

Abstract
IgE-sensitized rat basophilic leukemia (RBL)-2H3 mast cells have been shown to migrate towards antigen. In the present study we tried to identify the mechanism by which antigen causes mast cell migration. Antigen caused migration of RBL-2H3 cells at the concentration ranges of 1000-fold lower than those required for degranulation and the dose response was biphasic. This suggests that mast cells can detect very low concentration gradients of antigen (pg/ml ranges), which initiate migration until they degranulate near the origin of antigen, of which concentration is in the ng/ml ranges. Similar phenomenon was observed in human mast cells (HMCs) derived from CD34+ progenitors. As one mechanism of mast cell migration, we tested the involvement of sphingosine 1-phosphate (S1P). FcεRI-mediated cell migration was dependent on the production of S1P but independent of a S1P receptor or its signaling pathways as determined with S1P receptor antagonist VPC23019 and Gi protein inhibitor pertussis toxin (PTX). This indicated that the site of action of S1P produced by antigen stimulation was intracellular. However, S1P-induced mast cell migration was dependent on S1P receptor activation and inhibited by both VPC23019 and PTX. Cell migration towards antigen or extracellular S1P was dependent on the activation of the phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways, while only migration towards antigen was inhibited by the inhibitors of sphingosine kinase (SK) and phospholipase C (PLC) and intracellular calcium chelator BAPTA. In summary, our data suggest that the high affinity receptor for IgE (FcεRI)-mediated mast cell migration is dependent on the production of S1P but independent of S1P receptors. Cell migration mediated by either FcεRI or S1P receptors involves activation of both PI3K and MAPK.
Keywords: Mast cells, Migration, FcεRI, Sphingosine 1-phosphate, Sphingosine kinase, Signal transduction
1. Introduction
Mast cells are a major cell type that mediates allergic inflammatory diseases such as hay fever, atopic dermatitis and asthma by releasing inflammatory mediators [1]. Mast cells are derived from hematopoietic progenitor cells that home to tissue as committed progenitors [2,3]. Maturation and differentiation of mast cells occurs in tissue in response to local production of stem cell factor (SCF) [4]. Mature mast cells express high affinity receptors for IgE (FcεRI), crosslinking of which leads to tyrosine phosphorylation of receptor subunits by the activation of a Src family tyrosine kinase, Lyn and subsequent activation of Syk [5]. This phosphorylation cascade is followed by the recruitment of adaptor proteins including LAT, SLP-76 and Grb-2, which in turn activate downstream effector molecules such as guanine nucleotide exchange factors, phospholipases and protein and lipid kinases [6]. This cascade of events leads to the release of preformed and newly synthesized inflammatory mediators such as histamine, prostaglandins, leukotrienes and cytokines.
Mast cells have been shown to increase in their numbers in inflammation regions, suggesting that mature mast cells migrate to the inflammatory sites [7-10]. In fact, mast cells are recruited to the inflammatory regions by various chemokines such as MCP-1/CCL2, MIP-1α/CCL3, RANTES/CCL5, eotaxin/CCL11, and SDF-1α/CXCL12 via the corresponding receptors CCR1-5, CXCR1-2, and CXCR4 (reviewed in ref. [4]). In addition to these chemokines, mast cells are attracted by SCF and IgE [11]. IgE-sensitized mast cells have been shown to migrate towards the specific antigen via the mechanism depending on P38 MAPK, Rho-kinase/ROCK [12] and sphingosine kinase [13]. Cross-linking of FcεRI causes a rapid activation of sphingosine kinase, which leads to the production of S1P and Ca2+ mobilization [14-17]. S1P can act intracellularly or extracellularly after secretion. The FcεRI-mediated Ca2+ mobilization from the ER appears to be attributed to intracellular S1P because the blockade of S1P secretion via the ATP binding cassette (ABC) C1 transporter did not affect antigen-induced Ca2+ mobilization [18]. Although an intracellular target for S1P has not been identified yet, extracellular S1P is known to act through its G protein coupled receptors. Five subtypes of S1P receptors have been identified so far and the activation of these receptors leads to various cellular responses such as cell migration [19-23], cell proliferation and survival [24-26], Ca2+ mobilization [27-29] and inhibition of adenylate cyclase [28]. FcεRI-mediated mast cell migration and degranulation were shown to be mediated by the transactivation mechanism, in which S1P produced by crosslinking of FcεRI is secreted and binds S1P1 receptor on the mast cell surface to cause mast cell migration [30].
In the present study, we investigated the mechanism by which sensitized mast cells migrated towards antigen and demonstrated that antigen at very low concentrations could attract mast cells by a mechanism that was distinct from the one used by extracellular S1P. While both antigen and S1P use the PI3K and MAPK pathways to attract mast cells, only antigen requires the increase in cytosolic free Ca2+ to attract sensitized mast cells.
2. Materials and methods
2.1. Reagents
Pertussis toxin (PTX), LY294002, PD98059, U73122, U73343 and BAPTA were obtained from Sigma (St. Louis, MO); VPC23019 from Avanti (Alabaster, AL), sphingosine, N,N-dimethylsphingosine (DMS) and S1P were obtained from Biomol (Plymouth Meeting, PA). DNP-specific IgE (SPE-7) and DNP-HSA were obtained from Sigma (St. Louis, MO). Purified IgE-JK myeloma and polyclonal goat anti-human IgE were gifts from Dr. Robert G. Hamilton at Johns Hopkins University (Baltimore, MD) [31]. Materials for cell culture were obtained from GIBCO-BRL (Gaithersberg, MD).
2.2. Cell culture
RBL-2H3 cells were maintained as described previously [32]. RBL-2H3 cells were maintained as monolayer cultures in Minimum Essential Medium (MEM) supplemented with Earle’s salts, 15% fetal bovine serum (FBS), 2 mM glutamine, penicillin (1000 units/ml) and streptomycin (100 μg/ml). For the production of HMCs, CD34+ cells purified from peripheral blood was obtained from NHLBI PEGT Hematopoietic Cell Processing Core. Cells were thawed according to the instruction given by the NHLBI core. Cells (seeded at 2 × 105/ml) were cultured in Stem Pro-34 media (GIBCO) supplemented with IL-6 and stem cell factor (SCF) (both at 100 ng/ml), 2 mM glutamine, penicillin (1000 units/ml) and streptomycin (100 μg/ml) as previously described [33]. IL-3 (30 ng/ml) was also added during the first week of culture. One-half of the culture media was replaced weekly with fresh medium containing IL-6 and SCF. Cells were used for experiments after 6 weeks of culture.
2.3. Cell migration assay
RBL-2H3 cell migration was assayed by a modification of the Boyden chamber method [34] in microchemotaxis chambers (NeuroProbe, Gaithersburg, MD) containing polycarbonate membrane (NeuroProbe, Gaithersburg, MD) with a pore size of 8.0 μm. The membranes with no coating were placed between the chambers. The lower well of the chamber was filled with indicated concentrations of antigen or S1P in 28 μl MEM supplemented with 15% FBS. RBL-2H3 cells sensitized overnight with 50 ng/ml DNP-specific IgE were trypsinized, resuspended at a concentration of 1 × 106 cells/ml in MEM supplemented with 15% FBS and incubated for 1h for recovery. To investigate the effects of various inhibitors of signaling molecules, cells were preincubated with the indicated inhibitors in MEM supplemented with 15% FBS before placed in the upper well of the chamber (50 μl/well) and incubated at 37°C in 95% air and 5% CO2 for 4 hours. DMSO used to dissolve these inhibitors did not have any effects on FcεRI-mediated migration at 0.1 or 0.25% as indicated by the fact that the number of migrated RBL-2H3 cells stimulated with 0.5 μg/ml IgE in the absence or presence of 0.1 or 0.25% DMSO was 1414 ± 58 (mean ± SEM), 1288 ± 103 and 1353 ± 121, respectively. At the end of the incubation, the filters were removed and all non-migrated cells on the upper side of the filter were scraped off with a wet tissue paper. The migrated cells on the other side of the filter were fixed for 2 min with Fixative solution of HEMA 3 stain set and stained with solution 1 and 2 of HEMA 3 stain set each for 2 min (Fisher Scientific, Kalamazoo, MI). The number of stained cells was counted under Olympus CX21 microscope at a magnification of ×100. Experiments were performed in triplicates and repeated at least three times unless otherwise stated.
Migration of HMCs was assayed as described for RBL-2H3 cells with a slight modification. The membranes were coated with fibronectin (50 μg/ml) and placed between the chambers. The lower well of the chamber was filled with the indicated concentrations of α-IgE or S1P in 27 μl MEM supplemented with 15% FBS. HMCs sensitized overnight with human IgE (50 ng/ml) were collected, resuspended at a concentration of 1.5 × 106 cells/ml in MEM supplemented with 15% FBS and incubated for 1h for recovery. The cells were then placed in the upper well of the chamber (50 μl/well) and incubated at 37°C in 95% air and 5% CO2 for 3 hours. As HMCs migrated, some of them were attached to the bottom side of membrane and other cells fell into the lower wells of the chamber. At the end of the incubation, the filters were removed and processed as described for RBL-2H3 cells before the attached cells were counted. The cells collected in the bottom wells were also counted and included in the total number of cells migrated. Experiments were performed in triplicate and repeated at least three times.
2.4. Western blot analysis
RBL-2H3 cells sensitized overnight with DNP-specific IgE (50 ng/ml) were stimulated with the indicated concentrations of DNP-HSA in PIPES buffer (pH7.2), containing 25 mM PIPES, 110 mM NaCl, 5 mM KCl, 5.6 mM glucose, 0.4 mM MgCl2, 0.1% BSA and 1 mM CaCl2. At the end of the incubation, cells were washed twice with Tris-buffered saline and lysed by addition of ice-cold lysis buffer, containing 0.5% Triton X-100, 50 mM Tris (pH 8.0), 150 mM NaCl, 5 mM EDTA, 5 mM NaF, 1 mM Na3VO4, 10 μg/ml aprotinin and pepstatin A and 5 μg/ml leupeptin. Lysates were left on ice for 20 minutes and centrifuged for 30 minutes at 12,000 × g in a microcentrifuge at 4°C to remove nuclei. Proteins were separated by 4-20% SDS-polyacrylamide gels and transferred to PVDF membranes. Membranes were blocked with 5% non-fat dried milk in T-PBS (0.2% Tween 20 in PBS) and incubated overnight with antibody against phospho-ERK1/2 antibody (Santa Cruz, Santa Cruz, CA) to detect ERK phosphorylation. Subsequently, membranes were washed and incubated for 1 h with secondary antibody conjugated with horse radish peroxidase. Finally, membranes were washed and developed using an enhanced chemiluminescence system. For loading control, membranes were stripped with Restore stripping buffer (Pierce, Rockford, IL) for 6 min at room temperature and reprobed with ERK2 antibody (Santa Cruz, Santa Cruz, CA).
2.5. In vitro sphingosine kinase assay
Cells (5 × 106 cells/sample) sensitized overnight with DNP-specific IgE (50 ng/ml) were stimulated with the indicated concentrations of DNP-HSA in PIPES buffer (pH7.2), containing 25 mM PIPES, 110 mM NaCl, 5 mM KCl, 5.6 mM glucose, 0.4 mM MgCl2, 0.1% BSA and 1 mM CaCl2. At the end of the incubation, cells were lysed by freeze-thawing in 0.1 M phosphate buffer (pH 6.8) containing 10 mM MgCl2, 20% glycerol, 1 mM mercaptoethanol, 1 mM EDTA, 20 mM ZnCl2, 1 mM Na3VO4, 15 mM NaF, 10 μg/ml leupeptin and aprotinin, 1 mM PMSF and 0.5 mM deoxypyridoxine. Cytosolic fractions were prepared by ultracentrifugation at 105,000 × g for 90 min. Sphingosine kinase assay was performed by incubating the supernatant with 5 μM sphingosine, 0.25% Triton X-100, 10 mM MgCl2 and [32P]ATP (10 μCi, 1 mM) in total incubation volume of 50 μl for 15 min at 30 °C. The incubation was quenched by the addition of CHCl3/MeOH/cHCl (100:200:1, v/v) and the subsequent addition of CHCl3/0.1N HCl (1:1). The amount of radioactive S1P generated was determined by collection of the lower phase, silica gel thin layer chromatography using a BtOH/EtOH/CH3COOH/H20 (80:20:10:20) solvent system and autoradiography as previously described [14,35].
2.6. Hexosaminidase assay
Cells sensitized overnight with either DNP-specific IgE (50 ng/ml) or human IgE (50 ng/ml) were stimulated with the indicated concentrations of DNP-HSA or anti-IgE antibody in PIPES buffer (pH7.2), containing 25 mM PIPES, 110 mM NaCl, 5 mM KCl, 5.6 mM glucose, 0.4 mM MgCl2, 0.1% BSA and 1 mM CaCl2, at 37°C for 15 minutes. At the end of the incubation, aliquots (10 μl) of medium and cell lysates (in 500 μl 0.5% Triton X-100) were incubated with 10 μl of 1 mM p-nitrophenyl-N-acetyl-β-D-glucosaminide in 0.1 M sodium citrate buffer at 37°C for 1h. Finally, 250 μl of a 0.1M Na2CO3/0.1 M NaHCO3 buffer was added. Absorbance was read at 410 nm as described previously [36].
3. Results
3.1. RBL-2H3 cells migrate towards antigen at a concentration that is 1000-fold lower than that required for degranulation
It has been previously shown that sensitized mast cells, including murine [11-13,37] and human mast cells [38], migrate toward antigen. We were interested in the role of sphingosine kinase in FcεRI-mediated mast cell migration. As an initial study, we determined antigen dose-dependency of FcεRI-mediated migration. Antigen dose-dependently stimulated chemotaxis with maximum migration observed at 10 pg/ml and the dose response was biphasic. It was interesting to see that the antigen concentrations needed for cell migration were about 1000-fold lower than those needed for degranulation (Fig. 1A). This may indicate that a low concentration gradient of antigen may be detected by the sensitized mast cells leading to migration and as mast cells approach the source of antigen where the concentration gradient is high, they stop migrating and degranulate. Since S1P has been shown to be involved in antigen-induced mast cell migration [13], we tested extracellular S1P for mast cell migration. As expected from the previous reports [13,37], it attracted RBL-2H3 cells in a dose-dependent manner with maximum migration obtained at 10 nM (Fig 1B).
Fig. 1.

FcεRI- and S1P receptor-mediated migrations of RBL-2H3 cells and HMCs. Cells were sensitized overnight with 50 ng/ml DNP-IgE (A) or with 50 ng/ml human-IgE (C) and allowed to migrate toward the indicated concentrations of DNP-HSA (A), S1P (B,D) or anti-IgE (C) for 4 hrs for RBL-2H3 cells and 3 hrs for HMC. Migrated cell were fixed, stained and counted as described in Materials and Methods. FcεR-mediated release of β-hexosaminidase was measured in sensitized mast cells (A,C) at 37°C for 15 minutes as described in Materials and Methods. Values are means ± SEM obtained from at least three separate experiments, carried out in triplicates and asterisks indicate significant differences compared to the control cells at p<0.05 (*), 0.01 (**) and 0.001 (***) as determined by ANOVA and Tukey-Kramer multiple comparisons test.
The rodent system is not always reflected in the human system. Therefore, we were interested in the capability to migrate of HMCs derived from peripheral blood CD34+ cells. Similar to RBL-2H3 cells, sensitized HMCs migrated towards anti-IgE, at a concentration of about 1000-fold lower than that needed for degranulation (Fig. 1C). HMCs migrated towards extracellular S1P at concentrations higher than RBL-2H3 cells did, with maximum migration obtained at 100 nM (Fig. 1D).
3.2. FcεRI-mediated migration is independent of secreted S1P or S1P receptor signaling
It was previously suggested that antigen-induced migration of RBL-2H3 cells was dependent on FcεRI-mediated secretion of S1P [13]. In that study, the concentrations of antigen used to cause secretion of S1P and migration of RBL-2H3 cells were in an ng/ml range. Since the concentration range of antigen that caused migration of RBL-2H3 cells was about 1000-fold lower and the experimental conditions for migration were different in our experiments, we thought that the migration occurring at this very low concentration of antigen might be independent of S1P secretion.
If antigen-induced cell migration is dependent on the secretion of S1P, antigen-induced cell migration should be dependent on the S1P receptor activation. In order to test this hypothesis, we measured antigen- or S1P-mediated migration of RBL-2H3 cells in the presence or absence of VPC23019, a selective S1P receptor antagonist. As shown in Fig. 2A, VPC23019 as high as a 1 μM concentration did not affect antigen-induced cell migration, while completely inhibited extracellular S1P-induced migration of RBL-2H3 cells at all the concentrations tested (1 to 1000 nM), suggesting that antigen-induced mast cell migration was independent of S1P secretion. Since S1P was shown to activate the S1P1 receptor that is coupled to Gαi protein [13], we also tested whether antigen-induced mast cell migration would be dependent on Gαi protein. In agreement with our finding with the S1P receptor antagonist, antigen-induced cell migration was not inhibited by PTX, Gαi inhibitor while S1P-induced migration was completely inhibited, suggesting that antigen causes migration of RBL-2H3 cells via a mechanism that is different from extracellular S1P (Fig. 2B). HMCs were also tested for their dependency on S1P secretion when migration was stimulated via FcεRI. As shown in Figure 2C, FcεRI-mediated migration of HMCs was not inhibited by pretreatment with PTX, while S1P-induced migration was completely inhibited. Data indicated that FcεRI utilized the same mechanism of action in inducing mast cell migration in both RBL-2H3 cells and HMCs.
Fig. 2.

FcεRI-mediated mast cell migration is independent of S1P receptor activation. (A) RBL-2H3 cells sensitized overnight with 50 ng/ml DNP-IgE were preincubated with the indicated concentrations of VPC23019 (VPC) for 30 min and then allowed to migrate toward 0 (Con), 10 pg/ml of DNP-HSA or 10 nM S1P for 4 hrs. Values are mean ± SEM obtained from two separate experiments carried out in triplicates. (B,C) RBL-2H3 cells (B) or HMC (C) sensitized with 50 ng/ml DNP-IgE or 50 ng/ml human IgE, respectively, were preincubated with 100 ng/ml PTX or not for 4 hrs and allowed to migrate toward indicated concentrations of DNP-HSA, anti-IgE or S1P for 4hrs for RBL-2H3 cells and 3 hrs for HMC. Values are means ± SEM obtained from at least three separate experiments carried out in triplicates. The asterisks indicate significant differences compared to the control cells at p<0.05 (*), 0.01 (**) and 0.001 (***) and the pound signs indicate significant differences compared to the respective stimulants alone at p<0.05 (#), 0.01 (##) and 0.001 (###) as determined by ANOVA and Tukey-Kramer multiple comparisons test.
3.3. An increase in cytosolic free Ca2+ concentration is essential for FcεRI-mediated but not for S1P-mediated cell migration in RBL-2H3 cells
In order to identify signaling molecules that are involved in FcεRI- or S1P receptor-mediated mast cell migration, we measured Ag- or S1P-mediated migration of RBL-2H3 cells in the presence or absence of various inhibitors of signaling molecules, such as a PI3K inhibitor, LY294002, a MEK inhibitor, PD98059, a PLC inhibitor, U73122, an intracellular Ca2+ chelator BAPTA-AM and a sphingosine kinase inhibitor DMS (Figure 3). As shown in Fig. 3A, antigen-induced cell migration was inhibited by LY294002, PD98059, U73122, BAPTA-AM and DMS, indicating that antigen-induced chemotaxis was dependent on the activation of PI3K and MAPK as well as Ca2+ mobilization. Similarly, FcεRI-mediated migration of HMCs was inhibited by these inhibitors (data not shown). In contrast to the migration caused by antigen, S1P-induced cell migration was inhibited only by LY294002 and PD98059 but not by the inhibitors of Ca2+ mobilization (Fig. 3B). These data suggested that cell migration induced by both antigen and S1P was regulated by PI3K and MAPK, but only Ag-induced cell migration was dependent on an increase in the cytosolic free Ca2+ concentration.
Fig. 3.

FcεRI-mediated, not S1P receptor-mediated, mast cell migration is dependent on increases in cytosolic free Ca2+ concentration. RBL-2H3 cells were sensitized overnight with 50 ng/ml DNP-IgE and pre-incubated with none (-), 10 μM LY294002 (LY), 25 μM PD98059 (PD), 10 μM U73122 (U73), 20 μM BAPTA (BAP) or 5 μM DMS for 30 min then allowed to migrate toward 0 (Con), or 10 pg/ml DNP-HSA (A) and 0 (Con) or 10 nM S1P (B) for 4hrs. Values are means ± SEM obtained from at least three separate experiments (except for BAPTA in panel A which is from one experiment) carried out in triplicates. The asterisks indicate significant differences compared to the control cells at p<0.001 (***) and the pound signs indicate significant differences compared to the respective stimulants alone at p<0.001 (###) as determined by ANOVA and Tukey-Kramer multiple comparisons test.
Since antigen-induced mast cell migration was shown to be dependent on MAPK and sphingosine kinase, we tested whether these kinases were activated by DNP-HSA at the concentration where migration was activated. DNP-HSA caused activation of these kinases, in a time-dependent manner, at 10 pg/ml an optimum concentration for migration (Fig.4). Maximum phosphorylation of ERK was achieved 2.5 min after antigen stimulation, lasted 10 min and then started to diminish. Unlike ERK phosphorylation, antigen-stimulated activation of sphingosine kinase was slowly increased and until even 60 min after antigen stimulation, sphingosine kinase activity continued to increase. The time course of sphingosine kinase activation by 10 pg/ml antigen was different from that of 100 ng/ml antigen, which was biphasic and maximum activation was reached by 2.5 min [14]. The fact that ERK phosphorylation preceded sphingosine kinase activation may suggest that ERK is upstream of sphingosine kinase.
Fig. 4.

The low concentration of antigen (10 pg/ml) causes a rapid increase in ERK phosphorylation that precedes the activation of sphingosine kinase in RBL-2H3 cells. (A) RBL-2H3 cells sensitized overnight with 50 ng/ml DNP-IgE were stimulated with 10 pg/ml DNP-HSA for the indicated periods of time and total cell lysates were subject to Western blot analysis as described in Materials and Methods. Top panel shows a representative blot stained with anti-p-ERK and anti-ERK antibodies as indicated. Numbers shown above the blot is the ratio of density of p-ERK2 over that of ERK2. The mean values ± SEM obtained from three separate experiments are shown in the bottom panel. (B) For the measurements of sphingosine kinase activity, cells were sensitized overnight with 50 ng/ml DNP-IgE and stimulated with 10 pg/ml DNP-HSA for the indicated times as described in Materials and Methods. Values are mean ± SEM obtained from at least four separate experiments. Values are means ± SEM and ** indicates significant differences at p<0.01 in comparison to the values at 0 time point as determined by one-way analysis of variance (ANOVA) and Tukey-Kramer multiple comparisons test.
3.4. MAPK is upstream of sphingosine kinase in FcεRI-mediated migration of RBL-2H3 cells
Since we observed that sphingosine kinase, PI3K, MAPK, PLC, and Ca2+ mobilization were important for antigen-induced cell migration, we next attempted to determine whether any of these molecules have a linear relationship in the pathway or if they act independently of each other. We measured antigen-induced ERK phosphorylation (Fig. 5A) and sphingosine kinase activation (Fig. 5B) in the presence or absence of various inhibitors. As shown in Fig. 5, ERK phosphorylation by antigen stimulation for 2.5 min was inhibited only by a MEK inhibitor PD98059, whereas sphingosine kinase activity induced by antigen stimulation for 1 hr was inhibited by both PD98059 and DMS a sphingosine kinase inhibitor. Data indicated that as predicted by the differential time course for MAPK and sphingosine kinase, MAPK was upstream of sphingosine kinase and that PI3K acted independently of sphingosine kinase and MAPK or downstream of these two molecules.
Fig. 5.

FcεRI-stimulated sphingosine kinase activity is inhibited by MEK inhibitor PD98059 but MAPK activity is not inhibited by DMS in RBL-2H3 cells. RBL-2H3 cells were sensitized overnight with 50 ng/ml DNP-IgE and stimulated with 10 pg/ml DNP-HSA for 2.5 min to measure ERK activity (A) or for 60 min to measure sphingosine kinase activity (B) in the presence or absence of various inhibitors (100 ng/ml PTX for 4 hrs, 10 μM LY294002 (LY), 25 μM PD98059 (PD), 10 μM U73122 (U73) and 5 μM DMS for 30 min). (A) Top panel shows a representative blot stained with anti-p-ERK and anti-ERK antibodies as indicated. Numbers shown above the blot is the ratio of density of p-ERK2 over that of ERK2. The mean values ± SEM obtained from four separate experiments are shown in the bottom panel. (B) Values are means ± SEM from at least 4 separate experiments. The asterisks indicate significant differences compared to the control cells at p<0.001 (***) and the pound signs indicate significant differences compared to antigen alone at p<0.01 (##) or 0.001 (###) as determined by ANOVA and Tukey-Kramer multiple comparisons test.
3.5. IgE alone was able to attract both RBL-2H3 cells and HMCs probably via the FcεRI
Since some hyper-cytokinergic IgEs have been shown to stimulate mast cell migration [11,39] and we used a hyper-cytokinergic IgE (SPE-7) in this study, we tested the effects of IgE alone (SPE-7 or human IgE) on migration of RBL-2H3 cells and HMCs. As shown in Figure 6, SPE-7 and human IgE were able to cause migration of RBL-2H3 cells (Fig. 6A) and HMCs (Fig. 6B), respectively, in a dose-dependent manner. The hyper-cytokinergic effects of IgE, however, cannot explain the shift of the dose-response curve for FcεRI-mediated mast cell migration because cells were sensitized with IgE overnight and the effects of IgE appears to be subsided by the morning of the experiments as evidence by the fact that migration of RBL-2H3 cells (Fig. 1A) or HMCs (Fig. 1C) treated with IgE overnight with no addition of DNP-HSA or anti-IgE, respectively, was not significantly different from that observed in the cells with no treatment (at 0 μg/ml concentration of IgE in Fig. 6A or C). IgE-induced migration of RBL-2H3 cells were inhibited by the same inhibitors that inhibited antigen-induced migration (Fig. 3B). For example, IgE-mediated migration was not inhibited by PTX, indicating that IgE-mediated migration was independent of Gαi or S1P receptor stimulation. The migration was inhibited by inhibitors of PI3K, MEK, PLC and sphingosine kinase, but not inhibited by U73343, an inactive analog of PLC inhibitor U73122. The data might suggest that IgE causes mast cell migration through FcεRI as previously suggested [39].
Fig. 6.
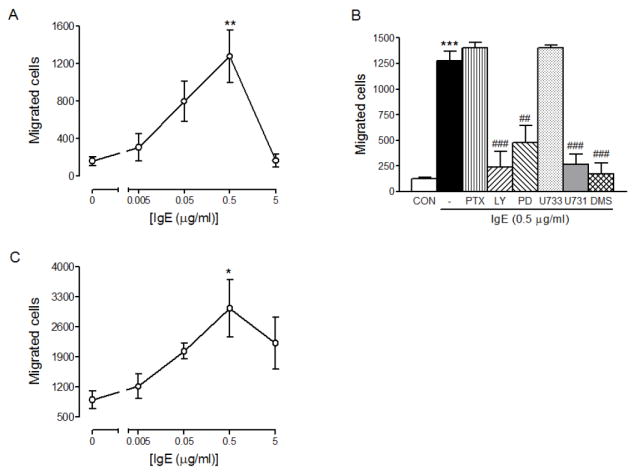
IgE alone can cause mast cell migration. RBL-2H3 cells (A) or HMCs (C) were allowed to migrate toward the indicated concentrations of mouse DNP-IgE (4 hrs) or human IgE (3 hrs), respectively. Migrated cell were fixed, stained and counted as described in Materials and Methods. Values are means ± SEM from at least 3 separate experiments carried out in triplicates. The asterisks indicate significant differences compared to the control cells at p<0.05 (*) or 0.01 (**) as determined by ANOVA and Tukey-Kramer multiple comparisons test. (B) To determine the inhibition of IgE-mediated migration of RBL-2H3 cells by various inhibitors, RBL-2H3 cells were pre-incubated with none (-), 10 μM LY294002 (LY), 25 μM PD98059 (PD), 10 μM U73343 (U733), 10 μM U73122 (U731 and 5 μM DMS for 30 min then allowed to migrate toward 0 (Con) or 10 pg/ml DNP-HSA for 4hrs. Values are means ± SEM from 2 separate experiments carried out in triplicates. The asterisks indicate significant differences compared to the control cells at p<0.001 (***) and the pound signs indicate significant differences compared to antigen alone at p<0.01 (##) or 0.001 (###) as determined by ANOVA and Tukey-Kramer multiple comparisons test.
4. Discussion
In this study, we have demonstrated that antigen (or anti-IgE) caused mast cell migration at 1000-fold lower concentrations than those required for degranulation (Fig. 1). The signaling pathway of FcεRI that leads to mast cell migration and its relationship with S1P receptor signaling are summarized in Figure 7. First of all, mast cell migration induced by the low concentrations of antigen was independent of S1P receptor signaling as demonstrated by that FcεRI-mediated mast cell migration was not inhibited by either a S1P receptor antagonist VPC23019 or an inhibitor of Gαi PTX, while S1P-mediated migration was completely inhibited by these two inhibitors. This is in contrast with the previous finding that FcεRI crosslinking led to the secretion of S1P that activated, in an autocrine manner, S1P receptors to cause mast cell migration [13]. One possible reason for this discrepancy could be due to the differences in the experimental conditions used to determine the migration of RBL-2H3 cells. In their studies, the migration of and secretion of S1P from RBL-2H3 cells were measured with 10 and 100 ng/ml DNP-HSA, respectively [13], while in our study, 10 pg/ml of DNP-HSA was an optimal concentration for migration (Fig. 1). In addition, we included FBS in both upper and lower chambers. FBS keeps cells in the best conditions but it should have not affected cell migration because there were no concentration gradients of FBS between upper and lower chambers. Sphingosine kinase was slowly activated by this concentration of antigen (Fig. 4B) and S1P, which was slowly produced, could have been quickly degraded before being secreted. In this experimental condition, we were able to reveal an intracellular role of S1P in mast cell migration.
Fig. 7.
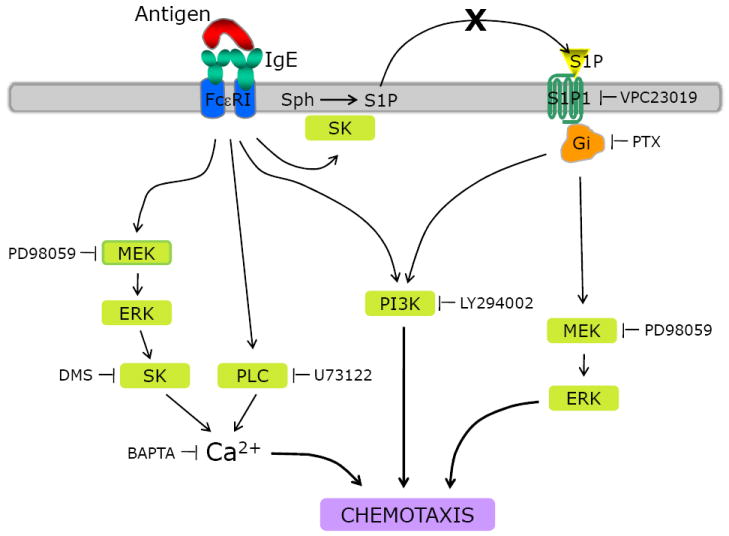
Proposed mechanism of mast cell migration induced by FcεRI or S1P receptor stimulation. Cross-linking of FcεRI induces ERK activation, which leads to the activation of sphingosine kinase to produce S1P. S1P acts intracellularly to increase cytosolic free Ca2+ concentrations. PLC is also activated by FcεRI and increases cytosolic free Ca2+ concentration. The increase in cytosolic Ca2+ is essential for FcεRI-mediated cell migration. Both PI3K and ERK are necessary for FcεRI- and S1P receptor-mediated migration but Ca2+ signaling is not required for S1P receptor-mediated migration. SK, sphingosine kinase.
FcεRI-mediated migration of mast cells appears to require the activation of MAPK, PI3K, sphingosine kinase, PLC and increases in cytosolic free Ca2+ concentrations (Figs. 3, 7). It is interesting to note that the inhibition of one signaling molecule led to almost complete inhibition of migration suggesting that all the signaling molecules tested are involved and essential for mast cell migration. However, not all of these molecules appear to be in the linear relationship. For example, although PI3K was previously suggested to be upstream of sphingosine kinase [40], FcεRI-mediated sphingosine kinase activity stimulated by a very low concentration of DNP-HSA (10 pg/ml) was not inhibited by LY294002, suggesting that PI3K and sphingosine kinase may act independently of each other. It is possible that small aggregates of FcεRI formed by low concentrations of antigen may cause the activation of a different array of signaling molecules. In fact, preferential signaling has been previously reported to occur by weak stimulation of FcεRI, although two concentrations of antigen were only 10-fold different (10 ng/ml vs. 100 ng/ml) [41,42]. In our experiments, we observed 1000-fold differences in the optimal concentrations of antigen for degranulation (10 ng/ml) and migration (10 pg/ml). Therefore, it is easier to assume that preferential signaling pathways could have been activated by the two concentrations of antigen.
In agreement with previous findings that ERK phosphorylates and activates sphingosine kinase 1 and 2 [43,44], FcεRI-mediated activation of sphingosine kinase is inhibited by a MEK inhibitor PD98059 suggesting that sphingosine kinase activation is dependent on the activation of ERK (Figs. 5, 7). Sphingosine kinase and PLC that increase cytosolic free Ca2+ concentrations appear to be necessary for FcεRI-mediated migration of mast cells, suggesting that the increase in cytosolic free Ca2+ concentration is absolutely required for the migration. This is also supported by the inhibition of migration by BAPTA, an intracellular Ca2+ chelator (Figs. 3, 7). However, if an increase in cytosolic free Ca2+ concentrations is sufficient to cause migration, a question arises as to why both of the enzymes are required. It is possible that, as demonstrated in our previous report where both S1P and IP3 are needed for FcεRI-mediated Ca2+ mobilization from the ER [32], the activation of either one of the enzymes is not sufficient to cause Ca2+ mobilization needed for migration. Another possibility is that Ca2+ influx is a major mechanism which increases total cytosolic free Ca2+ concentrations and these enzymes may regulate Ca2+ influx through a mechanism that is independent from Ca2+ mobilization from the ER. In fact, sphingosine kinase 2 was suggested to regulate Ca2+ influx without affecting Ca2+ mobilization from the ER [45] and a role of IP3 or phospholipase Cγ has been demonstrated to cause Ca2+ influx without Ca2+ release from the ER [46,47]. However, we cannot exclude a possibility that these pharmacological inhibitors such as DMS and U73122 may have some additional effects other than inhibition of these enzymes.
To summarize, mast cell migration induced by a very low concentration of antigen requires the activation of ERK and PI3K and increases in cytosolic free Ca2+ concentrations. Sphingosine kinase activation is mediated by ERK and independent of PI3K. In addition, S1P produced by FcεRI-activated sphingosine kinase appears to act intracellularly because FcεRI-migration was not inhibited by inhibitors of S1P receptors or Gαi. However, unlike FcεRI, S1P did not require increases in cytosolic free Ca2+ concentrations, while it did need activation of both PI3K and ERK to cause mast cell migration (Figs. 3, 7). Taken together, the data propose a major chemokine role of antigen and suggest that antigen may act as an initiating chemokine in the beginning of allergic reaction before other chemokines are released to recruit other inflammatory cells.
Acknowledgments
We would like to thank Ms. Janet Dorer for proofreading the manuscript.
Abbreviations
- IgE
Immunoglobulin
- FcεRI
high affinity receptor for IgE
- RBL-2H3
rat basophilic leukemia-2H3
- HMC
human mast cell
- DNP-HSA
dinitrophenyl-human serum albumin
- PI3K
phosphatidylinositol 3-kinase
- PLC
Phospholipase C
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- PTX
pertussis toxin
- S1P
sphingosine 1-phosphate
Footnotes
This work was supported by NIH Grant R01 HL68879.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wedemeyer J, Tsai M, Galli SJ. Curr Opin Immunol. 2000;12:624. doi: 10.1016/s0952-7915(00)00154-0. [DOI] [PubMed] [Google Scholar]
- 2.Austen KF, Boyce JA. Leuk Res. 2001;25:511. doi: 10.1016/s0145-2126(01)00030-3. [DOI] [PubMed] [Google Scholar]
- 3.Krishnaswamy G, Ajitawi O, Chi DS. Methods Mol Biol. 2006;315:13–34. doi: 10.1385/1-59259-967-2:013. [DOI] [PubMed] [Google Scholar]
- 4.Okayama Y, Kawakami T. Immunol Res. 2006;34:97. doi: 10.1385/IR:34:2:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner H, Kinet JP. Nature. 1999;402:B24–B30. doi: 10.1038/35037021. [DOI] [PubMed] [Google Scholar]
- 6.Rivera J. Curr Opin Immunol. 2002;14:688. doi: 10.1016/s0952-7915(02)00396-5. [DOI] [PubMed] [Google Scholar]
- 7.Huang B, Lei Z, Zhang GM, Li D, Song C, Li B, Liu Y, Yuan Y, Unkeless J, Xiong H, Feng ZH. Blood. 2008;112:1269. doi: 10.1182/blood-2008-03-147033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofman V, Lassalle S, Selva E, Kalem K, Steff A, Hebuterne X, Sicard D, Auberger P, Hofman P. J Clin Pathol. 2007;60:600. doi: 10.1136/jcp.2006.040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan J, Li Y, Qiu H, Lu G, Wu Z, Lin J, Zhang W. Int Ophthalmol. 2007;27:235. doi: 10.1007/s10792-007-9061-x. [DOI] [PubMed] [Google Scholar]
- 10.Esposito I, Friess H, Kappeler A, Shrikhande S, Kleeff J, Ramesh H, Zimmermann A, Buchler MW. Hum Pathol. 2001;32:1174. doi: 10.1053/hupa.2001.28947. [DOI] [PubMed] [Google Scholar]
- 11.Kitaura J, Kinoshita T, Matsumoto M, Chung S, Kawakami Y, Leitges M, Wu D, Lowell CA, Kawakami T. Blood. 2005;105:3222. doi: 10.1182/blood-2004-11-4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishizuka T, Okajima F, Ishiwara M, Iizuka K, Ichimonji I, Kawata T, Tsukagoshi H, Dobashi K, Nakazawa T, Mori M. J Immunol. 2001;167:2298. doi: 10.4049/jimmunol.167.4.2298. [DOI] [PubMed] [Google Scholar]
- 13.Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. J Exp Med. 2004;199:959. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi OH, Kim JH, Kinet JP. Nature. 1996;380:634. doi: 10.1038/380634a0. [DOI] [PubMed] [Google Scholar]
- 15.Urtz N, Olivera A, Bofill-Cardona E, Csonga R, Billich A, Mechtcheriakova D, Bornancin F, Woisetschlager M, Rivera J, Baumruker T. Mol Cell Biol. 2004;24:8765. doi: 10.1128/MCB.24.19.8765-8777.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melendez AJ, Khaw AK. J Biol Chem. 2002;277:17255. doi: 10.1074/jbc.M110944200. [DOI] [PubMed] [Google Scholar]
- 17.Ryu SD, Lee HS, Suk HY, Park CS, Choi OH. Cell Calcium. 2009;45:99. doi: 10.1016/j.ceca.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. PNAS. 2006;103:16394. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sadahira Y, Ruan F, Hakomori S, Igarashi Y. Proc Natl Acad Sci U S A. 1992;89:9686. doi: 10.1073/pnas.89.20.9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bornfeldt KE, Graves LM, Raines EW, Igarashi Y, Wayman G, Yamamura S, Yatomi Y, Sidhu JS, Krebs EG, Hakomori S. J Cell Biol. 1995;130:193. doi: 10.1083/jcb.130.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. The Journal of Experimental Medicine. 2004;199:959. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yokoo E, Yatomi Y, Takafuta T, Osada M, Okamoto Y, Ozaki Y. J Biochem (Tokyo) 2004;135:673. doi: 10.1093/jb/mvh081. [DOI] [PubMed] [Google Scholar]
- 23.Kon J, Sato K, Watanabe T, Tomura H, Kuwabara A, Kimura T, Tamama K, Ishizuka T, Murata N, Kanda T, Kobayashi I, Ohta H, Ui M, Okajima F. J Biol Chem. 1999;274:23940. doi: 10.1074/jbc.274.34.23940. [DOI] [PubMed] [Google Scholar]
- 24.Hla T, Lee MJ, Ancellin N, Paik JH, Kluk MJ. Science. 2001;294:1875. doi: 10.1126/science.1065323. [DOI] [PubMed] [Google Scholar]
- 25.Goodemote KA, Mattie ME, Berger A, Spiegel S. J Biol Chem. 1995;270:10272. doi: 10.1074/jbc.270.17.10272. [DOI] [PubMed] [Google Scholar]
- 26.Cuvillier O, Rosenthal DS, Smulson ME, Spiegel S. J Biol Chem. 1998;273:2910. doi: 10.1074/jbc.273.5.2910. [DOI] [PubMed] [Google Scholar]
- 27.An S, Bleu T, Zheng Y. Mol Pharmacol. 1999;55:787. [PubMed] [Google Scholar]
- 28.Ishii I, Ye X, Friedman B, Kawamura S, Contos JJ, Kingsbury MA, Yang AH, Zhang G, Brown JH, Chun J. J Biol Chem. 2002;277:25152. doi: 10.1074/jbc.M200137200. [DOI] [PubMed] [Google Scholar]
- 29.Sato K, Kon J, Tomura H, Osada M, Murata N, Kuwabara A, Watanabe T, Ohta H, Ui M, Okajima F. FEBS Lett. 1999;443:25. doi: 10.1016/s0014-5793(98)01676-7. [DOI] [PubMed] [Google Scholar]
- 30.Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. The Journal of Experimental Medicine. 2004;199:959. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton RG, Adkinson NF., Jr . Measurement of total serum immunoglobulin E and allergen-specific immunoglobulin E antibody. In: Rose H, de Macario E, Fahey J, Friedman H, Penn G, editors. Manual Clinical Immunology. American Society for Microbiology; Washington, DC: 1992. pp. 689–701. [Google Scholar]
- 32.Lee HS, Park CS, Lee YM, Suk HY, Clemons TCM, Choi OH. Cell Calcium. 2005;38:581. doi: 10.1016/j.ceca.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Kirshenbaum AS, Metcalfe DD. Methods Mol Biol. 2005;315:105. doi: 10.1385/1-59259-967-2:105. [DOI] [PubMed] [Google Scholar]
- 34.Wuyts A, Menten P, Van Osselaer N, Van Damme J. Methods Mol Biol. 2004;249:153. doi: 10.1385/1-59259-667-3:153. [DOI] [PubMed] [Google Scholar]
- 35.Olivera A, Rosenthal J, Spiegel S. Anal Biochem. 1994;223:306. doi: 10.1006/abio.1994.1589. [DOI] [PubMed] [Google Scholar]
- 36.Jones SV, Choi OH, Beaven MA. FEBS Lett. 1991;289:47. doi: 10.1016/0014-5793(91)80905-i. [DOI] [PubMed] [Google Scholar]
- 37.Jolly PS, Bektas M, Watterson KR, Sankala H, Payne SG, Milstien S, Spiegel S. Blood. 2005;105:4736. doi: 10.1182/blood-2004-12-4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oskeritzian CA, Alvarez SE, Hait NC, Price MM, Milstien S, Spiegel S. Blood. 2008;111:4193. doi: 10.1182/blood-2007-09-115451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitaura J, Song J, Tsai M, Asai K, Maeda-Yamamoto M, Mocsai A, Kawakami Y, Liu FT, Lowell CA, Barisas BG, Galli SJ, Kawakami T. PNAS. 2003;100:12911. doi: 10.1073/pnas.1735525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olivera A, Urtz N, Mizugishi K, Yamashita Y, Gilfillan AM, Furumoto Y, Gu H, Proia RL, Baumruker T, Rivera J. J Biol Chem. 2006;281:2515. doi: 10.1074/jbc.M508931200. [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez-Espinosa C, Odom S, Olivera A, Hobson JP, Martinez ME, Oliveira-Dos-Santos A, Barra L, Spiegel S, Penninger JM, Rivera J. J Exp Med. 2003;197:1453. doi: 10.1084/jem.20021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamasaki S, Ishikawa E, Kohno M, Saito T. Blood. 2004;103:3093. doi: 10.1182/blood-2003-08-2944. [DOI] [PubMed] [Google Scholar]
- 43.Pitson SM, Moretti PAB, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. EMBO J. 2003;22:5491. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S. J Biol Chem. 2007;282:12058. doi: 10.1074/jbc.M609559200. [DOI] [PubMed] [Google Scholar]
- 45.Olivera A, Mizugishi K, Tikhonova A, Ciaccia L, Odom S, Proia RL, Rivera J. Immunity. 2007;26:287. doi: 10.1016/j.immuni.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 46.van Rossum DB, Patterson RL, Kiselyov K, Boehning D, Barrow RK, Gill DL, Snyder SH. Proc Natl Acad Sci U S A. 2004;101:2323. doi: 10.1073/pnas.0308565100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patterson RL, van Rossum DB, Ford DL, Hurt KJ, Bae SS, Suh PG, Kurosaki T, Snyder SH, Gill DL. Cell. 2002;111:529. doi: 10.1016/s0092-8674(02)01045-0. [DOI] [PubMed] [Google Scholar]