

Abstract
Neutrophils are considered crucial effector cells in the pathophysiology of organ ischemia/reperfusion injury (IRI). Although neutrophil elastase (NE) accounts for a substantial portion of the neutrophil activity, the function of NE in liver IRI remains unclear. This study focuses on the role of NE in the mechanism of liver IRI. Partial warm ischemia was produced in the left and middle hepatic lobes of C57BL/6 mice for 90 min, followed by 6-24 h of reperfusion. Mice were treated with NE inhibitor (NEI; 2 mg/kg p.o.) at 60 min prior to the ischemia insult. NEI treatment significantly reduced serum ALT levels, as compared with controls. Histological examination of liver sections has revealed that unlike in controls, NEI treatment ameliorated hepatocellular damage and decreased local neutrophil infiltration, as assessed by MPO assay, naphtol AS-D chloroacetate esterase stains and immunohistochemistry (anti-Ly-6G). The expression of pro-inflammatory cytokines (tumor necrosis factor [TNF]-α, IL-6), and chemokines (CXCL-1, CXCL-2, CXCL-10) was significantly reduced in the NEI treatment group, along with diminished apoptosis by TUNEL staining and caspase-3 activity. In addition, TLR4 expression was diminished in NEI pretreated livers, which implies putative role of NE in the TLR4 signal transduction pathway. Thus, targeting NE represents a useful approach to prevent liver IRI, and hence to expand the organ donor pool and improve the overall success of liver transplantation.
Keywords: Neutrophil Elastase, Ischemia and Reperfusion Injury, Toll-like Receptor-4
Introduction
The ischemia/reperfusion injury (IRI), an exogenous Ag-independent inflammatory event remains an important problem in clinical transplantation. Indeed, severe damage at the time of organ retrieval, preservation, and reperfusion often leads to primary graft nonfunction and may adversely affect the development of both acute and chronic rejection (1). The mechanisms underlying liver IRI are complex, but are known to involve leukocyte accumulation and activation (neutrophils, Kupffer cells, and T cells), leading to the formation of reactive oxygen species (ROS), secretion of pro-inflammatory cytokines/chemokines, complement activation, and vascular cell adhesion molecule activation (2, 3). Recent studies suggest that the sentinel Toll-like receptor (TLR) system may trigger organ IRI (4-6). We have documented the essential role of TLR4 activation and endogenous TLR4 ligands in IR-mediated local inflammation leading to the hepatocellular damage (4, 7).
The neutrophil activation has been long considered the major effector mechanism in liver IRI (8-10). The rolling of leukocytes is an important prerequisite for adhesion and migration into tissues, and a two-step paradigm for leukocyte recruitment has been established (11). In addition to adhesion molecules, leukocyte proteases have also been implicated in the process of leukocyte migration through the vessel wall, due to their ability to disrupt endothelial cell junctional complexes (12) and degrade key components of the basement membrane. However, the interactions between leukocyte proteases and the liver inflammation cascade remain unknown.
Serine proteinases produced by polymorphonuclear neutrophils mediate tissue injury at the inflammatory sites. Neutrophil elastase (NE) is a 29-kDa glycoprotein chymotrypsin-like serine protease stored in azurophil granules in its active form until it is released following neutrophil exposure to the inflammatory stimuli. Once released, NE is potentially fully active because it functions optimally in a neutral environment (13, 14). As a result, excessive release of NE degrades elastin, collagens, laminins, and other extracellular matrix components of the endothelium, thereby leading to subsequent organ damage through endothelial cell injury (15, 16).
The present study was designed to examine the role of NE in a mouse liver warm IRI model. We found that treatment with NE inhibitor (NEI) not only attenuated otherwise fulminant liver damage and inflammatory cell recruitment resulting from IRI, but also downregulated the innate TLR4 signaling. These results suggest that targeting NE proteases represents a useful approach to prevent liver IRI in transplant recipients, and hence should expand the organ donor pool and improve the overall success of liver transplantation.
Materials and Methods
Animals
Male C57BL/6 mice (8-10 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were housed in the University of California Los Angeles animal facility under specific pathogen-free conditions. All animals received human care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of sciences and published by the National Institute of Health (NIH publication 86-23, revised 1985).
Liver IRI Model
We used an established mouse model of partial warm hepatic IRI (4, 17). Briefly, mice were anesthetized with isoflurane, injected with heparin (100 U/kg), and an atraumatic clip was used to interrupt the artery/portal venous blood supply to the left and middle liver lobes. After 90 min of partial warm ischemia, the clamp was removed, initiating hepatic reperfusion. To investigate the role of NE, we used NEI (Sigma; GW311616A). Previous studies have shown dose-dependent effects of orally given GW311616A in dogs. At 0.22 mg/kg, >50% inhibition of elastase activity was achieved at 6h, with activity returning towards normal relatively quickly. A single dose of 2 mg/kg rapidly abolished circulating enzyme activity, with >90% inhibition maintained for 4 days (18). Mice were orally treated with NEI (2mg/kg, p.o.) at 60 min prior to the ischemia insult, and then sacrificed at 6 or 24 h after reperfusion for serum and liver sampling. Serum alanine aminotransferase (sALT) levels, an indicator of hepatocellular injury, were measured in peripheral blood with an autoanalyzer (ANTECH Diagnostics). Liver specimens were fixed in 10% buffered formalin solution and embedded in paraffin. Liver paraffin sections (5-μm thick) were stained with hematoxylin and eosin, and then analyzed blindly. Sham controls underwent the same procedure, but without vascular occlusion.
Serum NE Activity
Serum NE activity was determined using N-methoxysuccinyl-Ala-Ala -Pro-Val-p-nitroanilide, a highly specific synthetic substrate for NE according to the method of Yoshimura et al. (19, 20). Briefly, serum sample was incubated with 0.1 M Tris–HCl buffer (pH 8.0) containing 0.5 M NaCl and 1 mM substrate at 37°C for 24 h and the amount of p-nitroanilide (pNA) liberated was measured spectrophotometrically at 405 nm and was considered as NE activity.
Liver Neutrophil Infiltration
Myeloperoxidase (MPO) assay
The activity of myeloperoxidase (MPO), an enzyme specific for polymorphonuclear neutrophils, was used as an index of hepatic neutrophil accumulation (21). Briefly, the frozen tissue was thawed and placed in iced 0.5% hexadecyltrimethyl- ammonium (Sigma-Aldrich) and 50 mM potassium phosphate buffer solution (Sigma-Aldrich, pH = 5.0). Each sample was homogenized and centrifuged at 15,000 rpm for 15 min at 4°C. Supernatants were then mixed with hydrogen peroxide-sodium acetate and tetramethyl- benzidine solutions (Sigma-Aldrich). The change in absorbance was measured by spectrophotometry at 655 nm in 1 min. One unit of MPO activity was defined as the quantity of enzyme degrading 1 μM peroxide/min at 25°C/g of tissue.
Naphtol AS-D chloroacetate esterase stain
The accumulation of activated neutrophils in 5-μm cryostat liver sections was assessed by staining for chloroacetate esterase, a neutrophil specific marker (Naphthol ASD-chloroacetate esterase kit, Sigma-Aldrich). Polymorphonuclear cells, identified by staining and morphology, were counted in 10 HPF/section under light microscopy (×400), and numbers of cells/HPF (mean±SEM) are shown.
Immunohistochemistry
Livers were snap frozen, and cryostat sections (5 μm) were fixed in acetone. Endogenous peroxidase activity was inhibited with 0.3% hydrogen peroxidase. Sections were then blocked with 10% normal goat serum. Primary rat mAb against mouse polymorphonuclear leukocytes (Ly-6G, 1A8; BD Pharmingen) was diluted (1/250 in 3% normal goat serum), and 100 μL was added to each section. The primary Ab was incubated for 60 min at room temperature. The secondary Ab, a biotinylated goat anti-rat immunoglobulin G (Vector; diluted 1:200), was incubated for 40 min at room temperature. Sections were incubated with immunoperoxidase (ABC Kit, Vector), washed, and developed with a 3,3′-diaminobenzidine kit (Vector). Slides were counterstained with hematoxylin, rinsed with ammonia, and tap water. Negative control was prepared by omission of primary Ab. Polymorphonuclear cells, identified by staining and morphology, were counted in 10 HPF/section under light microscopy (×400), and numbers of cells/HPF (mean±SEM) are shown.
Apoptosis assay
Apoptosis in 5-μm cryostat liver sections was detected by the TUNEL method using the In Situ Cell Death Detection Kit (Roche) according to the manufacturer's protocol. Negative control was prepared by omission of terminal transferase. Positive controls were generated by treatment with DNase I (1μg/ml for 10 min). The peroxidase activity was visualized with diaminobenzidine substrate, yielding a brown oxidation product; methyl green was used for counterstaining. TUNEL-positive cells were counted in 10 HPF/section under light microscopy (×400), and numbers of cells/HPF (mean±SEM) are shown.
Caspase-3 activity
Caspase-3 activity was determined in liver samples using the ApoAlert caspase-3 colorimetric assay kit (Clontech) following the manufacturer's instructions. Briefly, the tissue was homogenized on ice and centrifuged at 15,000g for 10 min (4°C). One hundred μg of total protein from the supernatant was used to measure caspase-3 activity (22). OD measurements at 405 nm were performed using a microplate reader (BioTeK Instruments). Caspase activity was expressed in units with 1 U being the amount of enzyme activity liberating 1 pmol of p-nitroanilide/min (23).
RNA Extraction and Reverse Transcriptase PCR
Total RNA was extracted from the liver using the TRIzol reagent (Life Technologies). Reverse transcription was performed using 5 μg of total RNA in a First-Strand cDNA Synthesis Kit (Fermentas). The cDNA product was amplified by PCR using primers specific for mouse cytokines and β-actin. Primers used in PCR were as follows: TNF-α sense (5′-AGC CCA CGT AGC AAA CCA CCA A-3′) and TNF-α antisense (5′-ACA CCC ATT CCC TTC ACA GAG CAA T-3′); IL-6 sense (5′-CAT CCA GTT GCC TTC TTG GGA-3′) and IL-6 antisense (5′-CAT TGG GAA ATT GGG GTA GGA AG-3′); CXCL-1 sense (5′-TGA GCT GCG CTG TCA GTG CCT-3′) and CXCL-1 antisense (5′-AGA AGC CAG CGT TCA CCA GA-3′); CXCL-2 sense (5′-GCT GGC CAC CAA CCA CCA GG-3′) and CXCL-2 antisense (5′-AGC GAG GCA CAT CAG GTA CG-3′); CXCL-10 sense (5′-ACC ATG AAC CCA AGT GCT GCC GTC-3′) and CXCL-10 antisense (5′-GCT TCA CTC CAG TTA AGG AGC CCT-3′); TLR-4 sense (5′-CAG CTT CAA TGG TGC CAT CA-3′) and TLR-4 antisense (5′-CTG CAA TCA AGA GTG CTG AG-3′) and β-actin sense (5′-GTG GGG CGC CCC AGG CAC CA-3′) and β-actin antisense (5′-CTC CTT AAT GTC ACG CAC GAT TTC-3′). PCR conditions for each primer couple were as follows: TNF-α, 94°C for 45 s, 55°C for 45 s, and 72°C for 60 s during 33 cycles; IL-6, 94°C for 45 s, 55°C for 45 s, and 72°C for 60 s during 33 cycles; CXCL-1 and CXCL-2, 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s during 35 cycles; CXCL-10, 95°C for 60 s, 63°C for 50 s, and 72°C for 50 s during 30 cycles; TLR-4, 94°C for 60 s, 56°C for 60 s, and 72°C for 60 s during 34 cycles; and β-actin, 95°C for 30 s, 57°C for 30 s, and 72°C for 60 s during 30 cycles. PCR products were analyzed in ethidium bromide-stained 1% agarose gel and scanned using Kodak Digital Science 1D Analysis software (version 2.0). To compare relative levels of each gene, all samples were normalized against the β-actin template cDNA ratio.
Statistical Analysis
All data are expressed as means ± SEM. Differences between experimental groups were analyzed using an unpaired 2-tailed Student t-test. All differences were considered statistically significant at the P value of <0.05.
Results
NE Activity Increases in Liver IRI
First, we measured serum NE activity in mice that underwent 90 min of warm ischemia followed by 24 h of reperfusion. As shown in Figure 1a, NE activity increased rapidly after reperfusion in IR-induced group, compared to sham-operated group without vascular occlusion. It peaked at 3 h (P<0.01), and then declined by 24 h to almost baseline.
Figure 1.
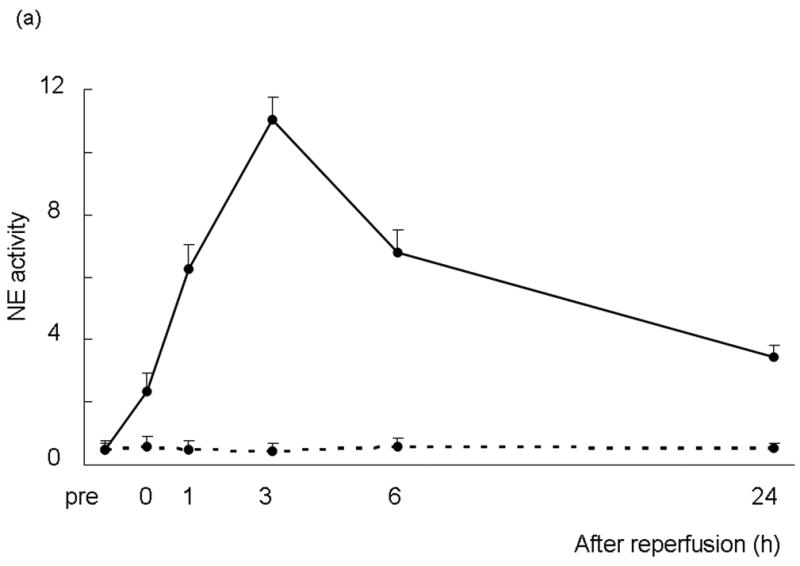
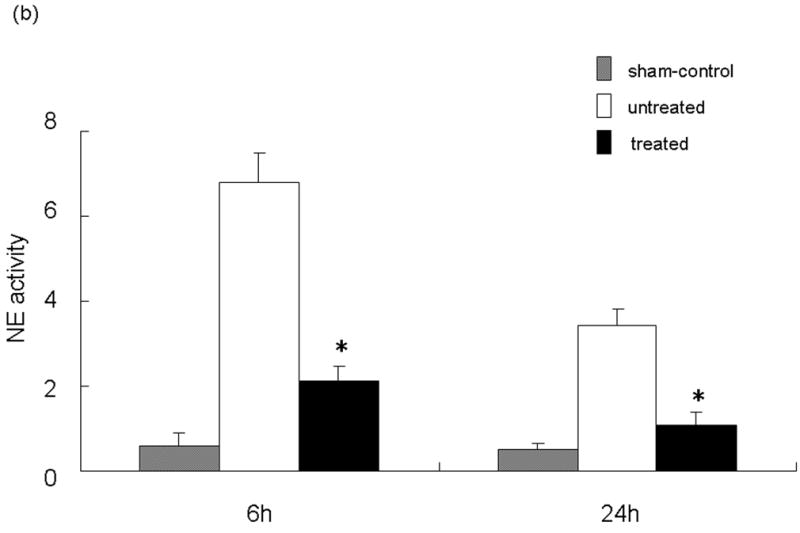
(a) NE activity in mouse serum at 0 - 24 h of liver reperfusion following 90 min of local warm ischemia. Note: NE activity increased rapidly after reperfusion in IR group (n=6, solid line), as compared with sham-operated group (n=3, dotted line). It peaked at 3 h, and then declined by 24 h of reperfusion. (b) NE activity after treatment with NE inhibitor (GW311616A; 2 mg/kg p.o.) at 60 min prior to the ischemia insult. Note: Treatment inhibited NE activity at both 6 and 24 h of reperfusion (*P<0.01; n=4-6/group). Means and SE are shown.
Inhibition of NE Activity Ameliorates Liver IRI
We analyzed the NE activity and hepatocellular function in our model. As shown in Figure 1b, NEI treatment significantly inhibited NE activity at both 6 and 24 h after reperfusion (p<0.01). In parallel, the inhibition of NE prevented IR-induced hepatocellular damage. The serum ALT levels were significantly suppressed at both 6 and 24 h after reperfusion in the treated group, as compared with controls ([6 h] 33650 ± 1896 vs 13510 ± 3404 and [24 h] 14483 ± 1985 vs 4810 ± 531; p<0.01; Figure 2a). These data correlated well with the histological criteria of the hepatocellular damage. Hence, untreated livers revealed severe lobular edema, congestion, ballooning and hepatocelluar necrosis. In contrast, treated livers showed good preservation of architecture and histological detail (Figure 2b).
Figure 2.
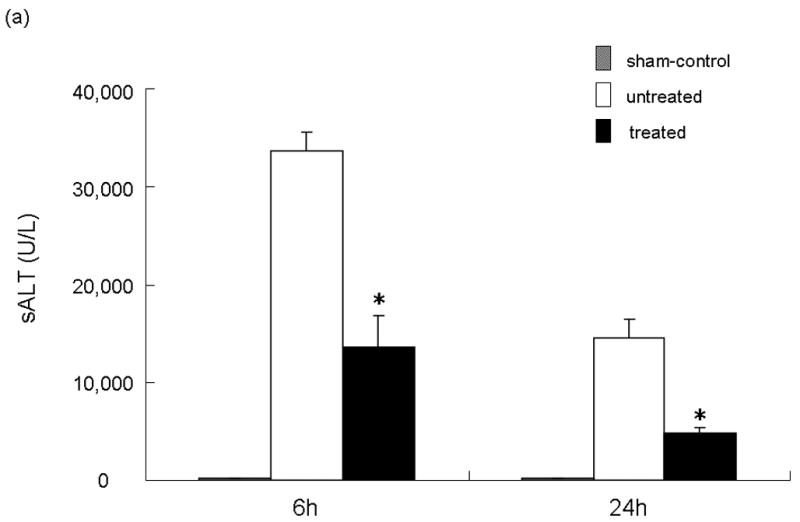
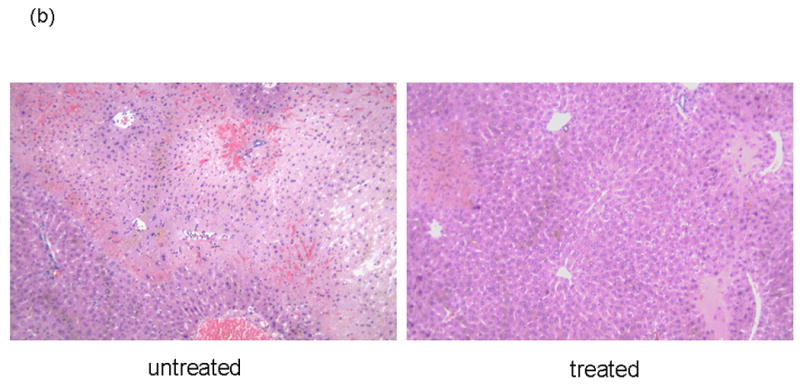
NEI treatment ameliorates liver IRI. (a) Hepatocellular function. Note: Serum ALT levels after liver warm ischemia (90 minutes) followed by reperfusion (6 and 24 h) were lower in NEI treatment group, as compared with untreated controls (*P<0.01; n=4-6/group). Means and SE are shown. (b) Representative liver histology (H&E staining; magnification ×100) of ischemic (90 min) liver lobes harvested after 6 h or reperfusion (n=4/group).
NEI Treatment Suppresses Neutrophil Infiltration
We performed the local MPO assay in livers at 6 h of reperfusion after 90 min of warm ischemia, as an index of neutrophil infiltration (Figure 3a). The MPO activity (U/g) was significantly suppressed in the treated group, as compared with controls (14.10 ± 3.12 vs 4.03 ± 0.49; p<0.05). The MPO activity is correlated with the number of activated neutrophils assessed by the naphtol AS-D chloroacetate esterase stains (24). Indeed, activated neutrophil accumulation in treated livers at 6 h was significantly decreased, as compared with controls (Figure 3b; 15.66 ± 2.96 vs 5.66 ± 2.33; p< 0.05). These results were correlated with the number of Ly-6G positive cells, a marker for neutrophil infiltration (Figure 3c; 34.56 ± 3.09 vs 15.47 ± 5.07; p< 0.05).
Figure 3.
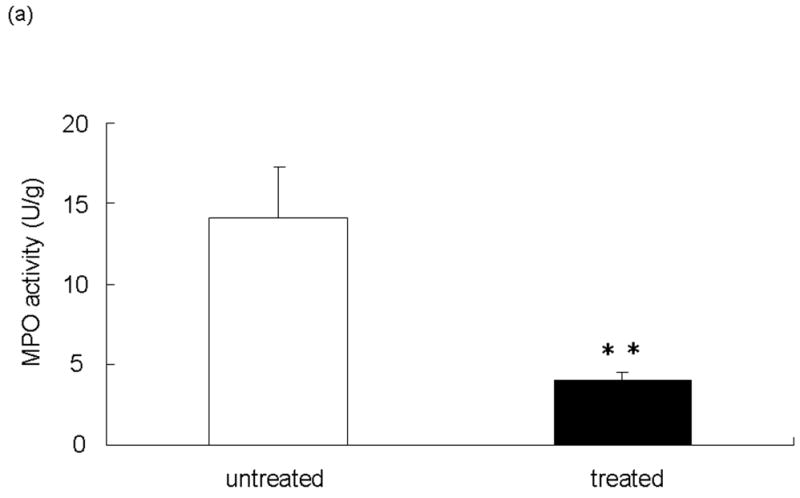
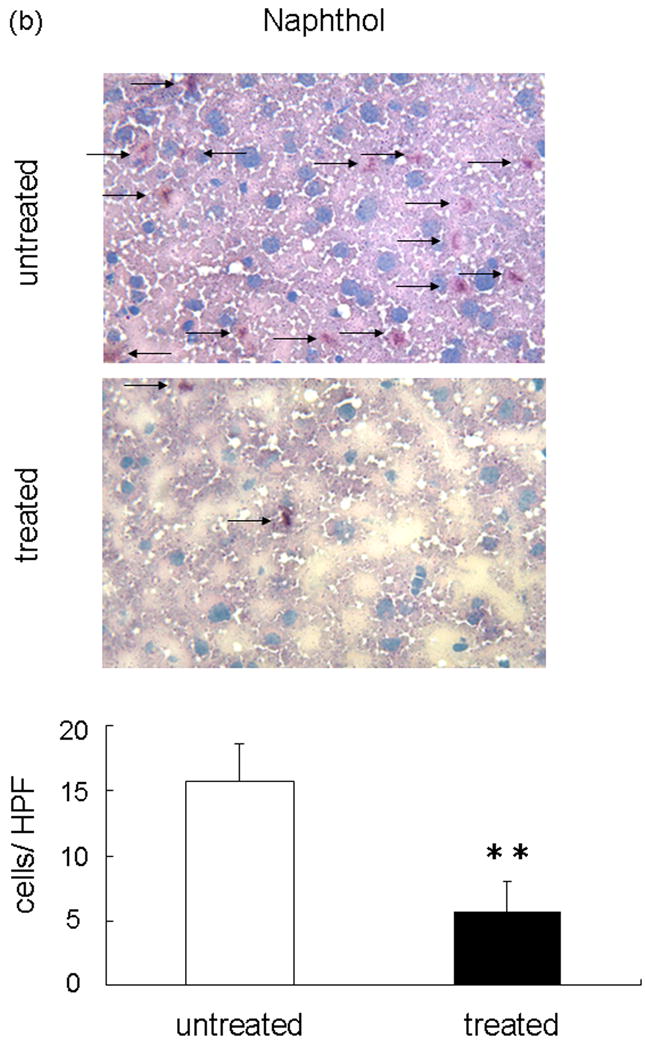
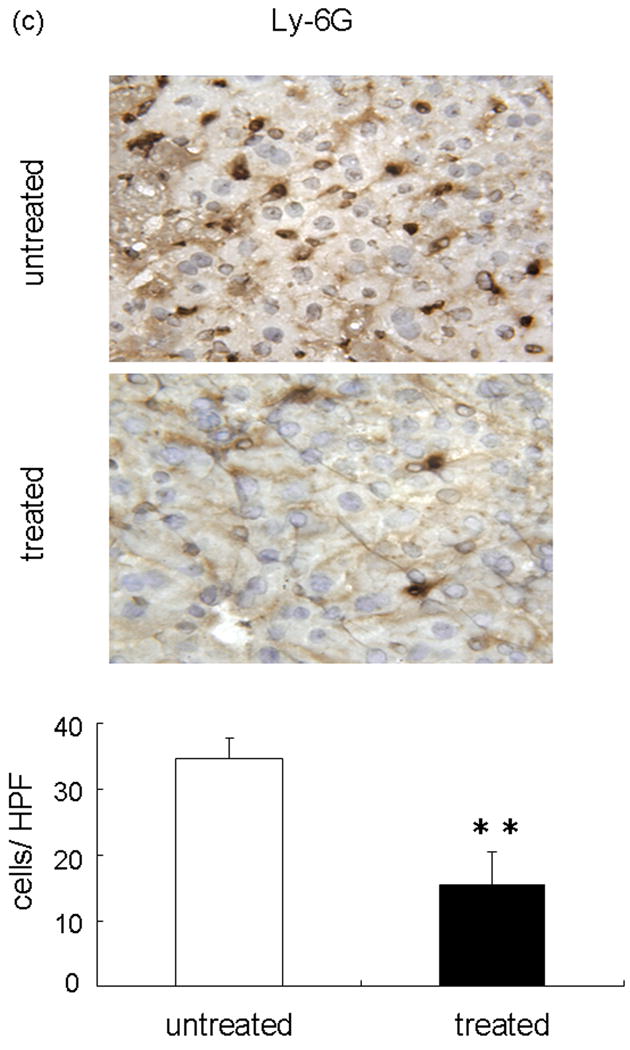
Neutrophil accumulation in ischemic liver lobes harvested at 6 h of reperfusion after 90 min of warm ischemia with or without NEI treatment. (a) MPO activity. (**P<0.05; n=6/group). (b) Upper panel: Representative liver sections stained by immunohistology using Naphthol ASD-chloroacetate esterase kit (×400 magnification). Lower panel: Quantitation of hepatic activated neutrophil accumulation by immunohistology. Note: Accumulation of polymorphonuclear cells (red spots, as shown by arrow) in treated livers was significantly decreased as compared with untreated controls (**P < 0.05; n = 3/group). (c) Upper panel: Representative liver sections stained by Ly-6G (×400 magnification). Lower panel: Quantitation of hepatic neutrophil infiltration by immunohistology. Note: Infiltration of polymorphonuclear cells (dark brown spots) in treated livers was significantly decreased as compared with untreated controls (**P < 0.05; n = 3/group). Means and SE are shown.
Inhibition of NE Activity Suppresses Pro-inflammatory Cytokines and Chemokines
We used RT-PCR to analyze pro-inflammatory cytokines (TNF-α, IL-6) and chemokines (CXCL-1 [KC] and CXCL-2 [macrophage inflammatory protein-2: MIP-2]) expression in the liver during IRI and calculated the ratio between post-IR and basal mRNA levels in each animal. Untreated group showed significantly increased induction ratios of TNF-α (TNF-α/β-actin mRNA: [6 h] 9.30±0.10 vs 5.15±0.61, p<0.05; [24 h] 6.70±0.47 vs 3.52±2.39, n=3/group) and IL-6 (IL-6/β-actin mRNA: [6 h] 1.00±0.13 vs 0.14±0.05, p<0.01; [24 h] 0.78±0.10 vs 0.14±0.03), as compared with treated group (Figure 4a). The CXC chemokines are considered to act predominantly on neutrophils (25). In mice, the two major CXC chemokines are cytokine-induced neutrophil chemoattractant (CXCL-1 [KC]) and (CXCL-2 [MIP-2]). Although no major variations were detected in CXCL-1 expression at 6 h (CXCL-1/β-actin mRNA: 4.10±0.42 vs 3.98±0.08), the expression at 24 h (CXCL-1/β-actin mRNA: 3.84±0.10 vs 1.55 ±0.17) was significantly reduced (p<0.01) between untreated and treated group (Figure 4b). In contrast, CXCL-2 expression was significantly reduced in the treated livers at both 6 h (CXCL-2/β-actin mRNA: 39.64 ± 2.82 vs 18.62 ± 4.84, p<0.05) and 24 h (CXCL-2/β-actin mRNA: 34.74 ± 2.94 vs 16.41 ± 2.74, p<0.01) after reperfusion.
Figure 4.
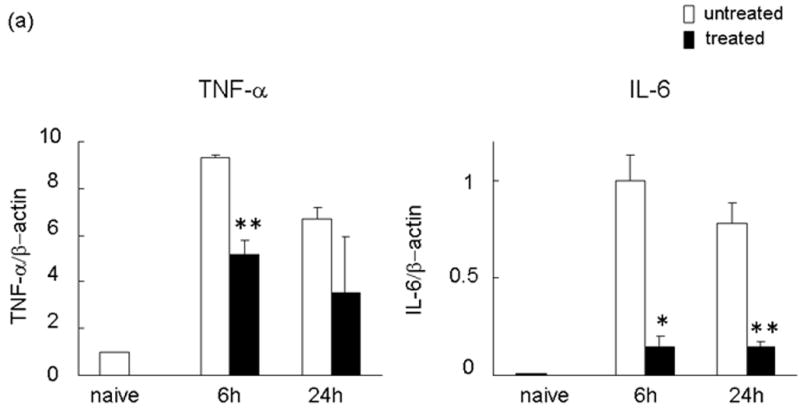
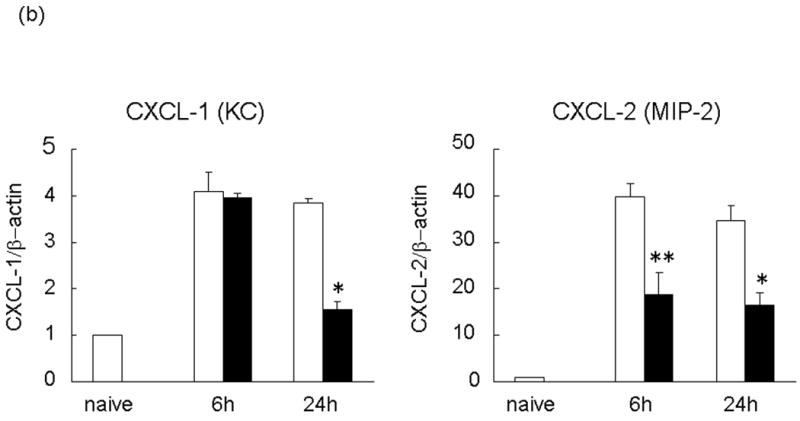
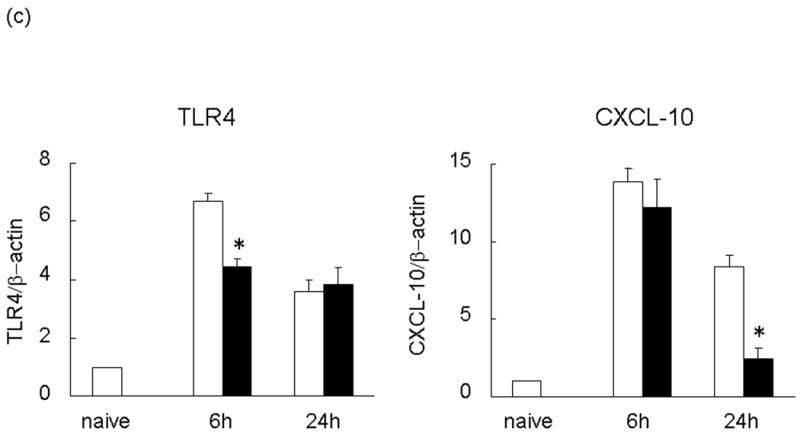
RT-PCR-assisted detection of pro-inflammatory mediators in IRI livers. (a) Cytokine gene expression. (b) Chemokine gene expression. (c) TLR4 and CXCL-10 gene expression in untreated and treated livers. Data were normalized to β-actin gene expression (*p < 0.01, **p < 0.05; n =3/group). Means and SE are shown.
Inhibition of NE Activity Suppresses CXCL-10 and TLR4 Expression
We measured CXCL-10 and TLR4 transcript levels in the ischemic liver lobes at 6 and 24 h after reperfusion following 90 min of warm ischemia. The expression of mRNA coding for TLR4 and CXCL-10 increased sharply in untreated group. Neutralization of NE was accompanied by the early inhibition of TLR4 expression at 6 h (TLR4/β-actin mRNA: 6.70±0.25 vs 4.45±0.24, p<0.01). This was followed by diminished CXCL-10 expression levels at 24 h (CXCL-10/β-actin mRNA: 8.40±0.71 vs 2.43±0.72, p<0.01; Figure 4c).
NEI Treatment Suppresses Apoptosis
We evaluated hepatocyte apoptosis by TUNEL staining of livers subjected to 90 min of warm ischemia and 6 h of reperfusion (Figure 5a). Augmented hepatocyte apoptosis (TUNEL positive cells/field) readily detectable in the untreated group remained diminished following NEI treatment (24.23 ± 5.31 vs 9.00 ± 2.18; p<0.05). Additionally, as shown in Figure 5b, the enzymatic activity of caspase-3, a proapoptotic marker, was significantly reduced at 6 h after reperfusion in the treated group, as compared with untreated controls (12.11 ± 1.53 vs. 25.86 ± 3.21 U/g, p<0.01).
Figure 5.
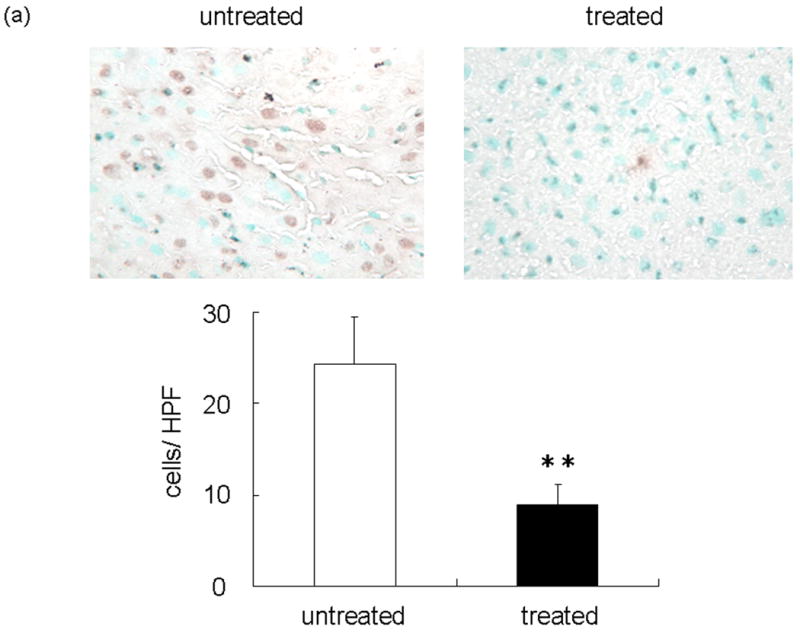
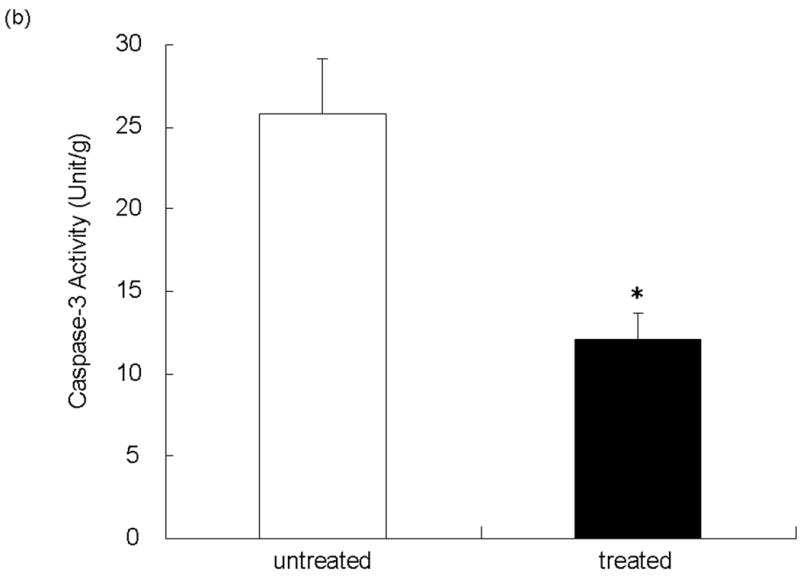
(a) Representative TUNEL-assisted detection of intrahepatic apoptosis in ischemic liver lobes harvested at 6 h after 90 min of reperfusion. Upper panel: Liver sections were stained by TUNEL method (×400 magnification). Lower panel: Quantitation of intrahepatic apoptosis by TUNEL method. Note: Untreated group with higher frequency of TUNEL positive cells (dark brown spots), as compared with NEI-treated group (**P < 0.05; n = 3/group). (b) Caspase-3 activity was significantly depressed in NEI-treated group as compared with untreated controls (*P < 0.01; n = 5/group). Means and SE are shown.
Discussion
Our results provide evidence for the role of NE in the pathogenesis of liver IRI. First, we showed that NE activity increases during the course of liver IRI. Second, inhibition of NE activity ameliorated IR-induced hepatocellular damage. We then showed that these beneficial effects after NEI treatment were accompanied by diminished local neutrophil infiltration, reduced expression of pro-inflammatory cytokines TNF-α/IL-6, chemokines CXCL-1/CXCL-2/CXCL-10, and TLR4. Hence, we conclude that targeting NE represents a useful approach to prevent IRI, which might improve the overall success of liver transplantation.
The protective effects of a neutrophil elastase inhibitor, Sivelestat, have been first documented in acute lung injury associated with systemic inflammation response, which may occur after infection, surgical intervention, traumatic or burn injury (26). Although some have shown the benefits of targeting NE, including Sivelestat in the liver injury models, the details remain unclear (27-29). Therefore, here we used commercially available NEI (GW311616A), with stronger inhibitory function than Sivelestat (18, 30), in an attempt to assess the role of NE in the mechanism of liver IRI. Indeed, treatment with GW311616A markedly inhibited serum ALT levels and prevented tissue damage at both 6 and 24 h after reperfusion, confirming the efficacy of that selective therapeutic approach to ameliorate warm ischemia-induced liver damage. This observation suggests several important interactions between NE and pro-inflammatory mediators at the local IRI site. As MPO activity was significantly decreased after GW311616A treatment, neutralization of NE may be sufficient to lessen the overall protease burden without the need for inhibition of all proteases.
Neutrophils can be triggered to express a number of mediators that can influence local inflammatory and immune responses. These include ROS, complement components, and proteases, as well as a variety of cytokines and chemokines (31). The C-X-C chemokines, CXCL-1 (KC), and CXCL-2 (MIP-2) are potent neutrophil chemoattractants, which are important in liver IRI (32, 33). Murine CXCL-2 (MIP-2) is a chemokine considered to be functionally analogous to human IL-8 and rat neutrophil chemoattractant (34) and is primarily induced by TNF-α (35). IL-6 is considered a marker for liver injury severity (36). Here, we have shown that administration of NEI suppressed expression of TNF-α, IL-6 and chemokines (CXCL-1 [KC], CXCL-2 [MIP-2]) in the liver.
Furthermore, TNF-α has a role as initiator of the apoptotic cascade, and apoptosis has been shown to be an important event after reperfusion, the severity of which correlates with the degree of hepatic injury (37). In this study, significantly increased frequency of TUNEL positive apoptotic cells during IRI noted in control untreated livers, accompanied by markedly elevated caspase-3 activity, became markedly diminished following neutralization of NE activity.
Although expressed by both hepatocytes and nonparenchymal cells, TLR4 signaling on Kupffer cells is critical in the pathogenesis of liver IRI (38). The activation of TLR4 on the surface of macrophages in the liver and spleen represents the critical initiating event preceding the inflammatory reaction induced by endotoxin or exotoxin (39, 40). The TLR4 signaling pathway begins with endotoxin in the blood binding to LPS-binding protein, which elicits LPS to anchor to CD14 molecule and combine with the extracellular TLR4 portion. This compound mediates LPS signaling, transducing extracellular inflammatory signal into cells, and with TNF-α mRNA initiating the inflammation response (41). During liver IR, the portal vein occlusion results in the congestion of intestinal wall, leading to the release of gut-derived molecules, including endotoxin into the blood stream. Thus, LPS is an obvious candidate for TLR4 activation, consistent with its role as an initiating factor in liver IRI (42-44). However, liver IRI may also develop in animal models under aseptic conditions (42). Moreover, we have shown that cold-preserved rat livers develop hepatocellular damage after reperfusion ex vivo (45). Thus, although gut-derived LPS contributes to IRI, it may not actually trigger the intrahepatic inflammation cascade that culminates in tissue damage. Indeed, endogenous TLR4 ligands generated during liver IRI may trigger local inflammation leading to the hepatocellular damage (4). It has been reported that IL-8 up-regulation by NE occurs in part through the cell surface membrane bound TLR4 in bronchial epithelium (46). Although NE may associate with increased expression of monocyte/macrophage TLR-4 in sepsis (47), the direct effect of NE on TLR4 could not be discarded in Leishmania infection (48). As the question remained as to how IR insult activates TLR4 system (4), our results imply that a direct effect of NE on TLR4 molecules cannot be discarded. Hence, the inhibition of NE at 3 h after reperfusion did suppress TLR4 expression at 6 h, followed at 24 h by diminished CXCL-10, the downstream TLR4 effector mediator.
Figure 6 depicts our working hypothesis on cross-talk between NE and the cascade of inflammatory mediators triggered in liver IR. Activated Kupffer cells and hepatocytes both produce pro-inflammatory cytokines, including TNF-α. The latter affects surrounding hepatocytes causing their apoptosis, and triggers neutrophil-attracting CXC chemokines to express adhesion molecules on vascular endothelial cells. Neutrophil adhesion to endothelial cells leads to transmigration from the vascular lumen into the liver parenchyma. The infiltrating neutrophil-derived NE induces inflammatory chemokine (CXCL-1, CXCL-2) expression by neutrophils. NEI-mediated depression of CXCL-2 and other chemokines prevents recruitment of neutrophils and macrophages, which in turn suppresses local inflammation response. It is plausible that NE may not only accelerate IR-mediated damage due to the feedback with recruited neutrophils, resulting in the direct injury to membrane components, but may also have some effect on TLR4 system by serving as a putative endogenous TLR4 ligand, and causing TLR4 up-regulation on Kupffer cells and hepatocytes.
Figure 6.
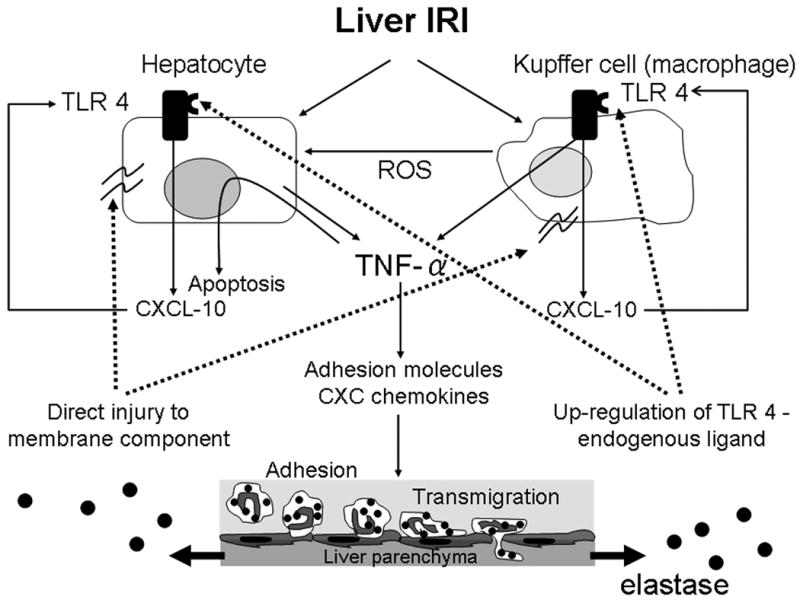
Cross-talk interactions between NE and local inflammation responses in liver IRI. Activated Kupffer cells and hepatocytes produce TNF-α. The latter affects surrounding hepatocytes causing their apoptosis, and triggers neutrophil-attracting CXC chemokines to express adhesion molecules on vascular endothelial cells. Neutrophil adhesion to endothelial cells leads to transmigration from the vascular lumen into the liver parenchyma and releases NE. NE accelerates IR-mediated damage due to the feedback with recruited neutrophils, resulting in the direct injury to membrane components. Moreover, NE may also serve as a putative endogenous TLR4 ligand, causing TLR4 up-regulation on Kupffer cells and hepatocytes.
In conclusion, our data indicate essential role of NE in the pathophysiology of liver IRI. NE contributes to the accumulation of neutrophils at inflamed site and the secretion of pro-inflammatory mediators. Thus, NEI treatment should be considered as a potential therapy for liver IRI. As inhibition of NE down-regulated innate TLR4 and its downstream CXCL-10 expression, this study suggests the previously unrecognized NE - TLR4 cross talk, and implies NE in the signal transduction pathway instrumental for liver IRI.
Acknowledgments
Funding sources: NIH Grants RO1 DK062357, AI23847, AI42223, and The Dumont Research Foundation.
Abbreviations
- ALT
alanine aminotransferase
- IRI
ischemia/reperfusion injury
- LPS
lipopolysaccharide
- MPO
myeloperoxidase
- NE
neutrophil elastase
- NEI
neutrophil elastase inhibitor
- TLR4
toll-like receptor 4
- TNF
tumor necrosis factor
- ROS
reactive oxygen species
References
- 1.Farmer DG, Amersi F, Busuttil RW, et al. Current concepts in ischemia and reperfusion injury in the liver. Transplant Rev. 2000;14:106. [Google Scholar]
- 2.Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury - a fresh look. Exp Mol Pathol. 2003;74:86. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 3.Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: Pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18:891. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 4.Zhai Y, Qiao B, Shen XD, et al. Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation. 2008;85:1016. doi: 10.1097/TP.0b013e3181684248. [DOI] [PubMed] [Google Scholar]
- 5.Zhai Y, Qiao B, Gao F, et al. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. 2008;47:199. doi: 10.1002/hep.21970. [DOI] [PubMed] [Google Scholar]
- 6.Wu HS, Zhang JX, Wang L, et al. Toll-like receptor 4 involvement in hepatic ischemia/reperfusion injury in mice. Hepatobiliary Pancreat Dis Int. 2004;3:250. [PubMed] [Google Scholar]
- 7.Zhai Y, Shen XD, O'Connell R, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 8.Jaeschke H. Chemokines and liver inflammation: the battle between pro- and anti-inflammatory mediators. Hepatology. 1997;25:252. doi: 10.1053/jhep.1997.v25.ajhep0250252. [DOI] [PubMed] [Google Scholar]
- 9.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. 1997;61:647. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 10.Jaeschke H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 11.Jaeschke H, Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006;26:912. doi: 10.1111/j.1478-3231.2006.01327.x. [DOI] [PubMed] [Google Scholar]
- 12.Moll T, Dejana E, Vestweber D. In vitro degradation of endothelial catenins by a neutrophil protease. J Cell Biol. 1998;140:403. doi: 10.1083/jcb.140.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janoff A, Scherer J. Mediators of inflammation in leukocyte lysosomes. IX. Elastinolytic activity in granules of human polymorphonuclear leukocytes. J Exp Med. 1968;128:1137. doi: 10.1084/jem.128.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janoff A. Elastase in tissue injury. Annu Rev Med. 1985;36:207. doi: 10.1146/annurev.me.36.020185.001231. [DOI] [PubMed] [Google Scholar]
- 15.Mainardi CL, Dixit SN, Kang AH. Degradation of type IV (basement membrane) collagen by a proteinase isolated from human polymorphonuclear leukocyte granules. J Biol Chem. 1980;255:5435. [PubMed] [Google Scholar]
- 16.Mainardi CL, Hasty DL, Seyer JM, et al. Specific cleavage of human type III collagen by human polymorphonuclear leukocyte elastase. J Biol Chem. 1980;255:12006. [PubMed] [Google Scholar]
- 17.Tsuchihashi S, Zhai Y, Bo Q, et al. Heme oxygenase-1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type-1 interferon signaling. Transplantation. 2007;83:1628. doi: 10.1097/01.tp.0000266917.39958.47. [DOI] [PubMed] [Google Scholar]
- 18.Macdonald SJ, Dowle MD, Harrison LA, et al. The discovery of a potent, intracellular, orally bioavailable, long duration inhibitor of human neutrophil elastase--GW311616A a development candidate. Bioorg Med Chem Lett. 2001;11:895. doi: 10.1016/s0960-894x(01)00078-6. [DOI] [PubMed] [Google Scholar]
- 19.Yoshimura K, Nakagawa S, Koyama S, et al. Roles of neutrophil elastase and superoxide anion in leukotriene B4-induced lung injury in rabbit. J Appl Physiol. 1994;76:91. doi: 10.1152/jappl.1994.76.1.91. [DOI] [PubMed] [Google Scholar]
- 20.Hagio T, Nakao S, Matsuoka H, et al. Inhibition of neutrophil elastase activity attenuates complement-mediated lung injury in the hamster. Eur J Pharmacol. 2001;426:131. doi: 10.1016/s0014-2999(01)01191-8. [DOI] [PubMed] [Google Scholar]
- 21.Tsuchihashi S, Livhits M, Zhai Y, et al. Basal rather than induced heme oxygenase-1 levels are crucial in the antioxidant cytoprotection. J Immunol. 2006;177:4749. doi: 10.4049/jimmunol.177.7.4749. [DOI] [PubMed] [Google Scholar]
- 22.Contreras JL, Vilatoba M, Eckstein C, et al. Caspase-8 and caspase-3 small interfering RNA decreases ischemia/reperfusion injury to the liver in mice. Surgery. 2004;136:390. doi: 10.1016/j.surg.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 23.Hamada T, Tsuchihashi S, Avanesyan A, et al. Cyclooxygenase-2 deficiency enhances Th2 immune responses and impairs neutrophil recruitment in hepatic ischemia/reperfusion injury. J Immunol. 2008;180:1843. doi: 10.4049/jimmunol.180.3.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada N, Okajima K, Murakami K, et al. Adenosine and selective A(2A) receptor agonists reduce ischemia/reperfusion injury of rat liver mainly by inhibiting leukocyte activation. J Pharmacol Exp Ther. 2000;294:1034. [PubMed] [Google Scholar]
- 25.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 26.Tamakuma S, Ogawa M, Aikawa N, et al. Relationship between neutrophil elastase and acute lung injury in humans. Pulm Pharmacol Ther. 2004;17:271. doi: 10.1016/j.pupt.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Soejima Y, Yanaga K, Nishizaki T, et al. Effect of specific neutrophil elastase inhibitor on ischemia/reperfusion injury in rat liver transplantation. J Surg Res. 1999;86:150. doi: 10.1006/jsre.1999.5661. [DOI] [PubMed] [Google Scholar]
- 28.Tomizawa N, Ohwada S, Ohya T, et al. The effect of neutrophil elastase inhibitor in hepatectomy with ischemia in dogs. J Surg Res. 1999;81:230. doi: 10.1006/jsre.1998.5499. [DOI] [PubMed] [Google Scholar]
- 29.Uchida Y, Kaibori M, Hijikawa T, et al. Protective effect of neutrophil elastase inhibitor ( FR136706) in lethal acute liver failure induced by D-galactosamine and lipopolysaccharide in rats. J Surg Res. 2008;145:57. doi: 10.1016/j.jss.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Kawabata K, Hagio T, Matsuoka S. The role of neutrophil elastase in acute lung injury. Eur J Pharmacol. 2002;451:1. doi: 10.1016/s0014-2999(02)02182-9. [DOI] [PubMed] [Google Scholar]
- 31.Scapini P, Lapinet-Vera JA, Gasperini S, et al. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- 32.Mosher B, Dean R, Harkema J, et al. Inhibition of kupffer cells reduced CXC chemokine production and liver injury. J Surg Res. 2001;99:201. doi: 10.1006/jsre.2001.6217. [DOI] [PubMed] [Google Scholar]
- 33.Lentsch AB, Yoshidome H, Cheadle WG, et al. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: Roles for macrophage inflammatory protein-2 and KC. Hepatology. 1998;27:563. doi: 10.1002/hep.510270440. [DOI] [PubMed] [Google Scholar]
- 34.Oppenheim JJ, Zachariae CO, Mukaida N, et al. Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu Rev Immunol. 1991;9:617. doi: 10.1146/annurev.iy.09.040191.003153. [DOI] [PubMed] [Google Scholar]
- 35.Tessier PA, Naccache PH, Clark-Lewis I, et al. Chemokine networks in vivo: involvement of C-X-C and C-C chemokines in neutrophil extravasation in vivo in response to TNF-alpha. J Immunol. 1997;159:3595. [PubMed] [Google Scholar]
- 36.Jin X, Zimmers TA, Perez EA, et al. Paradoxical effects of short- and long-term interleukin-6 exposure on liver injury and repair. Hepatology. 2006;43:474. doi: 10.1002/hep.21087. [DOI] [PubMed] [Google Scholar]
- 37.Rüdiger HA, Graf R, Clavien PA. Liver ischemia: apoptosis as a central mechanism of injury. J Invest Surg. 2003;16:149. [PubMed] [Google Scholar]
- 38.Tsung A, Hoffman RA, Izuishi K, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 39.Harju K, Glumoff V, Hallman M. Ontogeny of toll-like receptors TLR2 and TLR4 in mice. Pediatr Res. 2001;49:81. doi: 10.1203/00006450-200101000-00018. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 41.Faure E, Equils O, Sieling PA, et al. Bacterial lipopolysaccharide activates NF-kappaB through toll-like receptor 4 (TLR4) in cultured human dermal endothelial cells. J Bio Chem. 2000;275:11058. doi: 10.1074/jbc.275.15.11058. [DOI] [PubMed] [Google Scholar]
- 42.Arai M, Mochida S, Ohno A, et al. Selective bowel decontamination of recipients for prevention against liver injury following orthotopic liver transplantation: Evaluation with rat models. Hepatology. 1998;27:123. doi: 10.1002/hep.510270120. [DOI] [PubMed] [Google Scholar]
- 43.Fiorini RN, Shafizadeh SF, Polito C, et al. Anti-endotoxin monoclonal antibodies are protective against hepatic ischemia/reperfusion injury in steatotic mice. Am J Transplant. 2004;4:1567. doi: 10.1111/j.1600-6143.2004.00549.x. [DOI] [PubMed] [Google Scholar]
- 44.Tsoulfas G, Takahashi Y, Ganster RW, et al. Activation of the lipopolysaccharide signaling pathway in hepatic transplantation preservation injury. Transplantation. 2002;74:7. doi: 10.1097/00007890-200207150-00003. [DOI] [PubMed] [Google Scholar]
- 45.Amersi F, Buelow R, Kato H, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest. 1999;104:1631. doi: 10.1172/JCI7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Devaney JM, Greene CM, Taggart CC, et al. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Lett. 2003;544:129. doi: 10.1016/s0014-5793(03)00482-4. [DOI] [PubMed] [Google Scholar]
- 47.Tsujimoto H, Ono S, Majima T, et al. Neutrophil elastase, MIP-2, and TLR-4 expression during human and experimental sepsis. Shock. 2005;23:39. doi: 10.1097/01.shk.0000145936.31967.d7. [DOI] [PubMed] [Google Scholar]
- 48.Ribeiro-Gomes FL, Moniz-de-Souza MC, Alexandre-Moreira MS, et al. Neutrophils activate macrophages for intracellular killing of Leishmania major through recruitment of TLR4 by neutrophil elastase. J Immunol. 2007;179:3988. doi: 10.4049/jimmunol.179.6.3988. [DOI] [PubMed] [Google Scholar]