, 38.4; HRMS (ESI) m/z calc for C15H17ClN3O3 (M + H) 322.0958, found 322.0934.
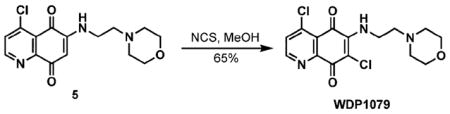
4,7-Dichloro-6-(2-morpholin-4-yl-ethylamino)quinoline-5,8-dione (WDP1079)
N-Chlorosuccinimide (18.7 mg, 0.14 mmol) was added to a solution of quinone 5 (45 mg, 0.14 mmol) in MeOH (14 mL). The reaction mixture was allowed to stir overnight at room temperature, concentrated and purified by chromatography on SiO2 (50% EtOAc–hexanes → 100% EtOAc → 10% MeOH–CH2Cl2) to yield WDP1079 as a red solid (32.4 mg, 0.09 mmol, 65%): Mp 152–154 °C; IR (neat, cm−1) 3211.4, 2959.8, 2850.8, 2828.9, 1674.6, 1602.5, 1553.3, 1324.5, 1200.8; 1H NMR (CDCl3, 300 MHz) δ 8.82 (d, 1H, J = 5.1 Hz), 7.58 (d, 1H, J = 5.4 Hz), 7.1 (bs, 1H), 3.98 (ddd, 2H, J = 5.4, 5.7, 5.7 Hz), 3.77 (dd, 4H, J = 4.5, 4.5 Hz), 2.68 (dd, 2H, J = 6.0, 6.0 Hz), 2.53 (dd, 4H, J = 4.5, 4.5 Hz); 13C NMR (CDCl3, 75 MHz) δ 178.3, 173.6, 153.8 (2C), 150.6, 144.8, 129.3 (2C), 123.7, 66.9, 56.6, 52.9, 40.9; HRMS (ESI) m/z calc for C15H16Cl2N3O3 (M + H) 356.0569, found 356.0546.
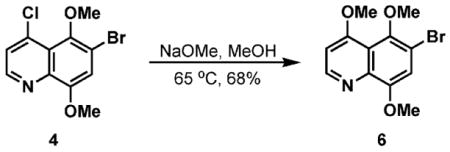
6-Bromo-4,5,8-trimethoxyquinoline (6)44
A 25 wt% NaOMe solution in MeOH (150 mL) was added to compound 4 (3.78 g, 12.5 mmol) in a round-bottomed flask and heated to 65 °C for 35 minutes. After cooling the reaction mixture, H2O (100 mL) was added and the resulting aqueous solution was neutralized with 2 N HCl. The aqueous layer was extracted with CH2Cl2 (3 × 150 mL) and the organic extracts were washed with H2O (100 mL), dried (MgSO4), and concentrated. The resulting residue was purified by chromatography on SiO2 (5% MeOH–CH2Cl2) to yield 6 as a yellow solid (2.53 g, 8.49 mmol, 68%): Mp 140–142 °C; IR (neat, cm−1) 3068.4, 2966.3, 2937.1, 1578, 1504.9, 1395.9, 1063; 1H NMR (CDCl3, 300 MHz) δ 8.71 (d, 1H, J = 5.1 Hz), 7.14 (s, 1H), 6.82 (d, 1H, J = 5.4 Hz), 4.03 (s, 3H), 4.01 (s, 3H), 3.81 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.5, 151.0, 149.6, 145.8, 140.0, 129.3, 117.4, 114.9, 112.8, 102.7, 61.7, 56.5, 56.4; HRMS (EI) m/z calc for C12H11BrNO3 (M – H) 295.9922, found 295.9919.

4-Methoxy-6-(2-morpholin-4-yl-ethylamino)quinoline-5,8-dione (7)
Quinoline 6 (1.7 g, 5.7 mmol) was dissolved in CH3CN (70 mL) and cooled in an ice bath. In a separate Erlenmeyer flask, CAN (12.5 g, 22.8 mmol) was dissolved in H2O (30 mL) then transferred to an addition funnel. The CAN solution was added dropwise to the CH3CN solution, and the mixture was allowed to stir for 4 h with slow warming to room temperature. The reaction mixture was then diluted with H2O (100 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried (MgSO4), filtered and concentrated in vacuo to yield the crude quinone, which was taken on to the next step without further purification.
The crude quinone (1.5 g) was dissolved in EtOH (120 mL) and CeCl3·7H2O (2.3 g, 6.27 mmol) was then added. The reaction mixture was allowed to stir until dissolution was obtained. 4-(2-Aminoethyl)morpholine (0.82 mL, 6.27 mmol) was then added at room temperature, which quickly turned the reaction mixture to a dark red color, and stirred overnight. The solvent was evaporated and the resulting residue was diluted with H2O (50 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried (MgSO4), filtered and concentrated in vacuo to yield a red residue. The crude material was purified by chromatography on SiO2 (10% MeOH–EtOAc → 10% MeOH–CH2Cl2) to yield 7 as an orange oil (633 mg, 2.0 mmol, 35% (2 steps)): IR (neat, cm−1) 3359.2, 2922.5, 2852.1, 1740.5, 1673.9, 1603.7, 1572.3, 1200.5; 1H NMR (CDCl3, 300 MHz) δ 8.83 (d, 1H, J = 6.0 Hz), 7.08 (d, 1H, J = 6.0 Hz), 6.69 (bs, 1H), 5.82 (s, 1H), 4.08 (s, 3H), 3.74 (dd, 4H, J = 4.5, 4.5 Hz), 3.22 (ddd, 2H, J = 5.4, 5.7, 5.7 Hz), 2.70 (dd, 2H, J = 6.0, 6.0 Hz), 2.50 (dd, 4H, J = 4.5, 4.5 Hz); 13C NMR (CDCl3, 75 MHz) δ 180.7, 180.2, 165.7, 155.7, 151.8, 148.1, 116.4, 109.8, 100.1, 66.8, 56.7, 55.3, 53.1, 38.2; HRMS (ESI) m/z calc for C16H20N3O4 (M + H) 318.1454, found 318.1439.

7-Fluoro-4-methoxy-6-(2-morpholin-4-yl-ethylamino)quinoline-5,8-dione (WDP1149)
Quinone 7 (47.5 mg, 0.15 mmol) was dissolved in CH3CN (3 mL) followed by the addition of 4 Å molecular sieves. Selectfluor® (106.3 mg, 0.3 mmol) was then added and the reaction mixture was stirred for 18 h at room temperature, transferred to a separatory funnel and diluted with H2O (5 mL). The aqueous layer was extracted with CHCl3 (3 × 5 mL) and the combined organic extracts were dried (MgSO4), filtered and concentrated in vacuo to yield an orange/red residue. The crude material was purified by chromatography on SiO2 (5% MeOH–CH2Cl2) to yield WDP1149 as an orange amorphous solid (10 mg, 0.03 mmol, 20%): IR (neat, cm−1) 3348.5, 2923.1, 2852.3, 1674.7, 1605.5, 1569.4, 1474.2, 1196.8; 1H NMR (CDCl3, 300 MHz) δ 8.82 (d, 1H, J = 6.0 Hz), 7.08 (d, 1H, J = 6.0 Hz), 6.39 (bs, 1H), 4.08 (s, 3H), 3.76 (dd, 4H, J = 4.5, 4.8 Hz), 3.72–3.66 (m, 2H), 2.66 (dd, 2H, J = 5.7, 6.0 Hz), 2.52 (dd, 4H, J = 4.5, 4.5 Hz); 13C NMR (CDCl3, 75 MHz) d 179.8 (d, J = 12.75 Hz), 172.9 (d, J = 15.75 Hz), 165.5, 155.8, 150.2 (d, J = 6.75 Hz), 141.3 (d, J = 247.5 Hz), 133.9, 115.5, 110.2, 66.8, 56.8, 53.1 (2C), 40.0 (d, J = 7.5 Hz); HRMS (ESI) m/z calc for C16H19FN3O4 (M + H) 336.1360, found 336.1366.

2,3-Diaminophenol (9)
2-Amino-3-nitrophenol 8 (2 g, 12.98 mmol) was dissolved in MeOH (100 mL) and 10% Pd/C (200 mg) in MeOH (5 mL), prepared in a separate flask, was added via pipette. The reaction mixture was allowed to stir under H2 (1 atm) for 5 h, upon which the starting material was consumed as observed by TLC. The reaction mixture was filtered through Celite and concentrated in vacuo to yield a brown solid (1.57 g). This product was taken on to the next step without further purification.

Quinoxalin-5-ol (10)45
2,3-Diaminophenol 9 (1.57 g, 12.6 mmol) was dissolved in a mixture of 4 M NaOAc (16 mL) and 2 M AcOH (24 mL) and heated to 60 °C. In a separate flask, sodium glyoxal bisulfite (3.5 g, 13.2 mmol) was dissolved in H2O (90 mL) and also heated to 60 °C. When both solutions reached ~60 °C, the 2,3-diaminophenol solution was then transferred by pipette to the sodium glyoxal bisulfite solution. The reaction mixture was allowed to stir at 60 °C for 1 h. After cooling the mixture to room temperature, the pH was adjusted to ~8 using 1 N NaOH. The resulting aqueous solution was extracted with EtOAc (8 × 100 mL), dried (MgSO4), filtered, and concentrated in vacuo to afford a brown solid. The crude material was purified by chromatography on SiO2 (10% EtOAc–hexanes → 50% EtOAc–hexanes) to yield 10 as a yellow solid (1.57 g, 10.7 mmol, 82% (2 steps)): Mp 100–102 °C; IR (neat, cm−1) 3300.4, 1620.7, 1577.9, 1498.4, 1258.4; 1H NMR (CDCl3, 600 MHz) δ 8.92 (d, 1H, J = 1.2 Hz), 8.74 (d, 1H, J = 1.8 Hz), 7.72 (d, 1H, J = 7.2 Hz), 7.68 (dd, 1H, J = 1.2, 8.4 Hz), 7.27 (dd, 1H, J = 1.2, 7.8 Hz); 13C NMR (CDCl3, 150 MHz) δ 152.0, 145.6, 143.2, 142.2, 132.9, 131.4, 119.7, 111.0; HRMS (EI) m/z calc for C8H6N2O (M) 146.0480, found 146.0483.

Quinoxaline-5,8-dione (11)46
Quinoxalin-5-ol 10 (49.5 mg, 0.34 mmol) was dissolved in CH3CN–H2O (1 mL/0.5 mL) and cooled in an ice bath. In a separate flask, [bis(trifluoroacetoxy)iodo]benzene (PIFA, 322 mg, 0.75 mmol) was dissolved in CH3CN–H2O (1 mL/0.5 mL) and added dropwise to the solution containing 10 at 0 °C. The reaction mixture was allowed to stir for 4 h, then diluted with H2O (5 mL). The aqueous solution was extracted with EtOAc (3 × 10 mL), dried (MgSO4), filtered, and concentrated in vacuo to afford a brownish solid. The crude material was purified by chromatography on SiO2 (20% EtOAc–hexanes → 50% EtOAc–hexanes → 100% EtOAc) to afford 11 as a yellow solid (25 mg, 0.16 mmol, 46%): Mp 171.7–172.5 °C; IR (neat, cm−1) 3044.4, 1673, 1603.1, 1313.9, 1086.6; 1H NMR (CDCl3, 600 MHz) δ 9.08 (s, 2H), 7.27 (s, 2H); 13C NMR (CDCl3, 150 MHz) δ 182.6, 149.1, 143.7, 138.7; HRMS (EI) m/z calc for C8H4N2O2 (M) 160.0273, found 160.0273.

6-(2-Morpholin-4-yl-ethylamino)quinoxaline-5,8-dione (12)
Quinone 11 (109 mg, 0.68 mmol) was dissolved in EtOH (17 mL) and CeCl3·7H2O (279 mg, 0.75 mmol) was added. The reaction mixture was stirred until dissolution was achieved. 4-(2-Aminoethyl)morpholine (0.098 mL, 0.75 mmol) was then added at room temperature upon which the mixture became dark (greenish) in color. The solution was stirred overnight, diluted with H2O (30 mL) and extracted with CHCl3 (3 × 75 mL). The resulting organic layers were dried (MgSO4), filtered and concentrated in vacuo to yield a crude residue. The crude material was purified by chromatography on SiO2 (5% MeOH–CH2Cl2) to yield 12 as a red solid (122 mg, 0.42 mmol, 62%): Mp 150–153 °C; IR (neat, cm−1) 3345.1, 2915.5, 2852.1, 1698.4, 1600.2, 1558.2, 1465.5; 1H NMR (CDCl3, 300 MHz) δ 8.99 (d, 1H, J = 2.4 Hz), 8.92 (d, 1H, J = 2.1 Hz), 6.74 (bs, 1H), 5.99 (s, 1H), 3.75 (dd, 4H, J = 4.5, 4.8 Hz), 3.29 (ddd, 2H, J = 5.7, 5.7, 5.7 Hz), 2.73 (dd, 2H, J = 6.0, 6.3 Hz), 2.52 (dd, 4H, J = 4.5, 4.8 Hz); 13C NMR (CDCl3, 75 MHz) δ 179.7, 179.6, 149.1, 148.0, 147.3, 145.6, 142.9, 102.0, 66.8, 55.3, 53.1, 38.6; HRMS (ESI) m/z calc for C14H17N4O3 (M + 1) 289.1301, found 289.1300.

6-Chloro-7-(2-morpholin-4-yl-ethylamino)quinoxaline-5,8-dione (WDP1263)
A solution of quinone 12 (42.8 mg, 0.15 mmol) in MeOH (15 mL) was treated with N-chlorosuccinimide (20.0 mg, 0.15 mmol). The reaction mixture was allowed to stir overnight at room temperature. The solvent was evaporated and the residue was purified by chromatography on SiO2 (50% EtOAc–hexanes → 100% EtOAc →10% MeOH–CH2Cl2) to yield WDP1263 as a dark red amorphous solid (33.5 mg, 0.104 mmol, 69%): IR (neat, cm−1) 3270, 2957.6, 2853.3, 1703.5, 1648.2, 1593.4, 1557.7, 1325.7; 1H NMR (CDCl3, 300 MHz) δ 9.0 (d, 1H, J = 2.1 Hz), 8.92 (d, 1H, J = 2.1 Hz), 7.22 (bs, 1H), 4.02 (ddd, 2H, J = 5.4, 5.7, 5.7 Hz), 3.76 (dd, 4H, J = 4.5, 4.5 Hz), 2.72 (dd, 2H, J = 5.7, 6.0 Hz), 2.55 (dd, 4H, J = 4.5, 4.8 Hz); 13C NMR (CDCl3, 75 MHz) δ 178.3 (2C), 149.1, 147.5 (2C), 145.1, 144.7, 142.3, 66.9, 56.4, 52.9, 40.97; HRMS (ESI) m/z calc for C14H16ClN4O3 (M + H) 323.0911, found 323.0883.
Acknowledgments
The authors thank the National Cancer Institute for grant support (CA78039). We are also grateful to Prof. Martin Field for providing the DYNAMO program. Support of the DARPA “Predicting Real Optimized Materials” project through ARO is gratefully acknowledged (W911NF-04-1-0243).
References
- 1.Sohn J, Kiburz B, Li Z, Deng L, Safi A, Pirrung MC, Rudolph J. J Med Chem. 2003;46:2580–2588. doi: 10.1021/jm0300835. [DOI] [PubMed] [Google Scholar]
- 2.Galaktionov K, Lee AK, Eckstein J, Draetta G, Meckler J, Loda M, Beach D. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 3.Lavecchia A, Cosconati S, Limongelli V, Novellino E. ChemMed-Chem. 2006;1:540–550. doi: 10.1002/cmdc.200500092. [DOI] [PubMed] [Google Scholar]
- 4.Brisson M, Nguyen T, Wipf P, Joo B, Day BW, Skoko JS, Schreiber EM, Foster C, Bansal P, Lazo JS. Mol Pharmacol. 2005;68:1810–1820. doi: 10.1124/mol.105.016360. [DOI] [PubMed] [Google Scholar]
- 5.Ham SW, Choe JI, Wang MF, Peyregne V, Carr BI. Bioorg Med Chem Lett. 2004;14:4103–4105. doi: 10.1016/j.bmcl.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Rudolph J. Antioxid Redox Signaling. 2005;7:761–767. doi: 10.1089/ars.2005.7.761. [DOI] [PubMed] [Google Scholar]
- 7.Ducruet AP, Vogt A, Wipf P, Lazo JS. Annu Rev Pharmacol Toxicol. 2005;45:725–750. doi: 10.1146/annurev.pharmtox.45.120403.100040. [DOI] [PubMed] [Google Scholar]
- 8.Lyon MA, Ducruet AP, Wipf P, Lazo JS. Nat Rev Drug Discovery. 2002;1:961–976. doi: 10.1038/nrd963. [DOI] [PubMed] [Google Scholar]
- 9.Kar S, Wang M, Ham SW, Carr BI. Biochem Pharmacol. 2006;72:1217–1227. doi: 10.1016/j.bcp.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 10.Park H, Carr BI, Li M. Bioorg Med Chem Lett. 2007;17:2351–2354. doi: 10.1016/j.bmcl.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 11.Aguirre G, Cerecetto H, Di Maio R, Gonzalez M, Alfaro MEM, Jaso A, Zarranz B, Ortega MA, Aldana I, Monge-Vega A. Bioorg Med Chem Lett. 2004;14:3835–3839. doi: 10.1016/j.bmcl.2004.04.088. [DOI] [PubMed] [Google Scholar]
- 12.Campos J, Nunez MdC, Rodrigues V, Entrena A, Hernandez-Alcoceba R, Fernandez F, Lacal JC, Gallo MA, Espinosa A. Eur J Med Chem. 2001;36:215–225. doi: 10.1016/s0223-5234(01)01219-3. [DOI] [PubMed] [Google Scholar]
- 13.Wardman P. Curr Med Chem. 2001;8:739–761. doi: 10.2174/0929867013372959. [DOI] [PubMed] [Google Scholar]
- 14.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 15.Fariss MW, Chan CB, Patel M, van Houten B, Orrenius S. Mol Interventions. 2005;5:94–111. doi: 10.1124/mi.5.2.7. [DOI] [PubMed] [Google Scholar]
- 16.Hoye AT, Davoren J, Wipf P, Fink MP, Kagan VE. Acc Chem Res. 2008;41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- 17.Edgar TF, Dixon DA, Reklaitis GV. Impact of advances in computing and communications technologies on chemical science and technology. National Academy Press; Washington, D. C: 1999. pp. 74–91. [PubMed] [Google Scholar]
- 18.Wang M, Hu X, Beratan DN, Yang W. J Am Chem Soc. 2006;128:3228–3232. doi: 10.1021/ja0572046. [DOI] [PubMed] [Google Scholar]
- 19.Keinan S, Hu X, Beratan DN, Yang W. J Phys Chem A. 2007;111:176–181. doi: 10.1021/jp0646168. [DOI] [PubMed] [Google Scholar]
- 20.Hu X, Beratan DN, Yang W. J Chem Phys. 2008 doi: 10.1063/1.2958255. in press. [DOI] [PubMed] [Google Scholar]
- 21.Xiao D, Yang W, Beratan DN. J Chem Phys. 2008 doi: 10.1063/1.2955756. [DOI] [PubMed] [Google Scholar]
- 22.Lilienfeld OAv, Lins RD, Rothlisberger U. Phys Rev Lett. 2005;95:153002. doi: 10.1103/PhysRevLett.95.153002. [DOI] [PubMed] [Google Scholar]
- 23.Koh SK, Ananthasuresh GK, Vishveshwara S. Int J Rob Res. 2005;24:109–130. [Google Scholar]
- 24.Land AH, Doig AG. Econometrica. 1960;28:497–520. [Google Scholar]
- 25.Ostrovsky GM, Achenie LEK, Sinha M. Comput Chem. 2002;26:645–660. doi: 10.1016/s0097-8485(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 26.Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 27.Field MJ, Albe M, Bret C, Proust-De Martin F, Thomas A. J Comput Chem. 2000;21:1088–1100. [Google Scholar]
- 28.Cassis R, Tapia R, Valderrama JA. Synth Commun. 1985;15:125–133. [Google Scholar]
- 29.Gomez-Bengoa E, Echavarren AM. J Org Chem. 1991;56:3497–3501. [Google Scholar]
- 30.Kitahara Y, Tamura F, Nishimura M, Kubo A. Tetrahedron. 1998;54:8421–8432. [Google Scholar]
- 31.Kitahara Y, Nakahara S, Yonezawa T, Nagatsu M, Shibano Y, Kubo A. Tetrahedron. 1997;53:17029–17038. [Google Scholar]
- 32.Wipf P, Joo B, Nguyen T, Lazo JS. Org Biomol Chem. 2004;2:2173–2174. doi: 10.1039/b408184f. [DOI] [PubMed] [Google Scholar]
- 33.Peng W, Shreeve JM. J Org Chem. 2005;70:5760–5763. doi: 10.1021/jo0506837. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt A, Shilabin AG, Nieger M. Org Biomol Chem. 2003;1:4342–4350. doi: 10.1039/b308412d. [DOI] [PubMed] [Google Scholar]
- 35.Freeman SK, Spoerri PE. J Org Chem. 1951;16:438–442. [Google Scholar]
- 36.Barret R, Daudon M. Tetrahedron Lett. 1990;31:4871–4872. [Google Scholar]
- 37.Kitahara Y, Nakahara S, Tanaka Y, Kubo A. Heterocycles. 1992;34:1623–1630. [Google Scholar]
- 38.Brisson M, Foster C, Wipf P, Joo B, Tomoko RJ, Nguyen T, Lazo JS. Mol Pharmacol. 2007;71:184–192. doi: 10.1124/mol.106.028589. [DOI] [PubMed] [Google Scholar]
- 39.Pick E, Keisari Y. Cell Immunol. 1981;59:301–318. doi: 10.1016/0008-8749(81)90411-1. [DOI] [PubMed] [Google Scholar]
- 40.Lazo JS, Nemoto K, Pestell KE, Cooley K, Southwick EC, Mitchell DA, Furey W, Gussio R, Zaharevitz DW, Joo B, Wipf P. Mol Pharmacol. 2002;61:720–728. doi: 10.1124/mol.61.4.720. [DOI] [PubMed] [Google Scholar]
- 41.Viehe HG, Janousek Z, Merenyi R, Stella L. Acc Chem Res. 1985;18:148–154. [Google Scholar]
- 42.Cassis R, Tapia R, Valderrama JA. Synth Commun. 1985;15:125–133. [Google Scholar]
- 43.Gomez-Bengoa E, Echavarren AM. J Org Chem. 1991;56:3497–3501. [Google Scholar]
- 44.Kitahara Y, Tamura F, Nishimura M, Kubo A. Tetrahedron. 1998;54:8421–8432. [Google Scholar]
- 45.Freeman SK, Spoerri PE. J Org Chem. 1951;16:438–442. [Google Scholar]
- 46.Kitahara Y, Nakahara S, Tanaka Y, Kubo A. Heterocycles. 1992;34:1623–1630. [Google Scholar]