

Abstract
Aberrant genetic alternations in human gliomas such as amplification of EGFR, mutation and/or deletion of tumor suppressor gene PTEN, and mutations of PIK3CA contribute to constitutive activation of the PI3K pathway. We investigated the potential anti-tumor activity of NVP-BEZ235, which is a novel dual PI3K/mTOR inhibitor, in gliomas. The compound suppressed glioma cell proliferation with IC50 values in the low nM range by specifically inhibiting the activity of target proteins, including Akt, S6K1, S6, and 4EBP1 in the PI3K/Akt/mTOR signaling pathway. NVP-BEZ235 treatment of glioma cell lines led to G1 cell cycle arrest, and induced autophagy. Furthermore, expression of the vascular endothelial growth factor (VEGF), which is an important angiogenic modulator in glioma cells, was significantly decreased, suggesting that NVP-BEZ235 may also exert an anti-angiogenic effect. Preclinical testing of the therapeutic efficacy of NVP-BEZ235 demonstrated that it significantly prolonged the survival of tumor bearing animals without causing any obvious toxicity. Tumor extracts harvested from animals post-treatment showed that the compound inhibited the activity of target proteins in the PI3K/Akt/mTOR cascade. Immunohistochemical analyses also showed a significant reduction in staining for VEGF von Willebrand factor (factor VIII) in NVP-BEZ235-treated tumor sections as compared with controls, further confirming that NVP-BEZ235 has an anti-angiogenic effect in vivo. We conclude from these findings that NVP-BEZ235 antagonizes PI3K and mTOR signaling, and induces cell cycle arrest, down-regulation of VEGF, and autophagy. These results warrant further development of NVP-BEZ235 for clinical trials for human gliomas or other advanced cancers with altered PI3K/Akt/mTOR signaling.
Keywords: small molecular mass inhibitor, PI3K, mTOR, autophagy, anti-angiogenesis
Introduction
The most frequent alterations observed in primary glioblastoma are amplification of the EGFR gene (1), mutation and deletion of the PTEN gene (2,3), and mutations of the PIK3CA gene (4). These findings suggest that such genetic alterations contribute to the uncontrolled PI3K activity, and thus the increased phosphorylation and activation of Akt. Because PI3K is an essential lipid kinase that controls many pathways involved in cell proliferation, apoptosis, and angiogenesis and plays a key role in glioma development and its aggressive behavior, multiple biotech and pharmaceutical companies and academic centers have mounted drug discovery efforts to develop targeted small-molecular mass kinase inhibitors of PI3K. Indeed, one PI3K modulator, LY294002, has already shown in vivo efficacy in the treatment of malignant glioma (5). Similarly, another PI3K modulator, wortmannin, showed marked anti-tumor activity both in vitro and in vivo (6). Recently, a novel class of wortmannin derivatives has been identified and shown greater chemical stability, lower toxicity, and greater anti-tumor activity than the parent compound (7). Cells with abnormalities in PTEN or amplification of PI3K are particularly sensitive to these inhibitors. Even more recently, a dual PI3K/mTOR inhibitor showed emergent efficacy in gliomas during laboratory testing (8).
Researchers at Novartis Institutes have developed a new class of PI3K inhibitors with multiple-target capabilities for Biomedical Research (NIBR) using a structure-based discovery approach. NVP-BEZ235 is an imidazo[4,5-c]quinoline derivative (Fig. 1) that inhibits PI3K and mTOR kinase activity by binding to the ATP binding cleft of these enzymes: IC50 = 4, 76, 7, 5 and 21 nM against p110α, β, γ, δ and mTOR, respectively. Further characterization of NVP-BEZ235 has shown that it poorly inhibits a representative panel of protein kinases in biochemical assays (9). The specificity against PI3K/mTOR observed in biochemical profiling has been further confirmed in cellular settings (9). Thus, NVP-BEZ235 is able to effectively and specifically block the dysfunctional activation of the PI3K/mTOR pathway in human tumor cell lines. In the present study, we demonstrated the effect of NVP-BEZ235 on PI3K/mTOR signaling and glioma cell growth in vitro as well as its therapeutic efficacy in vivo.
Figure 1. NVP-BEZ235 treatment inhibits glioma proliferation and attenuates PI3K/mTOR signaling pathway.

(A) Glioma cells in 96-well plates were treated with increasing concentrations of NVP-BEZ235 for 72 h and subjected to an SRB assay, as described in Materials and Methods. Plot depicts the percentage growth of NVP-BEZ235-treated cells compared with the growth of the vehicle-treated control cells. Each culture was performed in triplicate. The results shown are the arithmetic mean ± the standard deviation from a single experiment. Similar results were obtained from three independent experiments. U251 and U87 cells are used as PTEN negative cells and LN18 and LN229 as PTEN positive cells. (B) To evaluate the target inactivation by NVP-BEZ235 in a time-dependent manner, glioma cells were treated with a fixed concentration of NVP-BEZ235 72 h. At each indicated time point, cell extracts were subjected to immunoblotting analysis.
Materials and Methods
Cell lines and culture conditions
We used the following cell lines: U87, LN229, U251, LN428, D54 and LN18. All cell lines were maintained as monolayer cultures in DMEM/F12 supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (all from Life Technologies, Inc., Grand Island, NY).
Reagents
NVP-BEZ235 was provided by Novartis Pharma AG (Basel, Switzerland). For in-vitro use, NVP-BEZ235 was dissolved in DMSO (Sigma-Aldrich Corp., St. Louis, MO) to a concentration of 10 mmol/L, stored at –20°C, and further diluted to an appropriate final concentration in DMEM at the time of use.
Cell proliferation assay
The anti-proliferative effect of NVP-BEZ235 on cells growing in culture was determined using the sulforhodamine B (SRB) assay, as described previously (10).
Cell cycle and autophagy analyses
Glioma cells were plated in 60-mm plates and treated with NVP-BEZ235 for 72h. After that, cells were fixed in 70% ethanol in PBS and stored at −20° C for 24 h. Propidium Iodine staining of DNA was performed to determine the cell cycle distribution using a BD FACSCalibur Flow Cytometer, and CellQuest software (BD Biosciences, San Jose, CA). For autophagy analysis, cells were plated in a 6-well plate and treated with desired concentrations of NVP-BEZ235, for 72 h, after which media was removed and the cells were incubated in 1µg/ml Acridine Orange solution at 37°C for 15 minutes. Cells were then trypsinized, re-suspended in PBS, and analyzed for autophagy using a BD FACSCalibur Flow Cytometer, and CellQuest software (BD Biosciences, San Jose, CA).
Western blotting analysis
Glioma cells were serum-starved for 24 h prior to the incubation of 50 nM NVP-BEZ235 for an additional 6 h. Various concentrations of growth factors (IGF 100 ng/ml; EGF50 ng/ml; PDGF 50 ng/ml; VEGF 20 ng/ml) were then added to the cell cultures for 15 min. Cells were then harvested and extracts made for immunoblotting analysis. The membrane was probed with primary antibodies. Primary antibodies used in this study were phospho-Akt (Ser473), total Akt, phospho-MAPK (Thr202/Tyr204), total MAPK, phospho-S6K1, total S6K1, phospho-S6, and total S6 (Cell Signaling, Boston, MA). Anti-β-actin antibody was purchased from Sigma-Aldrich.
Intracranial animal model study
Male nude mice were purchased from the National Cancer Institute-Frederick Cancer Research and Development Center (Frederick, MD). Mice were maintained in a pathogen-free environment and used in accordance with the Animal Care and Use Guidelines of The University of Texas M. D. Anderson Cancer Center. Mice used for this study were 6 to 8 weeks old. In this study, 5 × 105 of U87 cells in DMEM/F12 serum-free media (SFM; 5 µL) were implanted intracranially in each mouse using a guide-screw system, as described previously (11). Four days after injection of the tumor cells, mice were randomized into three groups (10 animals per group). Mice in group 1 were given 25 mg/kg NVP-BEZ235 in 200 µl of NMP/PEG300 (1:9, v/v) solution, mice in group 2 were given 45 mg/kg NVP-BEZ235 in 200 µl of NMP/PEG300 (1:9, v/v) solution, and mice in group 3 were given the vehicle used for administration of NVP-BEZ235 (control). NVP-BEZ235 in all animals was administered via oral gavage and freshly prepared every day before adminstration to animals. Treatment frequency was once a day for 5 days with 2 days off between treatments, for a total duration of 4 weeks. Mice were monitored daily and euthanized when they became moribund. Then, the whole brain was extracted for rapid freezing in liquid nitrogen and storage at –70°C. Tumor volume was determined according to the formula: volume = [(length)2(width)2]/2.
ELISA assay and immunohistochemical staining
We performed human VEGF enzyme-linked immunosorbent assay (ELISA) analysis to quantify secretory VEGF165 in the conditioned media according to the manufacturer’s instructions (R & D Systems, Minneapolis, MN). Cells were treated with different concentrations of NVP-BEZ235 and VEGF was quantified in the conditioned media as described previously (12). Sections (5 µm thick) of formalin-fixed, paraffin-embedded whole brains from control vehicle and NVP-BEZ235 treated animal specimens were stained with anti Factor V111 antibodies (1:300 dilution; Santa Cruz Biotechnology). The level of expression of factor V111 was determined by standard immunohistochemical staining. The sections were visualized by using a diaminobenzidine substrate kit. The slides were examined under a bright-field microscope.
Statistical analyses
For the in vitro experiments, statistical analyses were done using a two-tailed Student’s t test. Data are given as the mean ± SD. The in vivo therapeutic efficacy of NVP-BEZ235 was assessed by plotting the Kaplan-Meier survival curves of animals, and group data were compared using the log-rank test.
Results
NVP-BEZ235 inhibits glioma proliferation and attenuates PI3K/Akt/mTOR signaling
The SRB assay to assess glioma cell growth demonstrated that NVP-BEZ235 effectively suppressed glioma cell growth at nM concentrations (Fig. 1A. We did observe a dose dependent growth inhibition in glioma cells tested in nM range of NVP-BEZ235 tested.
The activity of NVP-BEZ235 as a modulator of the PI3K/Akt/mTOR pathway was supported by Western blot analyses of different components of this signal pathway. NVP-BEZ235 treatment reduced the phosphorylation of Akt (Figure 1B) and also reduced the activation of intracellular Akt downstream targets, including p70S6K and pS6. However, NVP-BEZ235 did not interfere with the MAPK activation, suggesting that NVP-BEZ235 selectively blocks the PI3K/Akt pathway.
NVP-BEZ235 blocks growth factor-induced PI3K/Akt/mTOR signaling
To test if NVP-BEZ235 can effectively perturb growth factor-induced PI3K signaling, Cellular extracts from serum-starved glioma cell lines incubated in the presence or absence of 50 nM NVBEZ235 stimulated with or without several growth factors were subjected to immunobloting for the expression of phosphorylated and total Akt, S6K1, MAPK, and S6 proteins. In U87 cells, the phospho-Akt level was induced between un-stimulated and growth factor-stimulated samples. (Fig. 2) and NVP-BEZ235 significantly inhibited the phospho-Akt. In contrast, growth factor stimulation caused variable phospho-Akt activation in LN229 cells, a wt-PTEN cell line. Expression of phospho-S6K1 and S6K1 downstream target protein S6 was also evaluated by growth factor stimulation and NVP-BEZ235 blocked the mTOR activity in both cell lines, suggesting that NVP-BEZ235 is an effective antagonist of mTOR.
Figure 2. NVP-BEZ235 interrupts growth factor-induced PI3K/mTOR signaling.

Under basal conditions in serum-free medium (SFM), the PI3K signal as assessed by the phospho-Akt level was constitutively active in PTEN-negative U87 cells but not in wt-PTEN LN229 cells. Upon growth factor stimulation, levels of phospho-Akt in U87 cells remained unchanged, whereas a different pattern of phospho-Akt activation was noted in LN229 cells. Based on the extent of Akt activation, receptors for IGF-I seemed to be more abundant than receptors for EGF and PDGF in LN229 cells. The VEGF receptor appeared to be scarce in LN229 cells. The activity of Akt and S6K1, in both cell lines was significantly inhibited by NVP-BEZ235, suggesting its effectiveness on blocking the PI3K/mTOR signal pathway. Expression of the corresponding total target protein and actin was used as control.
NVP-BEZ235 causes cell cycle arrest and induces autophagy
We examined the effect of NVP-BEZ235 on glioma cell cycle progression. Results, shown in Figure 3A, revealed that NVP-BEZ235 treatment leads to G1 cell cycle arrest. In addition, studies have shown that anti-cancer drugs induce a type II programmed cell death, namely autophagy (13, 14). We also examined whether NVP-BEZ235 treatment induces autophagy in glioma cells as quantified AVO development by FACS using vital staining with an acridine orange fluorescence dye (15). The percentage of AVO-positive cells showing prominent red fluorescence significantly increased in cells treated with NVP-BEZ235 (Figure 3B) indicating that NVP-BEZ235 induced the development of AVOs in glioma cells tested. We also examined the expression of the LC3 proteins, a hallmark of cells undergoing autophagy (16, 17). Results from the immunoblotting analysis with anti-LC3 antibody shown in Figure 3C (bottom panel) indicate that LC3 is involved in NVP-BEZ235-induced autophagy and that NVP-BEZ235 stimulates the conversion of a fraction of LC3-I into LC3-II.
Figure 3. NVP-BEZ235 causes cell cycle arrest and induces autophagy.
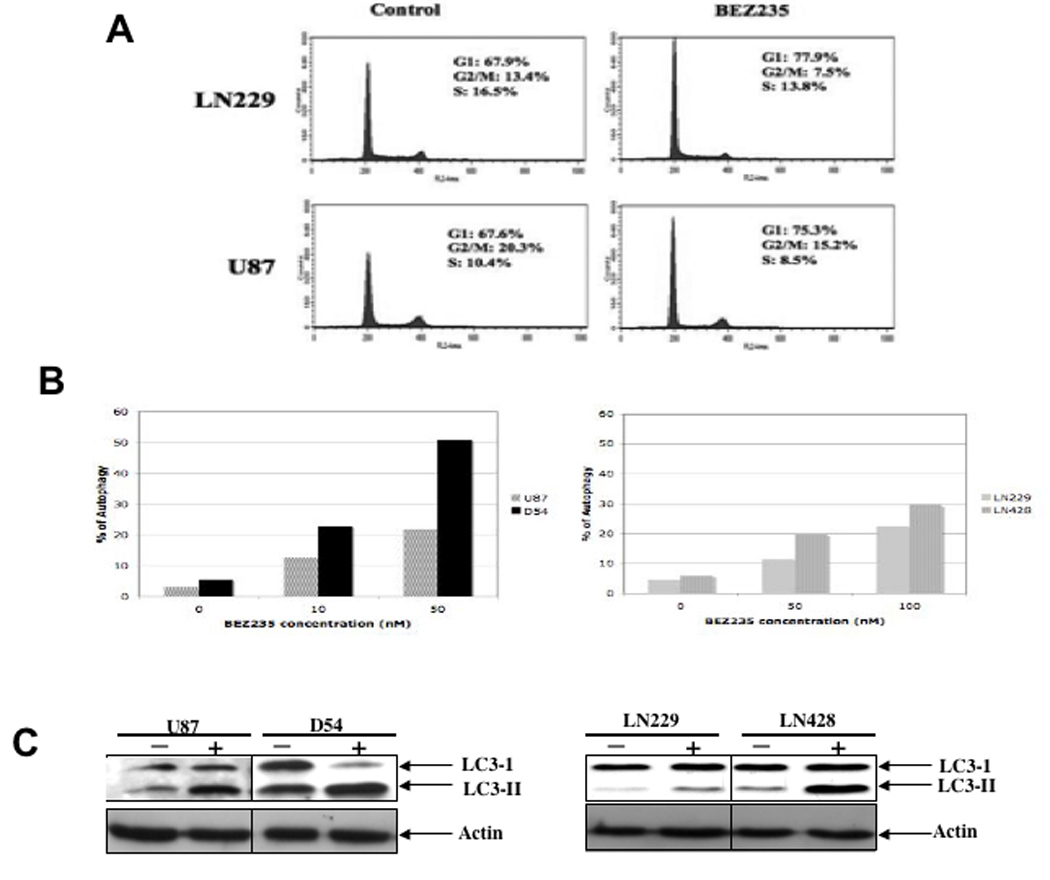
(A) Cell cycle analysis was performed on cells treated or untreated with 50 nM NVP-BEZ235 for 48 h. Cell cycle distribution was labeled on the histogram. NVP-BEZ235 treatment resulted in G1 cell cycle arrest. (B) Following NVP-BEZ235 treatment, glioma cells were either stained with acridine orange for FACS analysis, to assess autophagy, or harvested for immunoblotting analysis, to assess LC3-II expression. Top panel shows the results of FACS analysis of acridine-positive cells. It appears that glioma cells are prone to NVP-BEZ235-induced autophagy. Bottom panel shows LC3-II expression as an indication of autophagy in the (−) untreated and (+) NVP-BEZ235-treated cells.
NVP-BEZ235 prolongs survival of U87 intracranial tumor model
We evaluated the therapeutic efficacy of NVP-BEZ235 in vivo in an intracranial model in which the primary end point was animal survival (11).We used U87 glioma cell lines. The median survival was 28 days in vehicle control animals (Fig. 4A). In contrast, treatment with NVP-BEZ235 at doses of 25 mg/kg or 45 mg/kg extended the median survival of U87 xenograft animals by 7 and 14 days, respectively (P=0.025 and P=0.014 respectively) compared with vehicle-treated animals. Animal weights were measured during the course of the experiment and revealed minimal, non-statistically significant weight fluctuations in animals receiving NVP-BEZ235 as compared with controls (Figure 4.B), indicating that NVP-BEZ235 at the therapeutic dosages and schedule used in this study was well tolerated by the tumor bearing animals. In addition, the sizes of both untreated and treated tumors were measured; NVP-BEZ235-treated tumors were much smaller than vehicle-treated control tumors. The tumor volume after 4 weeks reached around 110 mm3 in control tumors, whereas the animals treated with NVP-BEZ235 at 25 mg/kg and 45 mg/kg showed tumor volumes of 70 mm3 and 30 mm3, respectively (Figure 4.C).
Figure 4. NVP-BEZ235 treatment prolongs survival of animals with intracranial xenografts.

(A) Top panel: The Kaplan-Meier survival curve for the U87 xenograft experiment, with a p value of 0.026. The p values in the plot determined by the log-rank test are for the comparison of the overall survival of the vehicle-treated mice with that of the NVP-BEZ235-treated mice. (B) Weight measurements of experimental animals ar regular intervals. (C) Tumor volumes of intracranial tumors were measured at the time of sacrifice. The mean tumor volume was reduced in treated animal with NVP-BEZ235. (D) Immunoblotting analyses to assess LC3-II expression after 2-week and 4-week tumor cell extracts following NVP-BEZ235 treatment. The increase in ratio of LC3-II/LC3-I is an indication of autophagy in NVP-BEZ235 treated tumors in comparison to vehicle treated animals. (E) Immunoblotting analyses of both the expression and activation of Akt and S6K1 in 2-week and 4-week tumors following NVP-BEZ235 treatment. Immunoblotting analyses demonstrated that NVP-BEZ235 inhibited the activity of S6K1 in both sets of tumors, as assessed by the level of corresponding phosphorylated protein.
The lysates of tumor tissue from animals treated with NVP-BEZ235 showed a dramatic change in the ratio of cytosolic LC3-I to membrane-bound LC3-II, a modification that is essential for the induction of autophagy, indicating that autophagy is involved in tumor growth retardation in vivo (Figure 4 D). We also examined the effect of NVP-BEZ235 on the components of the PI3K/mTOR signaling pathway in vivo. Tumor extracts from animals after 2 and 4 weeks of NVP-BEZ235 treatment (Figure 4 E) showed influence of NVP-BEZ235 on PI3K signaling after 4 weeks of administration, as evidenced by the decresaed phospho-Akt, phospho-S6K1 and phospho-S6 levels.
NVP-BEZ235 attenuates VEGF secretion and inhibits factor VIII expression
One of the important tumor suppressor functions of PTEN is to control tumor angiogenesis by regulating VEGF expression (17). Aberrant PI3K signaling due to impaired PTEN function plays important roles in normal vascular development and in tumor angiogenesis (18). We therefore examined whether NVP-BEZ235 can influence VEGF secretion in glioma cells and thereby interfere with tumor angiogenesis in vivo. U87 cells (19) were treated for 48 h with different concentrations of NVP-BEZ235 (Fig. 5 A) and levels of VEGF secreted in serum free media (SFM) was measured with an ELISA kit. Cells treated with NVP-BEZ235 showed a dose dependent decrease in VEGF secretion. Immunohistochemical analysis further showed a significant reduction in factor VIII–positive staining in NVP-BEZ235-treated tumor sections as compared with control sections, suggesting that NVP-BEZ235 may be involved in inhibiting tumor angiogenesis in vivo (Figure 5.B).
Figure 5. NVP-BEZ235 attenuates VEGF secretion and inhibits factor VIII expression VEGF expression in-vivo.

(A) NVP-BEZ235 attenuated VEGF secretion by at least 40% in U87 cells. The VEGF level in the serum-free medium was measured in triplicate and the experiment repeated at least twice to confirm results. The amount of secreted VEGF was rendered in pg/ml/105 cells/24 h, as described previously (26). (B) Detection of factor VIII. Staining of factor VIII was used to measure the effect of NVP-BEZ235 on tumor angiogenesis. A drastic reduction in factor VIII–positive staining was noted in the NVP-BEZ235-treated (45mg/Kg) tumor section as compared with staining results in control cells.
Discussion
PI3K is a pivotal lipid kinase involved in transmitting signals from various growth factors to promote cell growth (20). In cancers, including gliomas, PI3K–dependent activity is frequently elevated due to amplification (1) and/or gain-of-function mutations (4) of the PI3KCA gene, as well as loss of function of the PTEN tumor suppressor gene (2, 3). These observations have made PI3K an attractive therapeutic target for small-molecular mass inhibitors to be used in the treatment of cancers, including gliomas. In fact, several groups have already reported the anti-tumor efficacy of different forms of PI3K modulators (8, 10). Recently, a novel class of a dual pan-PI3K/ mTOR inhibitor, namely NVP-BEZ235, has been generated using a structure-based design approach.
As evidenced by the nM range of IC50 values obtained in our cellular studies, NVP-BEZ235 can effectively block glioma cell proliferation. The anti-proliferative activity of NVP-BEZ235 occurs partly through inhibition of cell cycle progression in glioma cells. Regardless of their PTEN status, glioma cells treated with BEZ235 showed G1 cell cycle arrest with no apparent induction of apoptosis. Thus, apoptosis could not have accounted for the potent effect of NVP-BEZ235 on glioma cell growth. Recently, several lines of evidence have demonstrated that small-molecular mass inhibitors such as imatinib (21), curcumin (22), and temozolomide (23) induce a type II programmed cell death, autophagy, which suggests that autophagy may be an response of tumor cells to the blockage of signals for survival. Indeed, in our study, NVP-BEZ235 caused significant autophagy, as shown by both FACS analysis for acridine-positive cells and the intensity of LC3-II expression, a cleaved product of LC3-I. Further, the extent of autophagy induced among glioma cell lines in our study substantiate their sensitivity to NVP-BEZ235, suggesting that the PI3K and mTOR signal nodes may regulate autophagy in glioma cells. Consistent with this observation, Takeuchi et al. recently showed that the combination of the PI3K inhibitor LY294002 with rapamycin synergistically augmented autophagy in glioma cells (24).
The therapeutic efficacy of NVP-BEZ235 was further substantiated in intracranial animal model in which the median survival was extended 14 days in animals treated with 45 mg/kg NVP-BEZ235, as opposed to survival in animals treated with vehicle control. An important characteristic of NVP-BEZ235 from the standpoint of its use in patients is its apparent safety. Animals that received BEZ235 not only survived longer than control animals but also tolerated the treatment well. As we observed, NVP-BEZ235 caused no abnormal fluctuation in weight throughout the period of the experiment, strongly suggesting the potential therapeutic benefit of this small-molecular mass inhibitor in cancer patients.
One important characteristic of human gliomas is their vascularity, which often results from the amplification of growth factor receptors, in particular EGFR (25), or from elevated levels of angiogenic factors such as VEGF (19). Recently, a clinically approved EGFR inhibitor gefitinib (Iressa) was found to decrease VEGF expression in squamous cell carcinoma cells, and this effect was found to be reversed by exogenous Akt expression (26). This suggests that the growth factor receptor/PI3K/Akt signal axis regulates VEGF expression and hence possibly tumor angiogenesis as well. Inhibition of the mTOR signal node by NVP-BEZ235 can also affect VEGF expression, because the hypoxic-inducible factor HIF-1α is a critical factor controlling the VEGF level and is a target of both mTOR signaling (27) and PTEN (28). Indeed, NVP-BEZ235 treatment led to a significant reduction in VEGF secretion in cells in cultures and greatly diminished the staining of factor VIII in tumor specimens from a xenograft animal model, a factor that has been used to measure angiogenesis (29) and predict tumor recurrence (30).
In summary, our results demonstrate the potential utility of a new class of dual PI3K/mTOR inhibitors for cancer management, including human gliomas. The oral administration of NVP-BEZ235 not only significantly prolonged survival in animals but also was also well tolerated by animals. The statistically significant therapeutic efficacy of NVP-BEZ235 is attributed to its concerted anti-tumor effects, including cell cycle arrest, autophagy induction, and anti-angiogenesis. Based on these findings, clinical use of NVP-BEZ235 in the treatment of glioma patients should be further explored.
Acknowledgement
This study was supported by the following grants: RO1 CA56041 (W.K.A.Y.), Goodwin Foundation, Accelerate Brain Cancer Cure (W.K.A.Y. and DK), and Cancer Center Support Grant (CA16672). We thank Verlene Henry and Jennifer Edge for animal studies and Beth Notzon for editing the manuscript.
References
- 1.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 4.Gallia GL, Rand V, Siu IM, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. 2006;4:709–714. doi: 10.1158/1541-7786.MCR-06-0172. [DOI] [PubMed] [Google Scholar]
- 5.Su JD, Mayo LD, Donner DB, Durden DL. PTEN and phosphatidylinositol 3’-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis: evidence for an effect on the tumor and endothelial compartment. Cancer Res. 2003;63:3585–3592. [PubMed] [Google Scholar]
- 6.Schultz RM, Merriman RL, Andis SL, et al. In vitro and in vivo antitumor activity of the phosphatidylinositol-3-kinase inhibitor, wortmannin. Anticancer Res. 1995;15:1135–1139. [PubMed] [Google Scholar]
- 7.Ihle NT, Williams R, Chow S, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3:763–772. [PubMed] [Google Scholar]
- 8.Fan QW, Knight ZA, Goldenberg DD, et al. A dual PI3kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 10.Rubinstein LV, Shoemaker RH, Paull KD, et al. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 11.Lal S, Lacroix M, Tofilon P, et al. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg. 2000;92:326–333. doi: 10.3171/jns.2000.92.2.0326. [DOI] [PubMed] [Google Scholar]
- 12.Koul D, Shen R, Garyali A, et al. MMAC/PTEN tumor suppressor gene regulates vascular endothelial growth factor-mediated angiogenesis in prostate cancer. Int J Oncol. 2002;21:469–475. [PubMed] [Google Scholar]
- 13.Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol. 2007;78:217–245. doi: 10.1016/S0070-2153(06)78006-1. [DOI] [PubMed] [Google Scholar]
- 14.Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy. 2006;2:85–90. doi: 10.4161/auto.2.2.2463. [DOI] [PubMed] [Google Scholar]
- 15.Kondo Y, Kanzawa T, Sawaya R, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 16.Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daido S, Kanzawa T, Yamamoto A, et al. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 18.Hamada K, Sasaki T, Koni PA, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005;19:2054–2065. doi: 10.1101/gad.1308805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ke LD, Shi YX, Im SA, Chen X, Yung WK. The relevance of cell proliferation, vascular endothelial growth factor, and basic fibroblast growth factor production to angiogenesis and tumorigenicity in human glioma cell lines. Clin Cancer Res. 2000;6:2562–2572. [PubMed] [Google Scholar]
- 20.Howes AL, Chiang GG, Lang ES, et al. The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Mol Cancer Ther. 2007;6:2505–2514. doi: 10.1158/1535-7163.MCT-06-0698. [DOI] [PubMed] [Google Scholar]
- 21.Ertmer A, Huber V, Gilch S, et al. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007;21:936–942. doi: 10.1038/sj.leu.2404606. [DOI] [PubMed] [Google Scholar]
- 22.Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 Signaling Pathways in Curcumin-Induced Autophagy. Autophagy. 2007;3:635–637. doi: 10.4161/auto.4916. [DOI] [PubMed] [Google Scholar]
- 23.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 24.Takeuchi H, Kondo Y, Fujiwara K, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 25.Mishima K, Johns TG, Luwor RB, et al. Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res. 2001;61:5349–5354. [PubMed] [Google Scholar]
- 26.Pore N, Jiang Z, Gupta A, et al. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006;66:3197–3204. doi: 10.1158/0008-5472.CAN-05-3090. [DOI] [PubMed] [Google Scholar]
- 27.Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomez-Manzano C, Fueyo J, Jiang H, et al. Mechanisms underlying PTEN regulation of vascular endothelial growth factor and angiogenesis. Ann Neurol. 2003;53:109–117. doi: 10.1002/ana.10396. [DOI] [PubMed] [Google Scholar]
- 29.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obermair A, Wanner C, Bilgi S, Speiser P, et al. Tumor angiogenesis in stage IB cervical cancer: correlation of microvessel density with survival. Am J Obstet Gynecol. 1998;178:314–319. doi: 10.1016/s0002-9378(98)80018-5. [DOI] [PubMed] [Google Scholar]