

Abstract
Analogs of the malaria therapeutic, artemisinin, possess in vitro and in vivo anti-cancer activity. In this study, two dimeric artemisinins (NSC724910 and 735847) were studied to determine their mechanism of action. Dimers were >1000 fold more active than monomer and treatment was associated with increased reactive oxygen species (ROS) and apoptosis induction. Dimer activity was inhibited by the anti-oxidant L-NAC, the iron chelator desferroxamine, and exogenous hemin. Similarly, induction of heme oxygenase (HMOX) with CoPPIX inhibited activity while inhibition of HMOX with SnPPIX enhanced it. These results emphasize the importance of iron, heme and ROS in activity. Microarray analysis of dimer treated cells identified DNA damage; iron/heme and cysteine/methionine metabolism, antioxidant response, and endoplasmic reticulum (ER) stress as affected pathways. Detection of an ER-stress response was relevant because in malaria, artemisinin inhibits pfATP6, the plasmodium orthologue of mammalian ER-resident SERCA Ca2+-ATPases. A comparative study of NSC735847 with thapsigargin, a specific SERCA inhibitor and ER-stress inducer showed similar behavior in terms of transcriptomic changes, induction of endogenous SERCA and ER calcium mobilization. However, thapsigargin had little effect on ROS production, modulated different ER-stress proteins and had greater potency against purified SERCA1. Furthermore, an inactive derivative of NSC735847 that lacked the endoperoxide had identical inhibitory activity against purified SERCA1, suggesting that direct inhibition of SERCA has little inference on overall cytotoxicity. In summary, these data implicate indirect ER-stress induction as a central mechanism of artemisinin dimer activity.
Keywords: Artemisinin dimer, reactive oxygen species (ROS), UPR, thapsigargin, SERCA
Introduction
Artemisinin (Qinghaosu), a traditional Chinese medicine, is an effective chemotherapeutic for the treatment of multi-drug resistant strains of malaria 1–3. More recently, semi-synthetic derivatives have been shown to have anti-cancer activity 4–6. Cancer cells exposed to artemisinin derivatives demonstrate decreased proliferation, increased levels of oxidative stress, induction of apoptosis, and inhibition of angiogenesis. Whereas monomeric forms have activity in the nanomolar range for treatment of malaria, activity versus tumor cells is in the upper micromolar range. Conversion of artemisinin to dimeric and trimeric forms was also shown to substantially enhance anti-cancer activity 5, 6.
The mechanism underlying pharmacological activity in both malaria and cancer is still the subject of debate 7,8. In malaria, the classical mechanism is thought to involve reaction of the endoperoxide bridge with free heme-iron liberated during degradation of hemoglobin inside the parasite food vacuole 9. Endoperoxide cleavage generates damaging reactive oxygen species (ROS) and carbon-centered radicals leading to parasite death. However, recent work suggests that artemisinins may also function as inhibitors of PfATP6, the Plasmodium falciparum orthologue of mammalian sarcoendoplasmic reticulum Ca2+-ATPases (SERCAs)9.
With respect to cancer, the current consensus regarding artemisinin activity involves indiscriminate generation of oxidative stress as a consequence of heme-mediated endoperoxide cleavage, leading to DNA damage and apoptosis 4. Indirect evidence to support this comes from studies of the NCI 60 cell line screen showing an inverse correlation between activity of artesunate (dihydroartemisinin hemisuccinate) and mRNA expression for anti-oxidant genes such as catalase, superoxide dismutase II, thioredoxin reductase, γ-glutamylcysteine synthase (γ-GCS) and several members of the glutathione-S-transferase (GST) family 4. Iron metabolism also plays a central role in the anti-cancer activity of artemisinin. In vitro and in vivo studies show that preloading cells with iron or inclusion of holotransferrin, enhances the activity of artemisinin derivatives 4, 10. Increased levels of iron within tumor cells relative to normal counterparts may provide a molecular basis for the high ‘therapeutic index’ observed by several authors 4, 10. The potential of artemisinin derivatives is further strengthened by anti-angiogenic activity in vitro and in vivo animal models, oral dosing inhibits vascularization of matrigel plugs 4. Activity has been shown to correlate with changes in expression of several angiogenesis related genes including HIF-1α, VEGFA/C and FGF2 11–14. Therefore, the ability of this well-characterized group of compounds to selectively induce apoptosis and inhibit angiogenesis makes them attractive candidates for clinical development.
However, several important questions remain regarding the mechanism of artemisinin-induced cell death, namely whether activity is dependent on definitive molecular targets. Here we present studies of the potent artemisinin dimers, NSC724910 and NSC735847, to further elucidate a mechanism of action. Results demonstrate that dimers are logarithmically more active than comparable monomeric forms and are associated with generation of ROS and rapid induction of apoptosis. We explored the potential of SERCA as a molecular target for the antitumor activity observed with artemisinin dimers. Comparator studies of dimer with thapsigargin, a specific SERCA inhibitor, demonstrated both agents mobilized calcium and inhibited SERCA enzymatic activity. Analysis of transcriptional changes demonstrated induction of ER stress related genes in a pattern similar for both agents. However, thapsigargin treatment did not induce ROS or oxidize SERCA cysteine residues. A deoxyartemisinin dimer, NSC735847DX, which is inactive in cytotoxicity assays and unable to generate ROS, was found to be equally potent to the parent compound, NSC735847, in inhibiting SERCA enzymatic activity. This provided evidence that direct inhibition of SERCA Ca2+-ATPase was not responsible for overall cytotoxicity. Therefore, ROS-mediated ER stress induction, independent of any direct SERCA inhibition, is likely an important component of artemisinin dimer cytotoxicity.
Materials and Methods
Materials
The artemisinin dimers, NSC724910, NSC735847 and NSC735847DX (Fig. 1A) were provided to the DTP Drug Repository (Developmental Therapeutics Program, DTCD, NCI, Rockville, MD; www.dtp.nci.nih.gov) by ElSohly Laboratories, Incorporated (Oxford, MS) and were prepared according to the scheme shown in the Supporting Information section, Figure 1. All remaining drugs were obtained from the DTP Drug Repository. All cell lines were from the Division of Cancer Treatment and Diagnosis Tumor Repository (Frederick, MD). Materials were from the following sources: cobalt protoporphyrin IX (CoPPIX), Sigma (St. Louis, MO); tin protoporphyrin IX (SnPPIX), Frontier Scientific (Logan, UT); cyclopiazonic acid, 2,5-di-(tert-butyl)-1,4-benzohydroquinone (BHQ), BAPTA/AM and EGTA/AM, Calbiochem (San Diego, CA); Fluo-3/AM, EMD Biosciences (San Diego, CA). Primary antibodies were from the following: anti-ATF6, anti-Calnexin, anti-Caspase 12 and anti-GRP78 BiP, Abcam, Inc. (Cambridge, MA), anti-HERPUD1, Novus Biologicals (Littleton, CO), anti-CD71/TFR-FITC, Serotec (Kingston, NH), anti-β-Actin, Sigma (St. Louis, MO). Unless otherwise indicated, all other chemicals and inhibitors were from Sigma (St. Louis, MO).
Figure 1.
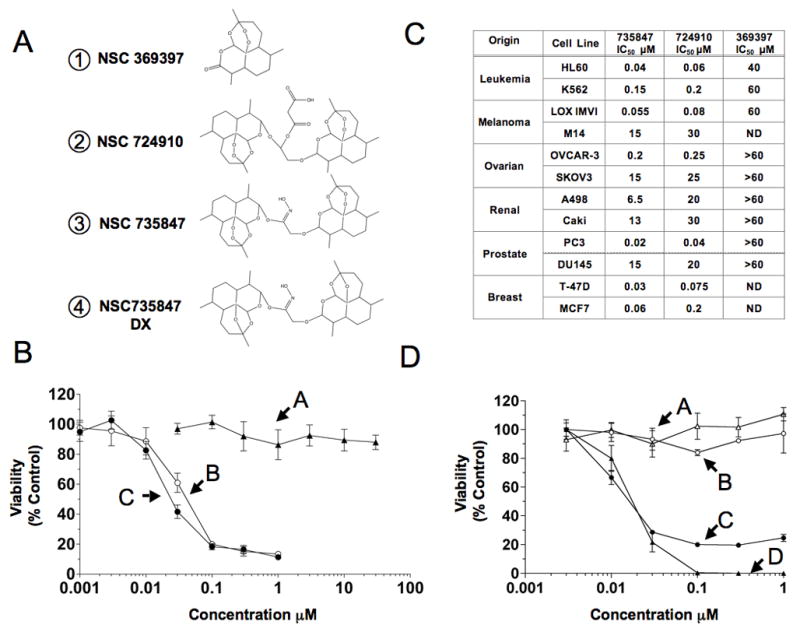
Artemisinin structure and activity. A) Structures of artemisinin monomer NSC 369397 (1), artemisinin dimers NSC 724910 (2), NSC 735847 (3), and deoxy-derivative of NSC 735847 (NSC 735847DX) (4). B) Activity of artemisinin monomer and dimers. PC-3 cells were incubated with NSC 369397 (A), NSC 724910 (B) or NSC 735847 (C) for 48 h before viability was determined. C) Activity of monomer and dimers in a panel of tumor cell lines following 48 h of exposure. IC50 was defined as the concentration of drug required to inhibit [14C]-leucine incorporation by 50% relative to control-treated cells. D) The importance of the endoperoxide bridge on dimer activity. HL-60 cells (triangles) or PC-3 cells (circles) were incubated for 24 h with either NSC 735847 or NSC 735847DX before protein synthesis was determined. HL-60 cells, NSC 735847 (D), NSC 735847DX (A); PC-3 cells, NSC 735847 (C), NSC 735847DX (B). [14C]-leucine cell viability assays were performed at least twice with triplicate determinations for each point and the data pooled.
[14C]-leucine cell viability assays
Adherent cells (2,500 in 100 μL) or non-adherent cells (10,000 in 100 μL) were placed into each well of a 96-well plate 24 h before treatment. Sample or buffer control (10 μL) was added to the appropriate wells and the plates were incubated at 37°C in a humidified CO2 incubator for the times indicated in the figure legends. For viability determinations based on the rate of protein synthesis, serum-containing media was replaced with serum-and leucine-free RPMI 1640 containing 0.03 μCi of [14C]-leucine and the samples processed as described in Stockwin et al. 15. The results are expressed as % [14C]-leucine incorporation into the control-treated cells. Experiments were performed at least twice with triplicate determinations for each point. The IC50 was defined as the concentration of drug required to inhibit protein synthesis (cell viability) by 50% relative to control-treated cells.
Western blotting
Treated cells were washed twice in PBS and lysed in RIPA-CHAPS buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1% CHAPS, 1% deoxycholate, 1x complete protease inhibitor). Lysates were sonicated, centrifuged to remove insoluble material and protein concentration determined using the BCA Protein assay. SDS-PAGE was performed using 20 μg protein on a 10% NUPAGE Bis-Tris gel with subsequent transfer to a PVDF membrane. Following overnight blocking in 2% Blotto/TBS-T, membranes were incubated with primary antibody following the manufacturer’s recommendations. Imaging and quantitation were performed using the Kodak Image Station 2000MM and Kodak Molecular Imaging software (Carestream Health, New Haven, CT).
Apoptosis and necrosis determination
The percentage of apoptotic and necrotic cells in culture was determined using the Vybrant Apoptosis Assay kit (Molecular Probes, Eugene, OR) comprising an annexin V-Alexa488 conjugate and propidium iodide as described by the manufacturer. Acquisition and analysis of data were performed using a FACScan flow cytometer (Becton-Dickinson, Franklin Lakes, NJ) controlled by Cellquest Pro Software.
Cell cycle analysis
Treated cells were harvested and washed once with PBS. The samples were resuspended in 5 mL PBS, and 5 mL cold 70% ethanol were added drop wise. After 5 min incubation, the cells were centrifuged, resuspended in 10 mL cold 70% ethanol and stored at 4°C for 1 h. The cells were washed twice with 5 mL PBS and resuspended in 1 mL PBS containing 100 μg/mL propidium iodide (Molecular Probes) and 100 μg/mL RNase A (Sigma). After 1 h at 37°C, cell cycle analysis was performed using the FL3-A channel on a FACScan flow cytometer.
Detection of ROS
Cells were harvested, washed once in PBS and resuspended in serum-containing media (1×106 cells/mL). CM-H2DCFDA (Molecular Probes), which reacts with peroxides to produce green fluorescence, was added to a final concentration of 10 μM. Following a 1–2 h labeling period, cells were aliquoted and incubated in the presence of varying drug concentrations for 1 h at 37°C. The cells were then transferred to FACS tubes and analyzed using the FL1 channel on a FACScan cytometer.
Fluorescence spectrophotometric measurement of [Ca2+]i
The intracellular calcium concentration [Ca2+]i was monitored by the change in fluorescence intensity of Fluo-3-loaded PC-3 cells according to Johnson et al. 16 and is further described in the Supporting Information section.
SERCA Ca2+-ATPase assays
Sarcoplasmic reticulum (SR), which contains high levels of SERCA1, was prepared from rat skeletal muscle. Skeletal muscle (200 g) was homogenized using a Waring blender in 200 ml buffer containing 0.1 M KCl, 0.1 mM EDTA and 20 mM MOPS (pH 7.4). Homogenate was centrifuged at 5,000 g for 20 min to remove cell debris. The pooled supernatant was centrifuged for 20 min at 11,800 g to pellet mitochondria and filtered through 6 layers of cheesecloth and solid KCl added to a final concentration of 0.6 M to dissolve myosin. After 20 min, the SR was pelleted at 23,500 rpm for 1 h, the pellets resuspended in 0.3 M sucrose/20 mM MOPS (pH 7.0) and centrifuged at 100,000 g for 30 min. The pellet was Dounce homogenized into suspension with 0.3 M sucrose/20 mM MOPS (pH 7.0), protein determined using BCA, aliquoted, snap frozen in liquid nitrogen and stored at −70°C. For the determination of oxidative modifications, a malemide derivative of a thiol reactive fluorescent molecule, ThioGlo1 (Calbiochem, Gibbstown, NJ), was used. Aliquots (50μg) of SR were treated with oxidizing agent overnight at 37°C. Protein samples were then labeled with saturating quantities of ThioGlo1 (1μg/ml). After 2 h, labeled SR protein was denatured in SDS gel loading buffer and 10μg protein resolved by SDS-PAGE. Fluorescence intensities of the major band (SERCA1) were quantified by UV transillumination followed by densitometry. For the determination of Ca2+-dependent ATPase activity a modified version of an assay described by Kijima et al. (1991) was used 17. A reaction mixture (0.515 ml final volume) containing 40 mM Hepes/KOH (pH 7.2), 1 mM ATP, 1 mM MgCl2, 0.2 mM NADH, 2 mM phosphoenolpyruvate, 3.5 units pyruvate kinase, 5 units lactate dehydrogenase and 15.5 μg/ml ATPase was used for each assay. The reaction was initiated with 100 μM CaCl2 and activity was followed over time (3–5 min) with data collection every 20 s. Activity determined in the presence of 60 μM EGTA was subtracted from the total to yield Ca2+-dependent activity. Thapsigargin, NSC735847 and NSC735847DX were added as solutions in DMSO with a final concentration of 2% DMSO.
Results
In vitro activity of artemsinin dimers
Two artemisinin dimers, NSC724910 and NSC735847, differing only in the linker moiety, were compared against artemisinin (NSC369397) for cytotoxicity (Fig. 1A). Consistent with published reports, they were >1500–3000 times more active than artemisinin in PC-3 cells, suggesting that the dimers have properties distinct from artemisinin (Fig. 1B) 4–6. The analysis was extended to a panel of tumor cell lines (Fig. 1C), confirming the increased activity of the dimers over artemisinin and demonstrating that activity for the dimers varies approximately 500-fold within the panel (IC50 range, 0.03–15μM for NSC735847 and 0.06–30μM for NSC724910), with NSC735847 having slightly better activity than NSC724910. The deoxy-derivative NSC735847DX was relatively inactive versus NSC735847 (IC50 > 1 μM compared to IC50 s of 20 nM for both HL-60 and PC-3 cells) (Fig. 1D), emphasizing the importance of the endoperoxide bridge in activity. It should also be noted that the linker moiety from NSC735847 and a monomer with attached linker were tested and determined to be inactive (see Supporting Information Fig. 2), indicating that this structure alone is not responsible for increased dimer activity.
Subsequently, the effect of artemisinin, NSC724910, NSC735847 and NSC735847DX on 1) the cell cycle 2) ROS production 3) apoptosis induction and 4) transferrin receptor expression was examined. Artemisinin was used at twice the concentration of the dimers to maintain molar equivalency and the established anti-cancer drugs, doxorubicin and cisplatin, were included as controls. The effects of the various drugs on cell cycle distribution (Fig. 2A) did not show dimer specific perturbations in HL-60 or PC-3 cells over increases in the sub-G0 apoptotic population. Both NSC724910 and NSC735847 markedly increased generation of intracellular peroxides in HL-60 cells as determined using the fluorescent ROS sensor, CM-H2DCFDA (Fig. 2B, left panel). The positive control, doxorubicin increased ROS, while cisplatin had little effect. Treatment of cells with the artemisinin dimers for 24 h resulted in significant increases in apoptosis (Fig. 2B, middle panel). FACS analysis of cells treated with the dimers revealed a profound decrease in levels of cell surface transferrin receptor (TFR1) expression after 24 h exposure in HL-60 cells (Fig. 2B, right panel). Consistent with the lack of cytotoxic activity (Fig. 1B and 1D), artemisinin and NSC735847DX had little effect in any of the above assays.
Figure 2.

Artemisinin dimers increase intracellular peroxides, reduce surface transferrin receptor (CD71) and induce apoptosis without phase-specific cell cycle arrest. A) Propidium iodide (PI) cell cycle analysis of HL-60 and PC-3 cells treated with drug for 48 h was performed as described in Materials and Methods. B) For drug-treated HL-60 cells; liberation of intracellular peroxides was determined using the fluorescent substrate CM-H2DCFDA, levels of apoptosis were analyzed by staining with annexin V/propidium iodide (AnV/PI) and changes in transferrin receptor I (αTFR, CD71) expression were monitored using a fluorochrome (FITC) conjugated antibody. For generation of intracellular peroxides, cells were incubated in drug and CM-H2DCFDA for 2 h prior to analysis, whereas for apoptosis and CD71 determination cells were incubated for 24 h in compound prior to analysis. C) Modulation of heme oxygenase (HMOX1) affects NSC735847 activity. HL-60 and PC-3 cells, pre-incubated for 3 h with the HMOX1 inducer, CoPPIX, or the HMOX1 inhibitor, SnPPIX, were exposed to NSC 735847 and changes in cell viability determined using [14C]-leucine incorporation. Left panel, HL-60 cells; Right panel, PC-3 cells. (1) NSC 735847 alone, (2) NSC 735847 + 30 μM CoPPIX, (3) NSC 735847 + 30 μM SnPPIX. [14C]-leucine cell viability assays were performed at least twice with triplicate determinations for each point and the data pooled. All other assays were replicated two to three times.
Evidence for the involvement of heme in anti-cancer activity came from experiments utilizing cobalt (CoPPIX) and tin protoporphyrin IX (SnPPIX), two molecules that modulate the activity of heme oxygenase (HMOX1) (Fig. 2C). CoPPIX, an inducer of HMOX, almost entirely abolished NSC735847 activity [IC50 35 (HL-60) and 20 nM (PC-3) in the absence and > 1000 nM in the presence of CoPPIX], suggesting that if the intracellular free heme pool is depleted, the drug is inactivated. Conversely, SnPPIX, an inhibitor of HMOX, enhanced the activity of NSC735847 in PC-3 cells (IC50 20 and 3 nM in the absence or presence of SnPPIX, respectively), probably by increasing the steady-state pool of intracellular heme. NSC735847 activity against HL-60 cells was not enhanced by SnPPIX, which may reflect low endogenous levels of HMOX1 in these cells. This data was supported by results showing that pre-treatment of cells with the iron chelator desferroxamine (DFX) or the addition of exogenous hemin markedly decreased activity of NSC735847 (Supporting Information Table 1 and Fig. 3).
Transcriptomic response to artemisinin
Microarray analysis of mRNA from PC-3 cells treated with NSC735847 was used to provide unbiased mechanistic insights. Cells were treated with 2μM NSC735847 for 24 h, followed by the isolation of mRNA and analysis using U133 plus 2.0 cDNA arrays. Pairwise analysis of control versus treated cDNA samples using a 3-fold cut-off, < 0.01 adjusted p-value, GC-RMA normalization with Benjamini and Hochberg false discovery estimation, revealed significant alterations in gene expression on treatment with the dimer. Drug treatment resulted in the modulation of 2,706 non-redundant transcripts, where 1,009 showed increased expression and 1,697 were downregulated. Gene ontogeny analysis of microarray data defined several pathways that appeared to be modulated by treatment with NSC735847. This included transcripts involved in DNA damage/apoptosis, cell cycle, iron/heme and methionine/cysteine metabolism and several stress response pathways, including the unfolded protein response (UPR) (Supporting Information, Table 2). This subset of genes was further scrutinized by performing Q-RT-PCR on cDNA from an extended panel of NSC735847 treated cell lines (Fig. 3A).
Figure 3.
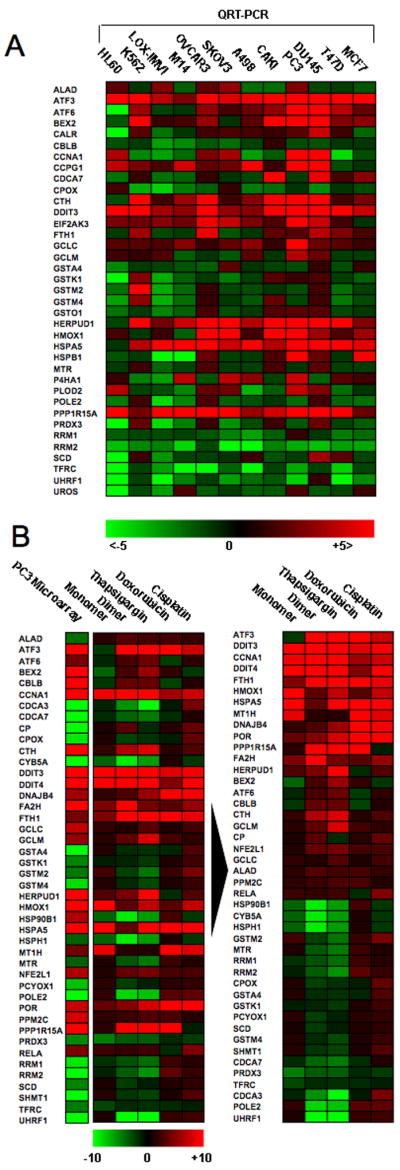
Real-time PCR analysis of selected transcripts identifies a conserved response to NSC735847 among different cell lines and illustrates that the response to the dimer and the SERCA inhibitor, thapsigargin, are almost identical. A) Total RNA was isolated from a panel of twelve cell lines treated with 2 μM NSC735847 for 24 h. Samples were reverse transcribed and analyzed by SYBR green real-time PCR for expression changes in selected transcripts previously identified by microarray analysis as modulated by treatment with NSC735847. Results are represented as a heat map with limits of +5 fold change (red), −5 fold change (green). B) Total RNA derived from PC-3 cells treated for 24 h with 2 μM artemisinin, 2 μM NSC735847, 0.2 μM thapsigargin, 2 μM doxorubicin or 2 μM cisplatin was isolated and reverse transcribed to cDNA. The left column shows results from the microarray analysis of dimer-treated PC-3 cells. The results were plotted (left panel) in terms of a heat map with limits of +10 change (red) and −10 fold change (green). Gene expression data was subjected to hierarchical cluster analysis (right panel) showing that the highest degree of homology between treatments was for NSC735847 and thapsigargin. Data shown is representative of that obtained over two replicate experiments using different cDNA samples where amplifications were measured in triplicate.
As with results obtained from the microarray analysis, RT-PCR analysis demonstrated consistent increases (12/12 cell lines) in ubiquitous DNA damage/stress response transcripts, including ATF3, DDIT3 and PPP1R15A/GADD34, inconsistent regulation of cell-cycle regulators (CCNA1, CDCA7, CCPG1, BEX2, POLE2) and consistent decreases in expression for the E3 ubiquitin-protein ligase UHRF1 (11/12 cell lines) and ribonucleotide reductases RRM1 and RRM2 (11/12 and 12/12 cell lines, respectively). With respect to iron/heme metabolism, RT-PCR data showed a decline (12/12 lines) in expression for the transferrin receptor (TFR1) following treatment, consistent with the data shown in Fig. 2, whereas levels of transcript coding for ferritin heavy chain (FTH) increased (7/12 lines). A further noteworthy result concerned upregulation of heme oxygenase, HMOX1 (10/12 lines), an enzyme involved in the catabolism of heme to bilirubin. Conversely, we observed a consistent downregulation in genes involved in heme biosynthesis, including coproporphyrinogen oxidase (CPOX, 10/12 lines), uroporphyrinogen III synthase (UROS, 9/12 lines) and aminolevulinate dehydratase (ALAD, 7/12 lines).
Although dimer treatment is associated with profound increases in oxidative stress, analysis of mRNA coding for several antioxidant genes failed to demonstrate conserved changes. Transcripts coding for catalase, myeloperoxidase and superoxide dismutase II as well as glutathione-S-transferase (GST) isoforms (GSTA4, GSTK1, GSTM2, GSTM4, GSTO1) all showed highly variable responses (Fig. 3A). However, transcripts involved in cysteine and glutathione synthesis, including γ-glutamylcysteine synthetase catalytic subunit (GCLC) (12/12 lines) and cystathionine gamma ligase (CTH) (11/12 lines) were upregulated in the majority of cell lines. Regarding ER stress and the unfolded protein response (UPR), increased expression of mRNA for the transcriptional regulator ATF6 was observed in 9/12 lines (Fig. 3A). The trend towards upregulation was also observed for the ER stress sensor PERK (EIF2AK3, 11/12 lines) and Bip/Grp78 (11/12 lines) and downstream effectors of the UPR such as HERPUD1 (11/12 lines), a protein involved in the degradation of misfolded ER proteins 18. Thus, results from transcriptome analysis demonstrate that, in the majority of lines, dimer treatment is associated with induction of UPR-related genes.
Activity of artemisinin dimers toward SERCA Ca2+-ATPase
Artemisinin monomers have been shown to inhibit PfATP6, the plasmodium orthologue of mammalian ER resident SERCA-type Ca2+ transporter 4. A study was undertaken to investigate whether artemisinin dimers share similarities with classical mammalian SERCA inhibitors such as thapsigargin 19, 20, which lack the artemisinin endoperoxide bridge 21. mRNA was isolated from PC-3 cells treated with several drugs, including artemisinin, NSC735847, thapsigargin, doxorubicin and cisplatin. Subsequently, quantitative RT-PCR was performed for a subset of transcripts identified as modulated by NSC735847 in the context of microarray analysis. As shown in Fig. 3B, there was a 93.2% concordance among genes modulated by the artemisinin dimer in microarray and quantitative RT-PCR experiments (41/44 genes), validating results from both techniques. Hierarchical clustering of drug treatment results provided visual proof of the high degree of homology between NSC735847 and thapsigargin (Fig. 3B, right panel). Statistically, 97.7% or 43/44 genes showed identical changes in expression; a compelling result given that this correlation superseded that observed between NSC735847 and artemisinin (65.9% or 29/44 genes).
We then examined the biological similarity between the artemisinin dimer and thapsigargin in more depth. First, the ability of dimer and thapsigargin to alter expression of key components of the UPR was investigated (Fig. 4A). Western blotting of treated cells determined 1) that NSC735847 had little effect on expression of native ATF6, whereas thapsigargin decreased the expression of ATF6 by 30–38%, 2) neither agent affected the expression of the ER chaperone HERPUD1, 3) both reduced the levels of native caspase 12 (29–44% and 39–42%, NSC735847 and thapsigargin, respectively), 4) only a high-dose (32 μM) of NSC735847 was capable of decreasing calnexin expression and 5) both increased the expression of Grp78 (HSPA5) (3.5–7.7 and 5.4–8.8 fold, NSC735847 and thapsigargin, respectively). Furthermore, the dimer was more effective than thapsigargin at producing ROS in both HL-60 and PC-3 cells (Fig. 4B). Also, pre-treatment of cells with the antioxidant, L-NAC, significantly attenuated the dimer activity while having little effect on thapsigargin activity (Fig. 4C, 2.0–2.5 versus 0.8–1 fold, NSC735847 and thapsigargin, respectively). Similarly, L-NAC was able to inhibit dimer mediated induction of Grp78 (Supporting Information, Fig. 4), reinforcing the role of ROS in ER-stress induction and anticancer activity.
Figure 4.
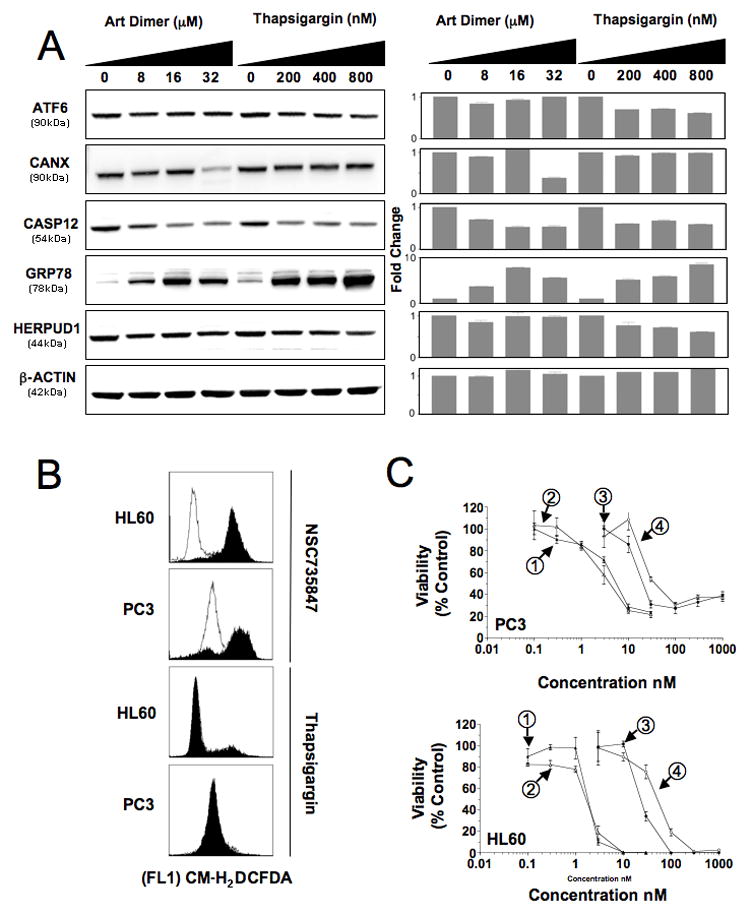
NSC735847 and thapsigargin have mechanistic differences. A) Lysates prepared from PC-3 cells treated with the indicated concentrations of NSC735847 or thapsigargin were probed for ER stress and UPR proteins. Left panel, western blot; right panel, densitometric analysis of the blots using Kodak Molecular Imaging software. Activating transcription factor 6 (ATF6), calnexin (CANX), caspase 12 (CASP12), heat shock 70 kDa protein 5 (HSPA5/GRP78) and homocysteine-responsive endoplasmic reticulum-resident ubiquitin-like domain member 1 (HERPUD1). Control, β-actin (ACTB). B) Generation of intracellular peroxides in HL-60 and PC-3 cells treated for 2 h with NSC 735847 (2 μM) or thapsigargin (1 μM) as determined using the fluorescent substrate CM-H2DCFDA. Solid histograms show drug treated cells, open histograms are control samples. C) The effect of the antioxidant L-NAC on the activity of NSC735847 and thapsigargin as determined by [14C]-leucine viability assays. Cells were pretreated with 11 mM L-NAC for 1 h before the addition of NSC735847 or thapsigargin and the incorporation of [14C]-leucine determined after 24 h. Top panel, PC-3; Bottom panel, HL-60 cells. Thapsigargin (1), thapsigargin + L-NAC (2), NSC735847 (3), NSC735847 + L-NAC (4). -[14C]-leucine cell viability assays were performed at least twice with triplicate determinations for each point and the data pooled. All other assays were replicated two to three times.
The effects of NSC735847 on both Ca2+ homeostasis and SERCA activity were also examined. Pre-incubation of PC-3 cells with the calcium chelators, BAPTA-AM/EGTA-AM, profoundly inhibited the cytotoxicity of both thapsigargin (Fig. 5A, bottom panel, IC50 5 nM versus 400 nM in the absence or presence of BAPTA-AM/EGTA-AM) and NSC735847 (Fig. 5A, top panel, IC50 15 nM versus > 300 nM, absence and presence of BAPTA-AM). Thapsigargin addition to cells has also been shown to induce a transient rise in cytosolic free Ca2+22, 23. PC-3 cells were loaded with Fluo-3 and the effect of NSC735847 on cytosolic free Ca2+ was compared to that of thapsigargin. As shown in Fig. 5B, thapsigargin caused an immediate rise in Ca2+ that declined to resting levels over approximately 200 sec. Similarly, NSC735847 caused an immediate rise in Ca2+, however this increase did not decline as noted for thapsigargin, but was sustained over the duration of the experiment. We then examined the effects of another inhibitor of the Ca2+-ATPases, chemically unrelated to thapsigargin, 2,5-di-(tert-butyl)-1,4-benzohydroquinone (BHQ), on the SERCA pump. As reported, BHQ also caused an immediate rise in Ca2+, but as with artemisinin dimer, this increase was sustained (Fig. 5B) 24, 25. These results implicate calcium mobilization as a critical event in dimer activity, further implicating an ER stress response.
Figure 5.
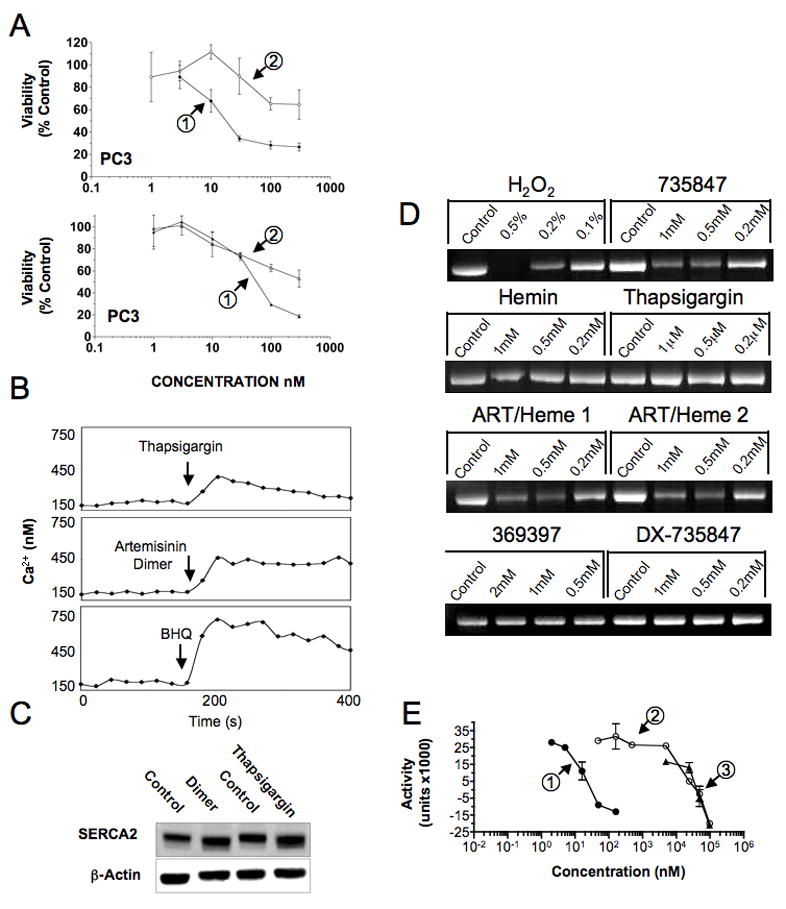
Exploring the role of SERCA Ca2+-ATPases in NSC735847 activity. A) Calcium chelation attenuates cytotoxicity of thapsigargin and NSC735847 as determined by [14C]-leucine viability assays. PC-3 cells were either preincubated with 100 μM BAPTA-AM for 1 h before addition of NSC735847 and the incubation continued for 23 h, or the cells were preincubated with 50 μM BAPTA-AM and 50 μM EGTA-AM for 1 h followed by the addition of thapsigargin for 9 h. Cells were then washed and the incubation continued for 14 h. Top panel, NSC735847 (1), NSC735847 + 100 μM BAPTA-AM (2); bottom panel, thapsigargin (1), thapsigargin + 50 μM BAPTA-AM and 50 μM EGTA-AM (2). B) Comparison of the effect of NSC735847 with that of the SERCA inhibitors, thapsigargin and BHQ, on the intracellular calcium concentration [Ca2+]i in PC-3 cells. Thapsigargin, NSC735847 and BHQ increase [Ca2+]i of PC-3 cells, loaded with Fluo-3. The tracings show the change in [Ca2+]i from the fluorescence of Fluo-3 loaded PC-3 cells in the presence of extracellular Ca2+ (1 mM). Fluo-3/AM fluorescence was recorded every 20 sec in duplicate and the readings averaged. C) Effects of treatment on expression of SERCA2. PC-3 cells were incubated for 24 h with NSC735847 (16 μM) or thapsigargin (100 nM) and total cell lysates probed by western blotting for expression of SERCA2 and β-actin. D) Dimer treatment induces oxidative modification of SERCA. Aliquots (50 μg) of SERCA were treated with increasing concentration of artemisinin, NSC735847, NSC735847DX or thapsigargin, and incubated over night at 37°C. Protein samples were labeled with ThioGlo1, a fluorescent reduced cysteine reactive molecule. After 2 h, labeled protein samples were resolved by SDS-PAGE and fluorescence intensities quantified by UV transillumination and densitometry. E) NSC735847 and NSC735847DX have superimposable SERCA inhibitory activity. Ca2+-dependent ATPase activity was measured with 15.5 μg/mL rat skeletal muscle SR at varying concentrations of thapsigargin (1), NSC735847 (2), or NSC735847DX (3) by a coupled enzyme assay. -[14C]-leucine cell viability assays were performed at least twice with triplicate determinations for each point and the data pooled. All other assays were replicated two to three times.
The direct effects of NSC735847 on SERCA with respect to 1) abundance changes, 2) oxidative modifications and 3) effects on Ca2+-ATPase activity were subsequently investigated. First, cells were treated with NSC735847 or thapsigargin for 24 h and probed for changes in expression of SERCA2 (Fig. 5C). Results demonstrated marked increases in SERCA2 expression following both NSC735847 and thapsigargin treatment. To address questions related to direct effects on SERCA protein, SR was isolated from rat skeletal muscle. This preparation was confirmed by mass spectrometry to be highly enriched for SERCA1 (results not shown). The extent of oxidative modification of SERCA after treatment was assessed using ThioGlo-1, a fluorescent compound that reacts selectively with reduced cysteines. Aliquots of SR were incubated with study agents, labeled with ThioGlo-1 and the extent of SERCA protein fluorescence determined after SDS-PAGE (Fig. 5D). Results demonstrated that NSC735847 decreased the amount of reduced cysteines in SERCA1, independent of the presence of heme. Conversely, artemisinin, NSC735847DX and thapsigargin had no effect on the quantity of reduced cysteines. Changes in SERCA Ca2+-ATPase activity after treatment were determined using SR in the context of an NADH-linked spectrophotometric assay. Results confirmed that thapsigargin was the most potent inhibitor with IC50 in the nanomolar range, whereas NSC735847 inhibited activity in the micromolar range (Fig. 5E). Unexpectedly, NSC735847DX had identical activity to the parent dimer in terms of SERCA inhibition. In this assay, artemisinin was also inactive against SERCA1 (results not shown).
Discussion
In this study, to confirm and extend previous reports, two artemisinin dimers NSC724910 and NSC735847, were investigated with respect to anti-cancer activity and mechanism of action. In cytotoxicity and apoptosis assays, the dimers were confirmed to be active in the sub-micromolar range, which is exponentially more active than artemisinin monomer, supporting previous activity differentials 5, 6. This fact provided the first evidence that artemisinin dimers have unique biological properties. The anti-malarial activity of artemisinin is thought to be mediated to some degree by ROS 9, 26. Interaction with Fe2+ or heme leads to bioreductive cleavage of the endoperoxide group and generation of carbon centered free radicals and ROS, which damage the Plasmodium parasite 27. Deoxyartemisinin derivatives are therefore devoid of anti-malarial activity 9. With respect to anti-cancer activity, in cells engineered to resist methotrexate and doxorubicin, artesunate is still active, whereas cells resistant to a direct oxidative stress such as H2O2, are resistant to artesunate 4, 28, 29. Here, the fact that cells treated with dimer experienced a rapid (<1 hr) increase in reactive oxygen species (ROS) production, combined with the ability of the anti-oxidant L-NAC to impair cytotoxicity and the inactivity of a deoxy-derivative (DX) of NSC 735847, served to reinforce the role of ROS as an effector. A correlation also existed between the extent of ROS generation for monomeric and dimeric forms with their respective IC50 values, reinforcing the suggestion that ROS is the primary effector. However, the basis for increased ROS production remains unclear; possible reasons include enhanced uptake of dimer relative to monomer, altered subcellular localization of dimer, or altered metabolism of the dimer compared to the monomer.
Several studies have illustrated the importance of iron/heme metabolism in artemisinin anti-cancer activity; e.g., in a fibrosarcoma model, co-administration of ferrous sulfate was an absolute requirement for dihydroartemisinin activity 4, while combining artemisinin derivatives with holotransferrin enhanced activity 10. Thus, experiments were performed to determine the relationship between heme and artemisinin dimer activity. Using a UV-Vis spectroscopic assay 30, NSC724910 and NSC735847 showed complete interaction with hemin at a ratio of 1:1 (data not shown). It should be noted, however, that in the cellular context, HMOX1 generates the antioxidant, bilirubin, which may impact the cells ability to tolerate the ROS generated by NSC735847 and that this may account for some aspect of the effect seen with CoPPIX and SnPPIX
Microarray analysis of NSC735847 treated cells provided insights into the transcripts and pathways perturbed by treatment. Interestingly, HMOX1 mRNA was upregulated after treatment, while transcripts involved in heme synthesis (CPOX, UROS and ALAD) were downregulated. Decreased expression of the transferrin receptor (TFR1) and elevated ferritin heavy chain (FTH) mRNA show that cellular control of iron metabolism is tightly regulated in treated cells. Gene expression analysis also identified changes for genes involved in DNA/RNA metabolism and repair (ATF3, DDIT3 and GADD34). This result was noteworthy given a recent report showing that artesunate was capable of inducing DNA damage and repair 31. This response is most likely a consequence of cellular adaptation to non-specific ROS production. The lack of conserved change for cell cycle regulators (CCNA1, CDCA7, CCPG1, BEX2 and POLE2) did not point to a defined arrest point, supporting data from initial experiments showing apoptosis without phase-specific blockade. However, artemisinin has recently been shown to induce G1-specific arrest in LNCAP prostate cancer cells through Sp1 mediated down-regulation of CDK4 32. Given that Sp1 is regulated by oxidative stress it is interesting to speculate whether arrest in this line is ROS driven33. Similarly, in pancreatic cancer lines, dihydroartemisinin has been shown to downregulate PCNA and upregulate cyclin D1, two molecular adaptations that also occur with oxidative stress 34, 35.
A central observation concerned altered expression of ATF6, EIF2AK2, GRP78 and HERPUD1 mRNA, suggesting existence of an unfolded protein response (UPR) and generalized ER stress. The UPR is a specialized ER localized stress response intended to prevent accumulation of damaged proteins 36. Once activated, the UPR increases protein-folding capacity while temporarily suspending translation and increasing the rate of ER associated protein degradation. The UPR is mediated by three sensors: IRE1, PERK and ATF6 37–39. Under resting conditions these factors bind the abundant ER chaperone Bip/Grp78 and are maintained in an inactive form. Upon accumulation of unfolded proteins in the ER, Bip/Grp78 (HSPA5) is released which results in the oligomerization of IRE1 and PERK and the translocation of ATF6 to the Golgi 40, 41. In the Golgi, ATF6 is sequentially cleaved by S1P serine protease and the metalloprotease S2P releasing the cytosolic domain, this domain is then translocated to the nucleus where transactivation of chaperone genes through interactions with ATF/cAMP (CRE) and ER stress response elements (ERSE) occurs 42, 43.
Identification of an ER stress response after dimer treatment is pertinent given that artemisinin has recently been shown to inhibit PfATP6, the P. falciparum orthologue of mammalian ER-resident Ca2+-ATPases (SERCAs) 4. In mammalian systems, inhibition of SERCA is recognized as one pathway leading to induction of ER stress 44, 45. Although the artemisinin monomer, artemisone, was previously shown to have little effect on mammalian SERCA 46, the increased potency of artemisinin dimers and their ability to induce UPR transcripts, necessitated a study into whether the dimers share similarities with specific SERCA inhibitors such as thapsigargin 19, 20.
Quantitative real-time PCR for a subset of genes first showed that NSC735847 and thapsigargin had near identical treatment profiles, surpassing the correlation found between artemisinin and the dimer. However, an examination of UPR-related changes by western blotting highlighted several differences between thapsigargin and NSC735847. Although dimer and thapsigargin both decreased protein expression of caspase 12 and enhanced GRP78, ATF6 cleavage was only seen with thapsigargin, while calnexin levels only declined with dimer. Cell based assays indicated that, unlike NSC735847, thapsigargin treatment did not induce ROS in the short-term and pre-treatment with L-NAC did not prevent thapsigargin cytotoxicity. Therefore, both NSC735847 and thapsigargin induce ER stress, but the effectors and pathways affected differ.
The activities of thapsigargin and NSC735847 were further differentiated by examining effects on cellular calcium homeostasis and SERCA. The cytotoxicity of both compounds was impaired using a calcium chelator, both agents promoted increases in cytosolic free Ca2+ and increased protein expression for SERCA2, however, direct effects of NSC735847 and thapsigargin on SERCA differed considerably. Using a preparation of SERCA1 isolated as sarcoplasmic reticulum from rat skeletal muscle, it was first shown that dimer, but not thapsigargin, was capable of modifying reduced cysteines on SERCA1, emphasizing the oxidative component of dimer activity. Crucially, thapsigargin was > 103 times more potent inhibitor of SERCA Ca2+-ATPase activity than NSC735847 or NSC735847DX, which had overlapping profiles.
Given that NSC735847 and NSC735847DX have identical inhibitory activity against SERCA1, the oxidative modifications that accompany the former compound treatment obviously have little effect on overall Ca2+ ATPase activity. This result fits with literature reports suggesting that SERCA can undergo substantial oxidative modification, without impacting function 47, 48. Furthermore, because both compounds have identical SERCA inhibitory activity and the deoxy-derivative has negligible anticancer activity, these data suggest that direct inhibition of SERCA plays a minimal role in ER stress induction and overall activity.
In conclusion, treatment of cells with artemisinin dimers is associated with ROS generation, a DNA-damage/ER stress response, calcium mobilization and the induction of apoptosis. In several regards, this mode of action is similar to the SERCA Ca2+-ATPase inhibitor thapsigargin. However, artemisinin dimer induced ER stress is not dependent on direct SERCA inhibition. We speculate that, like the observed DNA damage response, ER stress induction is likely a function of localized ROS generation. A preliminary investigation suggests that for a panel of classical anticancer agents, NSC735847 effectively synergizes with the proteasome inhibitor, bortezomib, and the toposisomerase inhibitors, etoposide and topotecan (results not shown). Therefore, the data generated in this study suggests that for artemisinin dimers, continued preclinical development with respect to disease indication, toxicology and pharmacokinetics appears warranted.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute. Disclosure of potential conflict of interest: authors M.A.E., W.G., and A.M.G. have a patent application covering the anticancer activity of the artemisinin dimers described herein.
Footnotes
Category: Carcinogenesis
In this study, we extend previous studies investigating the anti-cancer activity of dimeric artemisinins. We show that activity is critically dependent on an intact endoperoxide group and can be modulated by iron-chelators, exogenous heme and anti-oxidants. This provides compelling evidence that heme-catalzyed endoperoxide decay followed by reactive oxygen species (ROS) generation is a central mechanism. Dimer activity was also shown to induce an ER stress/DNA damage response. We then demonstrate that the ER-stress response induced by dimer is not SERCA-dependent, a key finding given that in malaria, artemisinins inhibit pfATP6, the orthologue of mammalian SERCA Ca2+ transporters.
Therefore, the work presented here suggests that artemisinin dimers catalyze the generation of ROS from cellular heme centers leading to SERCA-independent ER-stress induction and apoptosis. On this basis, continued pre-clinical evaluation of this group of compounds appears warranted.
References
- 1.White NJ. Qinghaosu (artemisinin): the price of success. Science. 2008;320:330–4. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 2.Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. Science. 1985;228:1049–55. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Wu YL. How Chinese scientists discovered qinghaosu (artemisinin) and developed its derivatives? What are the future perspectives? Med Trop (Mars) 1998;58:9–12. [PubMed] [Google Scholar]
- 4.Efferth T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr Drug Targets. 2006;7:407–21. doi: 10.2174/138945006776359412. [DOI] [PubMed] [Google Scholar]
- 5.Paik IH, Xie S, Shapiro TA, Labonte T, Narducci Sarjeant AA, Baege AC, Posner GH. Second generation, orally active, antimalarial, artemisinin-derived trioxane dimers with high stability, efficacy, and anticancer activity. J Med Chem. 2006;49:2731–4. doi: 10.1021/jm058288w. [DOI] [PubMed] [Google Scholar]
- 6.Nam W, Tak J, Ryu JK, Jung M, Yook JI, Kim HJ, Cha IH. Effects of artemisinin and its derivatives on growth inhibition and apoptosis of oral cancer cells. Head Neck. 2007;29:335–40. doi: 10.1002/hed.20524. [DOI] [PubMed] [Google Scholar]
- 7.Golenser J, Waknine JH, Krugliak M, Hunt NH, Grau GE. Current perspectives on the mechanism of action of artemisinins. Int J Parasitol. 2006;36:1427–41. doi: 10.1016/j.ijpara.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Efferth T. Willmar schwabe award 2006: antiplasmodial and antitumor activity of artemisinin--from bench to bedside. Planta Med. 2007;73:299–309. doi: 10.1055/s-2007-967138. [DOI] [PubMed] [Google Scholar]
- 9.O’Neill PM, Posner GH. A medicinal chemistry perspective on artemisinin and related endoperoxides. J Med Chem. 2004;47:2945–64. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 10.Singh NP, Lai H. Selective toxicity of dihydroartemisinin and holotransferrin toward human breast cancer cells. Life Sci. 2001;70:49–56. doi: 10.1016/s0024-3205(01)01372-8. [DOI] [PubMed] [Google Scholar]
- 11.Anfosso L, Efferth T, Albini A, Pfeffer U. Microarray expression profiles of angiogenesis-related genes predict tumor cell response to artemisinins. Pharmacogenomics J. 2006;6:269–78. doi: 10.1038/sj.tpj.6500371. [DOI] [PubMed] [Google Scholar]
- 12.McCarty MF. Turning an ‘Achilles’ Heel’ into an asset--activation of HIF-1alpha during angiostatic therapy will increase tumor sensitivity to iron-catalyzed oxidative damage. Med Hypotheses. 2003;61:509–11. doi: 10.1016/s0306-9877(03)00229-9. [DOI] [PubMed] [Google Scholar]
- 13.Chen HH, Zhou HJ, Wu GD, Lou XE. Inhibitory effects of artesunate on angiogenesis and on expressions of vascular endothelial growth factor and VEGF receptor KDR/flk-1. Pharmacology. 2004;71:1–9. doi: 10.1159/000076256. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Zhang B, Guo Y, Li G, Xie Q, Zhu B, Gao J, Chen Z. Artemisinin inhibits tumor lymphangiogenesis by suppression of vascular endothelial growth factor C. Pharmacology. 2008;82:148–55. doi: 10.1159/000148261. [DOI] [PubMed] [Google Scholar]
- 15.Stockwin LH, Bumke MA, Yu SX, Webb SP, Collins JR, Hollingshead MG, Newton DL. Proteomic analysis identifies oxidative stress induction by adaphostin. Clin Cancer Res. 2007;13:3667–81. doi: 10.1158/1078-0432.CCR-07-0025. [DOI] [PubMed] [Google Scholar]
- 16.Johnson AJ, Hsu AL, Lin HP, Song X, Chen CS. The cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2+-ATPases: a plausible link with its anti-tumour effect and cardiovascular risks. Biochem J. 2002;366:831–7. doi: 10.1042/BJ20020279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kijima Y, Ogunbunmi E, Fleischer S. Drug action of thapsigargin on the Ca2+ pump protein of sarcoplasmic reticulum. J Biol Chem. 1991;266:22912–8. [PubMed] [Google Scholar]
- 18.Schulze A, Standera S, Buerger E, Kikkert M, van Voorden S, Wiertz E, Koning F, Kloetzel PM, Seeger M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354:1021–7. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–61. doi: 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- 20.Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci. 1998;19:131–5. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- 21.Ridley RG. Malaria: to kill a parasite. Nature. 2003;424:887–9. doi: 10.1038/424887a. [DOI] [PubMed] [Google Scholar]
- 22.Takemura H, Hughes AR, Thastrup O, Putney JW., Jr Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J Biol Chem. 1989;264:12266–71. [PubMed] [Google Scholar]
- 23.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kass GE, Duddy SK, Moore GA, Orrenius S. 2,5-Di-(tert-butyl)-1,4-benzohydroquinone rapidly elevates cytosolic Ca2+ concentration by mobilizing the inositol 1,4,5-trisphosphate-sensitive Ca2+ pool. J Biol Chem. 1989;264:15192–8. [PubMed] [Google Scholar]
- 25.Mason MJ, Garcia-Rodriguez C, Grinstein S. Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5-di-(tert-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. J Biol Chem. 1991;266:20856–62. [PubMed] [Google Scholar]
- 26.Meshnick SR, Taylor TE, Kamchonwongpaisan S. Artemisinin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiol Rev. 1996;60:301–15. doi: 10.1128/mr.60.2.301-315.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cumming JN, Ploypradith P, Posner GH. Antimalarial activity of artemisinin (qinghaosu) and related trioxanes: mechanism(s) of action. Adv Pharmacol. 1997;37:253–97. doi: 10.1016/s1054-3589(08)60952-7. [DOI] [PubMed] [Google Scholar]
- 28.Efferth T, Dunstan H, Sauerbrey A, Miyachi H, Chitambar CR. The anti-malarial artesunate is also active against cancer. Int J Oncol. 2001;18:767–73. doi: 10.3892/ijo.18.4.767. [DOI] [PubMed] [Google Scholar]
- 29.Efferth T, Volm M. Glutathione-related enzymes contribute to resistance of tumor cells and low toxicity in normal organs to artesunate. In Vivo. 2005;19:225–32. [PubMed] [Google Scholar]
- 30.Bilia AR, Lazari D, Messori L, Taglioli V, Temperini C, Vincieri FF. Simple and rapid physico-chemical methods to examine action of antimalarial drugs with hemin: its application to Artemisia annua constituents. Life Sci. 2002;70:769–78. doi: 10.1016/s0024-3205(01)01447-3. [DOI] [PubMed] [Google Scholar]
- 31.Li PC, Lam E, Roos WP, Zdzienicka MZ, Kaina B, Efferth T. Artesunate derived from traditional Chinese medicine induces DNA damage and repair. Cancer Res. 2008;68:4347–51. doi: 10.1158/0008-5472.CAN-07-2970. [DOI] [PubMed] [Google Scholar]
- 32.Willoughby JA, Sr, Sundar SN, Cheung M, Tin AS, Modiano J, Firestone GL. Artemisinin blocks prostate cancer growth and cell cycle progression by disrupting Sp1 interactions with the cyclin-dependent kinase-4 (CDK4) promoter and inhibiting CDK4 gene expression. J Biol Chem. 2009;284:2203–13. doi: 10.1074/jbc.M804491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ammendola R, Mesuraca M, Russo T, Cimino F. The DNA-binding efficiency of Sp1 is affected by redox changes. Eur J Biochem. 1994;225:483–9. doi: 10.1111/j.1432-1033.1994.t01-1-00483.x. [DOI] [PubMed] [Google Scholar]
- 34.Chen H, Sun B, Pan S, Jiang H, Sun X. Dihydroartemisinin inhibits growth of pancreatic cancer cells in vitro and in vivo. Anticancer Drugs. 2009;20:131–40. doi: 10.1097/CAD.0b013e3283212ade. [DOI] [PubMed] [Google Scholar]
- 35.Barnouin K, Dubuisson ML, Child ES, Fernandez de Mattos S, Glassford J, Medema RH, Mann DJ, Lam EW. H2O2 induces a transient multi-phase cell cycle arrest in mouse fibroblasts through modulating cyclin D and p21Cip1 expression. J Biol Chem. 2002;277:13761–70. doi: 10.1074/jbc.M111123200. [DOI] [PubMed] [Google Scholar]
- 36.Schroder M. The unfolded protein response. Mol Biotechnol. 2006;34:279–90. doi: 10.1385/MB:34:2:279. [DOI] [PubMed] [Google Scholar]
- 37.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 38.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 39.Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, Iseki K, Wanaka A, Imaizumi K. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat Cell Biol. 2005;7:186–94. doi: 10.1038/ncb1213. [DOI] [PubMed] [Google Scholar]
- 40.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 41.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–20. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–9. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 44.Caspersen C, Pedersen PS, Treiman M. The sarco/endoplasmic reticulum calcium-ATPase 2b is an endoplasmic reticulum stress-inducible protein. J Biol Chem. 2000;275:22363–72. doi: 10.1074/jbc.M001569200. [DOI] [PubMed] [Google Scholar]
- 45.Hojmann Larsen A, Frandsen A, Treiman M. Upregulation of the SERCA-type Ca2+ pump activity in response to endoplasmic reticulum stress in PC12 cells. BMC Biochem. 2001;2:4. doi: 10.1186/1471-2091-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uhlemann AC, Cameron A, Eckstein-Ludwig U, Fischbarg J, Iserovich P, Zuniga FA, East M, Lee A, Brady L, Haynes RK, Krishna S. A single amino acid residue can determine the sensitivity of SERCAs to artemisinins. Nat Struct Mol Biol. 2005;12:628–9. doi: 10.1038/nsmb947. [DOI] [PubMed] [Google Scholar]
- 47.Sharov VS, Dremina ES, Galeva NA, Williams TD, Schoneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J. 2006;394:605–15. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viner RI, Ferrington DA, Huhmer AF, Bigelow DJ, Schoneich C. Accumulation of nitrotyrosine on the SERCA2a isoform of SR Ca-ATPase of rat skeletal muscle during aging: a peroxynitrite-mediated process? FEBS Lett. 1996;379:286–90. doi: 10.1016/0014-5793(95)01530-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.