

Abstract
The “Spanish influenza” of 1918 claimed an unprecedented number of lives, yet the determinants of virulence for this virus are still not fully understood. Here, we used functional genomics and an in vitro human lung epithelial cell infection model to define the global host transcriptional response to the eight-gene 1918 virus. To better understand the role of the 1918 virus NS1 gene, we also evaluated the host response to a reassortant 1918 virus containing the NS1 gene from A/Texas/36/91 (a seasonal isolate of human influenza virus), as well as the host response to a reassortant of A/Texas/36/91 containing the 1918 NS1 gene. Genomic analyses revealed that the 1918 virus blocked the transcription of multiple interferon-stimulated genes and also downregulated a network of genes associated with lipid metabolism. In contrast, the expression of genes encoding chemokines and cytokines, which serve to attract infiltrating immune cells, was upregulated. Viruses containing the NS1 gene from A/Texas/36/91 induced a significant increase in type I interferon signaling but did not repress lipid metabolism. The 1918 NS1 gene may therefore have contributed to the virulence of the 1918 pandemic virus by disrupting the innate immune response, inducing hypercytokinemia, and by blocking the transcription of certain lipid-based proinflammatory mediators that function as part of the host antiviral response.
The influenza pandemic of 1918 was the worst in recorded history, killing an estimated 50 million people (22). Nevertheless, the factors underlying the extreme virulence of this virus remain obscure. A significant step in unraveling this mystery came with the reconstruction of the 1918 virus using sequence data from various preserved, infected human lung tissue samples (48). Modern reverse genetics techniques have also made it possible to construct chimeric viruses, including viruses containing gene segments from the 1918 virus and from more recent human influenza virus isolates, to study the role of specific viral genes in virulence. Studies using such chimeric viruses indicate that multiple genes from the 1918 influenza virus may have contributed to its extreme lethality, including the hemagglutinin (HA), neuraminidase (NA), polymerase (PB1 and PB1-F2), and nonstructural 1 (NS1) genes (3, 12, 16, 21, 24, 39, 47, 49, 50).
The NS1 gene is of particular interest and is the subject of a considerable body of literature that describes a multitude of mechanisms by which NS1 may function and thereby contribute to pathogenesis (1, 4, 9, 17, 20, 25, 28, 31-36, 38, 42, 53). The NS1 gene of type A influenza virus encodes a protein of approximately 26.8 kDa, which functions as a homodimer during viral replication (10, 11, 18). The NS1 homodimer binds single- or double-stranded RNA (dsRNA) and has been reported to block the actions of key cellular proteins (including RIG-I, PKR, CPSF, PABPII, and 2′-5′ OAS) as well as to induce the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway (4, 9, 13, 17, 19, 28, 29, 32-36, 38, 41, 43, 44, 46, 54). More recently, it was found that NS1 inhibits induction of type I interferon (IFN) by binding to the E3 ubiquitin ligase TRIM25, required for optimal activation of the viral sensor RIG-I (14). NS1 may also selectively enhance the translation of viral mRNA transcripts by binding to cellular eIF4G1, and it is required for viral replication in the presence of an intact type I IFN response (1, 15).
We previously showed that a chimeric mouse-adapted A/WSN/33 influenza virus, containing the NS1 gene from the 1918 virus, blocked the expression of IFN-regulated genes more efficiently than the parental strain A/WSN/33 did, suggesting that the 1918 NS1 protein may be particularly adept at enabling the virus to evade host defense mechanisms (16). We now expand upon this earlier work by defining the global host transcriptional response to the eight-gene 1918 virus and to a chimeric 1918 virus containing the NS gene from influenza virus A/Texas/36/91 (a seasonal isolate of human H1N1 influenza virus), as well as the host response to a chimeric A/Texas/36/91 virus containing the 1918 NS1 gene. The current studies embody a significant advance over the previous study using the A/WSN/33 parent strain in part because of major improvements in microarray technology, human genome annotation, and genomics analysis software. We also sampled later time points as part of our experimental design and used a human-adapted parent virus, A/Texas/36/91, compared to the mouse-adapted A/WSN/33 virus. Human A549 lung epithelial cells (a type II pneumocyte-derived cell line) were used as the infection model. The use of cultured cells allowed us to analyze the host transcriptional response of infected cells in the absence of infiltrating immune cells that can confound analyses performed using lung tissues from infected animals. We report here that the 1918 NS1 gene may have contributed to the virulence of the 1918 virus by disrupting the innate immune response, inducing hypercytokinemia, and by blocking the transcription of certain lipid-based proinflammatory mediators. While the current studies were not intended to be mechanistic by design, they have led to important advances in our understanding about the host response to highly pathogenic influenza viruses and have also contributed to intriguing new hypotheses (mechanistic in nature) regarding the impact of cooperativity between viral gene segments on pathogenesis.
MATERIALS AND METHODS
Virus preparation.
For the A/Texas/36/91 (Tx/91) virus and a chimeric 1918 influenza virus containing the NS gene from influenza virus A/Texas/36/91 [Tx/91:NS(1918)], Madin-Darby canine kidney (MDCK) cells were cultured in growth medium containing minimum essential medium (MEM) and 5% fetal bovine serum (FBS). 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. The Tx/91 and Tx/91:1918(NS) viruses were generated from plasmid DNA by reverse genetics techniques (45). They were next propagated in the allantoic cavities of 7-day-old embryonated chicken eggs, concentrated, purified by high-speed centrifugation of infected allantoic fluid passed through a 30% sucrose density gradient, and suspended in calcium borate buffer. The sequences of both viruses were confirmed by real-time PCR (RT-PCR) and sequence analysis. For the 1918 and 1918:Tx/91(NS) viruses, MDCK cells and 293T cells were cultured in growth medium containing DMEM and 10% FBS. The 1918 and 1918:Tx/91(NS) viruses were generated from plasmid DNA by reverse genetics techniques (45) and sequenced. The rescued viruses were propagated in MDCK cells for 48 h. The supernatant was collected, and the virus titers were determined by plaque assay.
A549 cell infections.
For the Tx/91 and Tx/91:1918(NS) infections, A549 cells were propagated in MEM supplemented with 10% FBS and penicillin-streptomycin. Cells at low passage were seeded onto six-well plates and infected 2 days later at a multiplicity of infection (MOI) of 2. Mock-infected controls were treated with allantoic fluid instead of virus. Virus was allowed to bind to cells for 1 h at 4°C in serum-free infection medium supplemented with trypsin (1 μg/ml). Three replicate wells were used for each infection condition at each time point. Cells were lysed in solution D (4 M guanidinium thiocyanate, 25 mM sodium citrate, 0.5% sarcosyl, 0.1 M beta-mercaptoethanol) at 2, 6, and 24 h postinfection. Total RNA was extracted, purified, and checked for integrity as previously described (2). For the 1918 and 1918:Tx/91(NS) infections, A549 cells were propagated in DMEM supplemented with 10% FBS and penicillin-streptomycin. Cells were seeded onto six-well plates and used the second day. Mock-infected controls were treated only with DMEM containing 0.3% bovine serum albumin. Cells were infected with indicated viruses at an MOI of 2 in 500 μl of DMEM for 1 h and washed. DMEM (2 ml) with 0.3% bovine serum albumin was added to each well. Cells were lysed in 0.5 ml of solution D at 2, 6, and 24 h postinfection. Total RNA was extracted, purified, and checked for integrity as previously described (2). Three replicate wells were used for each infection condition at each time point.
Microarray analysis.
Biological replicates of infected cells were screened for influenza virus HA and NS1 mRNA levels using quantitative real-time PCR (qRT-PCR). For the 1918 and 1918:Tx/91(NS) infections, each individual sample was hybridized with pooled RNA from time-matched, mock-infected controls. For the Tx/91 and Tx/91:1918(NS) infections, total RNA from three biological replicates each was pooled in equivalent amounts. Thus, triplicate samples were combined to make one set of probes for microarray hybridization. Pooled RNA from each infection condition was hybridized with pooled RNA from mock-infected replicates. For all microarray experiments, Cy3- and Cy5-labeled cRNA probes were generated using the Agilent low RNA input linear amplification kit. Agilent human 4,000 by 44,000 microarrays were used according to the manufacturer's instructions in the Agilent 60-mer oligonucleotide microarray processing protocol.
Each microarray experiment was done with four technical replicates by reversing dye hybridization for experimental and reference samples. Slides were scanned with an Agilent DNA microarray scanner, and image data were processed using Agilent Feature Extractor software, which also performed error modeling. All data were subsequently uploaded into Rosetta Resolver for data analysis. In accordance with proposed MIAME (minimum information about a microarray experiment) standards (5, 6), all data described in this report, including sample information, intensity measurements, microarray content, and slide hybridization conditions, are publically available at http://viromics.washington.edu.
The Rosetta Resolver system performs a squeeze operation that creates ratio profiles by combining replicates while applying error weighting. The error weighting consists of adjusting for additive and multiplicative noise. A P value that represents the probability that a gene is differentially expressed is generated. In this study, a threshold P value of 0.01 was used to identify genes that were significantly differentially expressed. The Rosetta Resolver system then combines ratio profiles to create ratio experiments using an error-weighted average as described previously (45a). For each microarray experiment, the calculation of mean ratios between expression levels of each gene in the analyzed sample pair, standard deviations, and P values was performed using Rosetta Resolver.
To perform direct comparisons between the different viruses, we utilized the reratio tool in Rosetta Resolver. This tool creates new ratio experiments from two or more existing ratio experiments that share a common reference, therefore removing the reference and focusing on the difference between two conditions. Direct comparisons were created using the reratio tool, resulting in 24-h 1918 versus 1918:Tx/91(NS) and 24-h Tx/91 versus Tx/91:1918(NS), respectively.
Rosetta Resolver was also used to compare the expression levels of genes in infected cells versus the corresponding mock-infected cells for each experiment. Specifically, genes identified as being significantly differentially expressed in our direct comparisons of each virus pair were then analyzed to measure their expression versus mock-infected cells. In the latter analysis, we filtered out any genes that were less than twofold different in expression (P < 0.01) relative to mock-infected controls.
IPA.
Interactive gene networks were generated through the use of Ingenuity Pathways Analysis (IPA) (Ingenuity Systems, Redwood City, CA). Networks were generated using a data set containing gene identifiers and corresponding expression values. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. Differentially expressed genes were defined as having a ≥2-fold change in expression (P ≤ 0.01). These genes, termed focus genes, were overlaid onto a global molecular network developed from information contained in the Ingenuity Pathways Knowledge Base. Networks of focus genes were then algorithmically generated on the basis of their connectivity. The IPA Functional Analysis identified the biological functions that were most significant to entire data sets or particular networks. The genes from the data set or network that met the criteria for differential expression and that were associated with biological functions in the Ingenuity Pathways Knowledge Base, were considered for the analysis. The right-tailed Fisher exact test was used to calculate a P value determining the probability that each biological function assigned to that data set or network was due to chance alone. In the right-tailed Fisher exact test, only overrepresented functions—those that have more function-eligible molecules than expected by chance—are significant. Underrepresented functions (“left-tailed” P values) are not shown. Fisher's exact test, which is computationally less expensive and widely used, was chosen over other types of P value calculations because the random model was used for the statistical null model. The Benjamini-Hochberg method of multiple testing correction returns adjusted P values and enables one to control the noise in certain functional analysis results. This corrected P value can be interpreted as an upper bound for the expected fraction of false-positive results. For example, if the threshold is 0.01, one can expect that the fraction of false-positive results among the significant functions is less than 1%. We used IPA software (Ingenuity Systems, Redwood City, CA) for the Benjamini-Hochberg formula and statistical algorithms.
IPA networks are graphical representations of the molecular relationships between genes or gene products. Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). The intensity of the node color indicates the degree of up- or downregulation.
qRT-PCR.
We used qRT-PCR to validate certain transcriptional changes found by microarray analysis and to validate the extent of replication of live viruses in each biological replicate experiment. Total RNA samples were treated with DNase using DNA-free DNase treatment and removal reagents (Ambion, Inc., Austin, TX). Reverse transcription was performed using TaqMan reverse transcription reagents (Applied Biosystems, Foster City, CA). Primer and probe sets for each of the target sequences were chosen from the Applied Biosystems assays-on-demand product list. qRT-PCR was performed on an ABI 7500 real-time PCR system, using TaqMan chemistry. Each target was run in quadruplicate, with 20-μl reaction mixture volumes of TaqMan 2× PCR universal master mix. rRNA (18S) was chosen as the endogenous control to normalize quantification of the target within the Applied Biosystems Sequence Detections Software version 1.3. Quantification of each gene, relative to the calibrator, was calculated using the 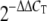 equation (30).
equation (30).
Western blot.
Following influenza virus infection, cells were lysed at the indicated times postinfection in disruption buffer (0.5% Triton X-100, 50 mM KCl, 50 mM NaCl, 20 mM Tris-HCl [pH 7.5], 1 mM EDTA, 10% glycerol, 1× Complete protease inhibitor [Roche, Indianapolis, IN], 25 mM β-glycerophosphate, 1 mM Na3VO4). Lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride paper. Immunoblots were blocked for 1 h in phosphate-buffered saline (PBS) containing 0.5% Tween 20 and 5% nonfat dry milk, washed in PBS containing 0.05% Tween 20, and incubated at 4°C overnight with rabbit anti-influenza virus NS1 or nucleoprotein (NP) antibody or a mouse antibody recognizing actin (MP Biochemicals, Irvine, CA) in PBS containing 0.5% Tween 20 and 1% nonfat dry milk. The membranes were washed, incubated for 2 h with horseradish peroxidase-conjugated donkey anti-mouse or anti-rabbit immunoglobulin G (Jackson Immunoresearch, West Grove, PA), and bound antibodies were detected with ECL Western blotting detection reagent (Amersham Biosciences/GE Healthcare).
RESULTS
Replication kinetics of parental and chimeric viruses.
To define the host transcriptional response to the 1918 influenza virus and to determine the role of the NS1 gene in inducing this response, we made use of a panel of wild-type (WT) and chimeric viruses. These viruses included the eight-gene 1918 pandemic H1N1 virus, A/Texas/36/91 (a seasonal isolate of human H1N1 influenza virus), and two chimeric viruses in which the NS gene segment of the 1918 and A/Texas/36/91 viruses was interchanged to generate 1918:Tx/91(NS) (the 1918 virus with the A/Texas/36/91 NS1 gene) and Tx/91:1918(NS) (the A/Texas/36/91 virus with the 1918 NS1 gene).
To determine whether these influenza viruses had similar replication kinetics, we infected A549 lung epithelial cells at an MOI of 2 and harvested total RNA at 2, 6, and 24 h postinfection. Quantitative real-time PCR was then used to measure viral NS1 and HA RNA levels at each time point relative to mock-infected cells. We found that the 1918 virus produced approximately 1 to 2 log units more viral RNA than did the 1918:Tx/91(NS) virus at each time point (Fig. 1A and B). Both viruses showed robust replication patterns, with viral NS1 and HA RNA levels increasing consistently over time. Thus, although these viruses have been reported to replicate at similar levels in normal human bronchial epithelial cells, a primary multicellular culture system (39), substituting the Tx/91 NS gene into the 1918 virus appeared to lower the amount of viral RNA produced in A549 cells. We next compared viral NS1 and NP protein levels for the 1918 and 1918:Tx/91(NS) viruses (Fig. 1E). By 24 h postinfection, viral protein expression was significant for each of the viruses, despite the differences in viral RNA levels observed between these viruses at 24 h.
FIG. 1.

Viral NS1 (A and C) and HA (B and D) transcript levels at 2, 6, and 24 hours postinfection in A549 human lung epithelial cells as measured by quantitative real-time PCR. Average log10 ratio values were calculated by averaging threshold cycle (CT) values from four technical replicates in each case and using the 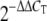 equation. Endogenous 18S rRNA was used as an internal control for each assay. (E) Western blot showing viral NS1 and NP protein expression at 2, 6, and 24 h postinfection (h p.i.). Actin was used as a loading control.
equation. Endogenous 18S rRNA was used as an internal control for each assay. (E) Western blot showing viral NS1 and NP protein expression at 2, 6, and 24 h postinfection (h p.i.). Actin was used as a loading control.
The Tx/91 and Tx/91:1918(NS) viruses also replicated efficiently in A549 cells, with viral RNA levels steadily increasing between 2 and 24 h. Substituting the 1918 NS gene into the Tx/91 virus did not have an effect on replication, as measured by plaque assay. At 2 h postinfection, the Tx/91 and Tx/91:1918(NS) viruses had plaque assay titers of 11.5 × 104 and 3.5 × 104 PFU/ml, respectively. At 24 h postinfection, the Tx/91 and Tx/91:1918(NS) viruses had plaque assay titers of 2.5 × 107 and 4.0 × 107 PFU/ml, respectively. Viral NS1 and HA RNA levels were nearly equivalent for both viruses at each time point (Fig. 1C and D, respectively).
Global host transcriptional response to parental and chimeric viruses.
After confirming that each virus was able to establish a robust infection in A549 cells, we next analyzed global gene expression patterns in response to infection. For these experiments, A549 cells were infected in triplicate using an MOI of 2. Total RNA was then harvested at 2, 6, and 24 h postinfection, and Agilent 4,000 × 44,000 human oligonucleotide microarrays were used to compare cellular expression patterns of infected cells with those of mock-infected cells at each time point.
Global gene expression patterns induced by the 1918 and 1918:Tx/91(NS) influenza viruses were particularly distinct at the 6- and 24-h time points, with the 1918 virus inducing considerably more gene expression changes than the 1918:Tx/91(NS) virus did (Fig. 2A). The greater host transcriptional response to the 1918 virus may have been due in part to its higher level of viral RNA expression; however, at the 2-h time point, when the difference in viral RNA levels was the greatest between the 1918 and 1918:Tx/91(NS) viruses, the cellular gene expression profiles were quite similar. Conversely, at the 24-h time point, when the viral RNA levels for the two viruses were more similar, the differences in cellular gene expression patterns were more pronounced. Therefore, there did not appear to be a strong correlation between viral RNA levels and the number of differentially expressed genes. Rather, differences in the host response appear to have been driven by the NS1 gene expressed by the virus. Because NS1 protein expression was high for both the 1918 and 1918:Tx/91(NS) viruses at 24 h postinfection, we focused on this time point for many of our microarray analyses.
FIG. 2.

Global host transcriptional response to infection. Signature genes were defined as having greater than twofold differential expression compared with expression in mock-infected cells (P < 0.01). (A) Cells infected with the 1918 influenza virus versus mock-infected cells and 1918:Tx/91(NS)- versus mock-infected cells. A total of 6,910 signature genes are shown. The three replicates are shown for each virus at each time point. (B) Tx/91-infected versus mock-infected cells and Tx/91:1918(NS)- versus mock-infected cells. A total of 7,701 signature genes are shown. Increases (red) or decreases (green) in expression level are indicated by colors.
Global gene expression patterns induced by the Tx/91 and Tx/91:1918(NS) viruses were more similar to one another (Fig. 2B), with the largest number of gene expression changes occurring at the 6- and 24-h time points. Because these viruses replicated to similar levels in A549 cells, differences in the host response again appear to have been driven by the NS1 gene expressed by the virus. Surprisingly, adding the 1918 NS1 gene to the Tx/91 virus had less of an impact on the host response than adding the Tx/91 NS1 gene to the 1918 virus did. This suggests that the NS1 gene of the 1918 virus may be maximally functional only in the context of the complete 1918 viral genome. While we cannot exclude the possibility that cooperativity between viral gene segments affected NS1 function and/or the global gene expression patterns shown in Fig. 2, it is known that viruses lacking a functional NS1 protein cannot replicate efficiently in IFN-competent systems (15). Because we saw an increase in viral mRNA levels over time for all four viruses, appreciable levels of viral proteins expressed at 24 h for the 1918 and 1918:Tx/91(NS) viruses, and an increase in viral titer over time for the WT Tx/91 and Tx/91:1918(NS) viruses, we can assume that the NS1 proteins in all four viruses are functional within their larger constellations of viral gene segments.
Direct comparison of the host transcriptional response to parental and chimeric viruses.
To further analyze the gene expression data for differences in the host response that could be attributed to the NS1 gene, we performed one-way analysis of variance (ANOVA) on data generated from the 24-h time point of the 1918 and 1918:Tx/91(NS) experiments. Our ANOVA was generated based on three biological replicates from each group. [Note that these biological replicates were not pooled for analysis, whereas the WT Tx/91 and Tx/91:1918(NS) replicates were pooled for analysis. The latter set were pooled only after it was established, using qRT-PCR, that the replicates contained equivalent viral mRNA levels and had equivalent levels expression of various cellular genes.] From the resulting list of significant (ANOVA P ≤ 0.01) genes, we utilized in silico analysis (described in Materials and Methods) to further filter gene expression patterns induced by the 1918 and 1918:Tx/91(NS) viruses. We selected genes that were at least twofold different in their expression in this direct comparison and then used Ingenuity Pathways Analysis to group those genes according to their function. This allowed us to determine whether genes within a given functional category were expressed at a higher level in response to one virus compared to the other virus and to identify categories containing large numbers of differentially expressed genes (Fig. 3A). This approach is particularly useful for depicting general trends and highlighting differences in the host response to each virus, which was one of the primary purposes of our analyses. However, the results must be interpreted carefully since the direct comparison does not provide information about gene expression changes relative to mock-infected cells. Therefore, genes identified as being differentially expressed in direct comparisons were also analyzed to measure their expression values relative to mock-infected cells (see Fig. 2, 4B, 5B, and 6B) (also see Table S1 in the supplemental material).
FIG. 3.

Functional categories of differentially expressed genes as generated using Ingenuity Pathways Analysis. (A) Direct comparison of the 1918 influenza virus versus 1918:Tx/91(NS). (B) Direct comparison of Tx/91 versus Tx/91:1918(NS). Genes from cells infected with influenza virus containing the 1918 NS gene segment are shown in black, while genes from cells infected with influenza virus containing the Tx/91 NS gene segment are shown in gray.
FIG. 4.

Regulation of IFN-stimulated gene expression in A549 cells infected with the 1918 or 1918:Tx/9(NS) influenza virus at 24 h postinfection. IFN-stimulated genes suppressed by the 1918 virus are shown. (A) Network diagram showing a subset of IFN-stimulated genes that were greater than 2-fold more downregulated by the 1918 virus compared to the 1918:Tx/91(NS) virus. Edges depicted as solid lines between nodes in the network represent direct interactions, whereas dashed lines represent indirect interactions. Darker shading indicates a greater degree of suppression. (B) Expression of 12 IFN-stimulated genes as measured by quantitative real-time PCR. Average log10 ratio values were calculated by averaging CT values from four technical replicates in each case and using the  equation. Endogenous 18S rRNA was used as an internal control for each assay.
equation. Endogenous 18S rRNA was used as an internal control for each assay.
FIG. 5.

Regulation of immune response genes. (A) IPA network diagram generated using genes from the in silico comparison of the 1918 and 1918:Tx/91(NS) influenza viruses (P < 0.01). Genes that were expressed at a higher level in cells infected with the 1918 influenza virus are shown in yellow, and genes expressed at a higher level in cells infected with the 1918:Tx/91(NS) influenza virus are shown in blue. Only direct interactions are shown. (B) Rosetta Resolver heat map of genes contained in the network depicted in panel A. Gene expression changes are relative to mock-infected cells.
FIG. 6.

Regulation of lipid metabolism gene expression in A549 cells infected with the 1918 or 1918:Tx/91(NS) influenza virus at 24 h postinfection. (A) Network of lipid metabolism genes identified by the in silico comparison of the 1918 and 1918:Tx/91(NS) viruses in A549 cells at 24 h postinfection (P < 0.01). Genes that were expressed at a higher level in cells infected with the 1918 virus are shown in yellow, and genes expressed at a higher level in cells infected with 1918:Tx/91(NS) are shown in blue. Only direct interactions are shown. (B) Expression of 11 lipid metabolism genes suppressed by the 1918 virus as measured by quantitative real-time PCR. Average log10 ratio values were calculated by averaging CT values from four technical replicates in each case and using the  equation. Endogenous 18S rRNA was used as an internal control for each assay.
equation. Endogenous 18S rRNA was used as an internal control for each assay.
Overall, this analysis revealed that both viruses induced gene expression changes in numerous functional categories. Genes associated with several of these categories, including cell signaling and immune response, tended to be more highly expressed in cells infected with the 1918 virus. Unexpectedly, genes associated with cell turnover (including the categories of cell cycle, cell death, and cell growth and proliferation) and with metabolic pathways (including the categories of amino acid, carbohydrate, and lipid metabolism) tended to be more highly expressed in cells infected with 1918:Tx/91(NS). Thus, even though the 1918 virus induced more gene expression changes overall than 1918:Tx/91 did (Fig. 2A), Tx/91 NS1 preferentially induced the expression of specific functional categories of genes. This may be due to structural differences in the two NS1 proteins, which may contribute to differential virus-host factor interactions during the course of infection.
Due to the pooling of biological replicates in the Tx/91 and Tx/91:1918(NS) experiments, we next went directly to the in silico analysis to compare the gene expression patterns induced by the Tx/91 and Tx/91:1918(NS) viruses. Consistent with the findings described above, we found that genes associated with cell turnover and metabolic pathways tended to be expressed at higher levels in cells infected with the Tx/91 virus (Fig. 3B). Since a similar trend was observed in cells infected with 1918:Tx/91(NS), this host response appears to be correlated with viral expression of the Tx/91 NS1 gene. In contrast, genes associated with the category of immune response also tended to be expressed at higher levels in cells infected with the Tx/91 virus. Therefore, the expression of such genes was elevated in response to both the parental 1918 and Tx/91 viruses compared with the response to the chimeric viruses. Again, the context of viral genes in which NS1 is present appears to impact the functioning of the NS1 protein and the virus-host interactions responsible for the induction of immune response genes.
Regulation of IFN-stimulated and immune function genes.
We next used IPA to generate interactive networks of the genes appearing in the functional categories shown in Fig. 3. The top-scoring network generated from the gene lists distinguishing the 1918 and 1918:Tx/91(NS) viruses was a network of IFN-stimulated genes (Fig. 4). This is consistent with the fact that the 1918:Tx/91(NS) virus induced more than eight times the amount of beta interferon than the 1918 virus at 24 h postinfection, as measured by microarray. All of the genes in the network in Fig. 4A were expressed at a higher level in cells infected with 1918:Tx/91(NS) than in cells infected with the 1918 virus at 24 h, highlighting a striking functional difference between the two viruses. Thus, while immune response genes in general were expressed at higher levels in response to infection with the parental viruses, the expression of IFN-stimulated genes in particular was repressed by the 1918 virus. This is consistent with our earlier report that the NS1 gene of the 1918 virus may act to antagonize IFN-stimulated gene expression (16).
To extend this finding, we used qRT-PCR to measure the expression of a panel of 12 IFN-stimulated genes (Fig. 4B). The expression of all but one of these genes (OAS2) was significantly upregulated in cells infected with the 1918:Tx/91(NS) virus relative to mock-infected controls. In contrast, all but one of these genes (IFI44) was downregulated in cells infected with the 1918 virus compared with that observed in mock-infected cells, indicating that the 1918 NS1 protein suppressed even low-level, constitutive expression of these genes. We did not observe the same pattern of IFN-stimulated gene downregulation by the WT Tx/91 virus. Although the mechanism underlying the inhibition of IFN-stimulated gene expression by the 1918 NS1 protein cannot be directly ascertained by our analyses, amino acid sequences and structural differences between NS1 proteins and the consequent differences in virus-host factor interactions are likely to contribute to this effect. Because the viral polymerase complexes are identical in the 1918 and 1918:Tx/91(NS) viruses, it is unlikely that differences exist between the two viruses in their respective abilities to undergo cap snatching in the nucleus (excluding this as a probable mechanism for the differences in IFN-stimulated gene expression observed between them). In addition, a number of the genes repressed by the 1918 NS1 protein, including RIG-I (DDX58), PKR, and OAS1/OAS2, have been reported to interact directly (or through a dsRNA intermediate) with the NS1 protein during infection, and it is possible that in some cases NS1 may impose an additional block on the activities of these proteins at the posttranslational level (4, 17, 19, 28, 32-34, 38, 46).
As noted, immune response genes in general were expressed at higher levels in cells infected by the 1918 virus than in cells infected by the Tx/91:1918(NS) virus, and these genes also formed a high-scoring IPA network (Fig. 5A). This network included genes associated with chemokine signaling (CCL2, CCL19, CCL21, CCR10, and LGALS3), cytokine signaling (FES and NFATC4), lymphocyte activation (CD4, LCK, PTPRCAP, SELL, SELPLG, SLAMF1, THY1, CD79A, and SPI1), and neutrophil activation (ELA2, MMP9, NCF1, and SELL). Importantly, during an in vivo infection, the increased expression of these genes may serve to attract infiltrating immune cells to the site of infection and to increase proinflammatory processes and subsequent lung pathology. Expression relative to mock-infected cells of the genes from Fig. 5A is shown in Fig. 5B. The fact that the 1918 virus paradoxically upregulated many chemokine genes but downregulated many IFN-stimulated genes is one of the intriguing findings of our study and challenges the traditional dogma that IFN is required for the cell to signal to immune infiltrates.
Regulation of lipid metabolism genes.
Perhaps most intriguingly, our examination of high-scoring networks generated from the gene lists distinguishing the 1918 and 1918:Tx/91(NS) viruses revealed an extensive, direct network of lipid metabolism genes that were expressed at higher levels in cells infected with the 1918:Tx/91(NS) virus than in cells infected with the 1918 virus (Fig. 6A). This network contained key activators of cell proliferation and differentiation, including PIK3R1, PLCE1, RARB, and SIRT1. Cell proliferation is typically associated with an increased de novo synthesis of fatty acids, phospholipids, and cholesterol to build cellular membrane components, and there was an increase in the expression of genes involved in these pathways.
To extend this finding, we used qRT-PCR to measure the expression of a panel of 11 lipid metabolism genes (Fig. 6B). The expression of all but one of these genes (RGS2) was significantly downregulated in cells infected with the 1918 virus relative to mock-infected controls. In contrast, the 1918:Tx/91(NS) virus induced the expression of some of these genes relative to mock-infected cells. In every case, the 1918 virus significantly suppressed the expression of these genes relative to the 1918:Tx/91(NS) virus. As some of the genes represented in Fig. 6 function in proinflammatory (and potentially antiviral) pathways, it is possible that an early suppression of these genes contributes to the severe lethality associated with the 1918 virus.
Cells infected with 1918:Tx/91(NS) also had higher levels of expression of genes encoding proinflammatory lipid mediators (including BDKRB2, LTA4H, PLA2G4A, PTGS2, and VIM) and genes encoding different representatives of the ATP-binding cassette family of proteins (ABC transporters) (Fig. 6). Certain members of this family, including ABCB1, are reported to stimulate dendritic cell activation (40). Several CYP family members were also induced by the 1918:Tx/91(NS) virus, including CYP24A1, which is reported to induce monocyte and macrophage differentiation, and CYP3A4, which aids in the oxidative catabolism of lipids (7, 8). Thus, the 1918 NS1 protein may also play a role in inhibiting the transcription of certain lipid-based proinflammatory mediators that function as part of the host antiviral response. Consistent with this idea, the expression of many of the same genes related to lipid metabolism were also expressed at a higher level in cells infected with the Tx/91 virus than in cells infected with Tx/91:1918(NS) (Fig. 3). These included CYP family genes, genes encoding proinflammatory mediators, and cholesterol biosynthesis genes.
DISCUSSION
NS1, the immune response, and IFN-stimulated gene expression.
This is the first study to use a human lung epithelial cell infection model to define the global cellular transcriptional response to the eight-gene 1918 pandemic influenza virus. The role of NS1 as a virulence factor for both the 1918 virus and for seasonal influenza virus isolates has been the subject of some debate, and our use of WT and chimeric viruses allowed us to identify NS1-induced gene expression changes that likely contribute to virulence in vivo. Gene expression changes associated with the immune response were particularly influenced by the NS1 gene expressed by the virus. However, we found that a larger number of immune response genes were induced in response to each WT virus than were induced by their chimeric counterparts, suggesting that the ability of NS1 to modulate the host response is also dependent upon the larger constellation of viral gene segments present. Although this is likely the case, unique differences in the function of the 1918 NS1 protein were also revealed through the use of chimeric viruses, particularly with regard to the effect of the 1918 NS1 on the expression of genes associated with cytokine signaling, IFN-stimulated gene expression, and lipid metabolism.
As an example, viruses expressing the 1918 NS1 gene induced the expression of numerous proinflammatory chemokine and cytokine genes, many of which serve to attract and activate infiltrating immune cells that have proinflammatory or other antiviral functions. An increase in the inflammatory response mediated by such gene expression changes can have a direct impact on lung pathology. In contrast, despite the large number of immune response genes induced by the 1918 virus, the expression of many IFN-stimulated genes was suppressed. Infection with the chimeric 1918:Tx/91(NS) virus, on the other hand, resulted in the induction of the same IFN-stimulated genes, providing additional evidence that the 1918 NS1 protein is a particularly potent IFN antagonist.
Our data suggest that cells infected with influenza virus rely first on the early upregulation of IFN-stimulated genes as part of the antiviral response. Should this early IFN response be blocked by NS1, as in the case of infection with the 1918 virus, the cells in turn may upregulate the expression of chemokine and cytokine genes in an effort to attract infiltrating immune cells to the site of infection. The suppression of IFN-stimulated gene expression by the 1918 NS1 protein could therefore contribute to disease by providing the virus with additional time to replicate and by causing infected cells to secrete excessive levels of proinflammatory chemokines and cytokines in an effort to attract additional infiltrating immune cells. The resulting combination of increased viral load, abundant cytokine signaling, and immune cell infiltration may trigger an exaggerated immune response characterized by hypercytokinemia and severe tissue damage. In fact, macaques experimentally infected with the 1918 virus exhibit these specific clinical signs (23).
Of the four viruses used for these studies, only the 1918 virus caused cytopathic effects (CPE) in A549 cells; the other viruses were not associated with CPE, even at 24 h postinfection (data not shown). The 1918 virus caused CPE at 24 h, but not at 2 or 6 h postinfection. These observations are consistent with reports describing the pathologies induced by these viruses in mice, with the 1918 virus being the most lethal in mice (39).
Transcriptional profiling of cells infected with viruses expressing the Tx/91 NS1 revealed consistently higher levels of gene expression in the categories of cellular growth and proliferation and cell death. A simultaneous upregulation of genes in these functional categories may indicate increased cell turnover, indicative of recovery from infection. Various metabolic pathways were also preferentially induced in cells infected with viruses expressing the Tx/91 NS1. Taken together, our data suggest that cells infected with viruses expressing the Tx/91 NS1 began to recover from infection more quickly than cells infected with viruses expressing the 1918 NS1 gene, the former showing signs of recovery at 24 h after infection. In contrast, gene expression changes in cells infected with viruses expressing the 1918 NS1 gene suggest decreased cell turnover and a general shutdown of a broad range of metabolic pathways at this time point. This may explain our observation that only cells infected with the 1918 virus exhibited CPE. When looked at in the context of the immune response, our data suggest that lung epithelial cells able to mount an effective IFN response against influenza virus are more likely to continue metabolizing and proliferating, which in turn would serve to minimize tissue damage.
NS1 and lipid metabolism.
Genomic analyses of certain other RNA viruses, such as hepatitis C virus and human immunodeficiency virus, have shown that pathways involved in lipid metabolism are significantly altered during infection (27, 51, 52). Our previous analyses have also revealed a subset of lipid metabolism and lipoprotein signaling genes that are downregulated in the lungs or bronchi of mice or macaques infected with the 1918 virus, but not in animals infected with a less pathogenic influenza virus (22, 23). Data from our current study suggest that this downregulation of genes associated with lipid metabolism is due to a novel function of the NS1 protein. This downregulation of lipid metabolism can also result in increased pathogenesis, as certain lipid effectors activate dendritic cells or stimulate the differentiation of monocytes and macrophages, and others are highly proinflammatory or otherwise active in signaling to the immune response.
In A549 cells, infection with viruses expressing the 1918 NS1 gene resulted in the downregulation of a large subset of genes involved in lipid metabolism, whereas this effect was not seen in cells infected with viruses expressing the Tx/91 NS1. These downregulated genes included many genes needed for cholesterol synthesis and for the synthesis of other structural components of the phospholipid membrane, which would likely be required for the increased cell proliferation response displayed by cells infected with viruses expressing the Tx/91 NS1. It is possible that the 1918 NS1 protein exerts its effects on lipid metabolism by blocking the expression of certain transcription factors that control a wide variety of lipid metabolism genes. For example, SREBF1 and P53 were both downregulated by the 1918 virus. It is unclear whether suppression of these genes required a direct interaction with NS1, or whether their downregulation simply reflected the larger metabolic shutdown in these cells.
It is possible that the ability of the 1918 virus to modulate lipid metabolism contributed to the differences in viral RNA and protein levels observed at the various time points between the 1918 and 1918:Tx/91(NS) viruses. Furthermore, only the 1918 virus produced noticeable CPE at 24 h (data not shown). Because the ability of NS1 to suppress lipid metabolism is a novel discovery, many additional studies will be needed to elucidate the mechanism and the broader biological impact associated with this observation.
We chose to use the A549 transformed human lung cell line because it is a well-established and well-characterized model for influenza virus infection. These cells offer advantages including ease of manipulation and reproducibility between experiments (as opposed to primary human lung cells). Nevertheless, as type II pneumocytes, they represent only one type of cell targeted by influenza virus during a natural infection. While cellular homogeneity is an advantage when it comes to interpreting microarray data, the use of any in vitro approach also has inherent limitations (including the lack of an intact immune system and/or chromosomal aberrations in the cell line chosen). Therefore, our results from the current studies should be interpreted and extrapolated with these limitations in mind. In the future, these studies could be extended to a primary cell model, which could offer advantages including greater physiological relevance, differentiated cell types, and a higher percentage of cells infected.
Mechanisms of differential NS1 function.
There are many reports describing the purported mechanisms by which NS1 functions (1, 4, 9, 17, 20, 25, 28, 31-36, 38, 42, 53), and different regions of the protein mediate specific activities. The N terminus of NS1 binds and sequesters dsRNA, which may thereby block the activation of RIG-I, 2′-5′ OAS, PKR, or other dsRNA-activated proteins. The C terminus of NS1 can block the activity of the nuclear proteins PABPII and CPSF, which prevent the processing and export of mRNA transcripts. The interaction between NS1 and CPSF appears to be stabilized by the viral polymerase complex (specifically PA) and by the viral NP protein, again suggesting that at least some NS1 functions are dependent upon viral gene constellation (26). More recently, an SH3-binding motif on the NS1 C terminus was shown to interact with the p85β subunit of PI3K, which in turn activates the PI3K/Akt pathway in order to mediate anti-apoptotic signaling responses (13, 29, 43, 44, 54). Also, as stated previously, it was recently shown that NS1 inhibits induction of type I IFN by binding and inhibiting the E3 ubiquitin ligase TRIM25 (14). The authors of this latter study showed that the 1918 and several other influenza virus NS1 proteins bound to TRIM25, but the data were not quantitative. It is possible that the 1918 NS1 protein is a highly potent inhibitor of TRIM25.
In addition to viral gene constellation effects, differences in the function of the 1918 and Tx/91 NS1 proteins are likely due to differences in amino acid sequence. The amino acid sequences of the 1918 NS1 and Tx/91 proteins are different at 24 (out of 230) positions, with the distal C terminus showing a slight enrichment in sequence differences, including differences in the PDZ ligand domain and PABPII-binding site (both contained in positions 223 to 230) (37). The dsRNA-binding domain (positions 23 to 43) is identical for the two proteins, as is the CPSF-binding domain (positions 103, 106, and 183 to 188) (25). Differences in PABPII or PDZ protein interactions at the C termini of the 1918 and Tx/91 NS1 proteins could account mechanistically for the range of cellular processes affected. For example, PABPII is thought to enhance the maturation and nuclear export of nearly all RNA polymerase II-dependent pre-mRNA transcripts produced in the nucleus (9). An inhibition of PABPII function by the 1918 NS1 protein could, in theory, result in the shutdown of gene expression across a broad range of cellular metabolic pathways. A549 cells infected with the 1918 virus showed precisely this pattern of expression at 24 h. It is possible that amino acid sequence differences in the PABPII-binding site of the 1918 NS1 protein allow for a stronger binding affinity to PABPII, resulting in a greater shutdown of PABPII function than is caused by binding of the Tx/91 NS1 to PABPII.
Alternatively, the 1918 and Texas NS1 proteins may have different capacities to bind cellular PDZ-binding proteins. The terminal four amino acids of NS1 comprise a PDZ domain ligand of the X-S/T-X-V type that may contribute to virulence by perturbing a broad range of cellular signaling patterns (20). This is thought to occur via binding of up to 30 different human PDZ-binding proteins. The identity of the amino acid at position 227 of NS1 appears to be of particular importance. A comparison of 1,196 NS1 proteins from avian, human, swine, and equine influenza viruses reveals that only the 1918 pandemic virus contains a K at position 227 (37). Infections using a mouse infection model also showed that a K at position 227 is associated with significantly greater weight loss, lung pathology, viral dissemination throughout the lungs, and mortality (20). It is therefore possible that the 1918 and Tx/91 NS1 proteins attract different PDZ-binding proteins, which in turn could affect a wide array of cellular signaling pathways.
A comparison of the NEP amino acid sequences for the Tx/91 and 1918 viruses showed differences at 6 out of 121 positions. These differences were scattered uniformly throughout the length of the protein. We cannot exclude the possibility that differences in the respective NEP proteins accounted for some of the differences in gene expression observed. However, NEP is an alternative splice product of the NS gene segment and is expressed at much lower abundance than NS1. In addition, NEP has not previously been shown to impact IFN signaling, whereas this is a primary function attributed to NS1.
In conclusion, our use of functional genomics and WT and chimeric viruses have allowed us to demonstrate that the NS1 proteins of highly pathogenic and attenuated viruses differ in their effects on cellular gene expression. The unique ability of the 1918 NS1 protein to downregulate the expression of IFN-stimulated genes and genes associated with lipid metabolism, while inducing the expression of chemokines and cytokine genes, may have a profound effect on disease outcome.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service grant P01AI058113 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print on 12 August 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aragón, T., S. de la Luna, I. Novoa, L. Carrasco, J. Ortin, and A. Nieto. 2000. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol. Cell. Biol. 20:6259-6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baas, T., C. R. Baskin, D. L. Diamond, A. García-Sastre, H. Bielefeldt-Ohmann, T. M. Tumpey, M. J. Thomas, V. S. Carter, T. H. Teal, N. Van Hoeven, S. C. Proll, J. M. Jacobs, Z. R. Caldwell, M. A. Gritsenko, R. R. Hukkanen, D. G. Camp, R. D. Smith, and M. G. Katze. 2006. Integrated molecular signature of disease: analysis of influenza virus-infected macaques through functional genomics and proteomics. J. Virol. 80:10813-10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basler, C. F., A. H. Reid, J. K. Dybing, T. A. Janczewski, T. G. Fanning, H. Zheng, M. Salvatore, M. L. Perdue, D. E. Swayne, A. García-Sastre, P. Palese, and J. K. Taubenberger. 2001. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl. Acad. Sci. USA 98:2746-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmann, M., A. García-Sastre, E. Carnero, H. Pehamberger, K. Wolff, P. Palese, and T. Muster. 2000. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 74:6203-6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brazma, A., T. Freeman, K. Gardiner, S. Weissman, T. Werner, and B. Korn. 2004. Report on the thirteenth international workshop on the identification and functional, evolutionary and expression analysis of transcribed sequences: comparative and functional genomics workshop. Cytogenet. Genome Res. 105:11-17. [DOI] [PubMed] [Google Scholar]
- 6.Brazma, A., P. Hingamp, J. Quackenbush, G. Sherlock, P. Spellman, C. Stoeckert, J. Aach, W. Ansorge, C. A. Ball, H. C. Causton, T. Gaasterland, P. Glenisson, F. C. P. Holstege, I. F. Kim, V. Markowitz, J. C. Matese, H. Parkinson, A. Robinson, U. Sarkans, S. Schulze-Kremer, J. Stewart, R. Taylor, J. Vilo, and M. Vingron. 2001. Minimum information about a microarray experiment (MIAME) toward standards for microarray data. Nat. Genet. 29:365-371. [DOI] [PubMed] [Google Scholar]
- 7.Brian, W. R., M. A. Sari, M. Iwasaki, T. Shimada, L. S. Kaminsky, and F. P. Guengerich. 1990. Catalytic activities of human liver cytochrome P-450 IIIA4 expressed in Saccharomyces cerevisiae. Biochemistry 29:11280-11292. [DOI] [PubMed] [Google Scholar]
- 8.Chen, K. S., J. M. Prahl, and H. F. DeLuca. 1993. Isolation and expression of human 1,25-dihydroxyvitamin D3 24-hydroxylase cDNA. Proc. Natl. Acad. Sci. USA 90:4543-4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, Z., Y. Li, and R. M. Krug. 1999. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 18:2273-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chien, C. Y., R. Tejero, Y. Huang, D. E. Zimmerman, C. B. Rios, R. M. Krug, and G. T. Montelione. 1997. A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat. Struct. Biol. 4:891-895. [DOI] [PubMed] [Google Scholar]
- 11.Chien, C. Y., Y. Xu, R. Xiao, J. M. Aramini, P. V. Sahasrabudhe, R. M. Krug, and G. T. Montelione. 2004. Biophysical characterization of the complex between double-stranded RNA and the N-terminal domain of the NS1 protein from influenza A virus: evidence for a novel RNA-binding mode. Biochemistry 43:1950-1962. [DOI] [PubMed] [Google Scholar]
- 12.Conenello, G. M., D. Zamarin, L. A. Perrone, T. Tumpey, and P. Palese. 2007. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehrhardt, C., T. Wolff, S. Pleschka, O. Planz, W. Beermann, J. G. Bode, M. Schmolke, and S. Ludwig. 2007. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 81:3058-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gack, M. U., R. A. Albrecht, T. Urano, K. S. Inn, I. C. Huang, E. Carnero, M. Farzan, S. Inoue, J. U. Jung, and A. Garcia-Sastre. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Sastre, A., A. Egorov, D. Matassov, S. Brandt, D. E. Levy, J. E. Durbin, P. Palese, and T. Muster. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324-330. [DOI] [PubMed] [Google Scholar]
- 16.Geiss, G. K., M. Salvatore, T. M. Tumpey, V. S. Carter, X. Wang, C. F. Basler, J. K. Taubenberger, R. E. Bumgarner, P. Palese, M. G. Katze, and A. García-Sastre. 2002. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl. Acad. Sci. USA 99:10736-10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo, Z., L. M. Chen, H. Zeng, J. A. Gomez, J. Plowden, T. Fujita, J. M. Katz, R. O. Donis, and S. Sambhara. 2007. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 36:263-269. [DOI] [PubMed] [Google Scholar]
- 18.Hatada, E., and R. Fukuda. 1992. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 73:3325-3329. [DOI] [PubMed] [Google Scholar]
- 19.Hatada, E., S. Saito, and R. Fukuda. 1999. Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J. Virol. 73:2425-2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson, D., M. J. Hossain, D. Hickman, D. R. Perez, and R. A. Lamb. 2008. A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 105:4381-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kash, J. C., C. F. Basler, A. García-Sastre, V. Carter, R. Billharz, D. E. Swayne, R. M. Przygodzki, J. K. Taubenberger, M. G. Katze, and T. M. Tumpey. 2004. Global host immune response: pathogenesis and transcriptional profiling of type A influenza viruses expressing the hemagglutinin and neuraminidase genes from the 1918 pandemic virus. J. Virol. 78:9499-9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kash, J. C., T. M. Tumpey, S. C. Proll, V. Carter, O. Perwitasari, M. J. Thomas, C. F. Basler, P. Palese, J. K. Taubenberger, A. García-Sastre, D. E. Swayne, and M. G. Katze. 2006. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443:578-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobasa, D., S. M. Jones, K. Shinya, J. C. Kash, J. Copps, H. Ebihara, Y. Hatta, J. H. Kim, P. Halfmann, M. Hatta, F. Feldmann, J. B. Alimonti, L. Fernando, Y. Li, M. G. Katze, H. Feldmann, and Y. Kawaoka. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319-323. [DOI] [PubMed] [Google Scholar]
- 24.Kobasa, D., A. Takada, K. Shinya, M. Hatta, P. Halfmann, S. Theriault, H. Suzuki, H. Nishimura, K. Mitamura, N. Sugaya, T. Usui, T. Murata, Y. Maeda, S. Watanabe, M. Suresh, T. Suzuki, Y. Suzuki, H. Feldmann, and Y. Kawaoka. 2004. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 431:703-707. [DOI] [PubMed] [Google Scholar]
- 25.Kochs, G., A. García-Sastre, and L. Martínez-Sobrido. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81:7011-7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuo, R. L., and R. M. Krug. 2009. Influenza A virus polymerase is an integral component of the CPSF30-NS1A protein complex in infected cells. J. Virol. 83:1611-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lederer, S. L., K. A. Walters, S. Proll, B. Paeper, S. Robinzon, L. Boix, N. Fausto, J. Bruix, and M. G. Katze. 2006. Distinct cellular responses differentiating alcohol- and hepatitis C virus-induced liver cirrhosis. Virol. J. 3:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, S., J. Y. Min, R. M. Krug, and G. C. Sen. 2006. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13-21. [DOI] [PubMed] [Google Scholar]
- 29.Li, Y., D. H. Anderson, Q. Liu, and Y. Zhou. 2008. Mechanism of influenza A virus NS1 protein interaction with the p85beta, but not the p85alpha, subunit of phosphatidylinositol 3-kinase (PI3K) and up-regulation of PI3K activity. J. Biol. Chem. 283:23397-23409. [DOI] [PubMed] [Google Scholar]
-
30.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the
 method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar] - 31.Melén, K., L. Kinnunen, R. Fagerlund, N. Ikonen, K. Y. Twu, R. M. Krug, and I. Julkunen. 2007. Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J. Virol. 81:5995-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mibayashi, M., L. Martinez-Sobrido, Y. M. Loo, W. B. Cardenas, M. Gale, Jr., and A. Garcia-Sastre. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81:514-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Min, J. Y., and R. M. Krug. 2006. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo(A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 103:7100-7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min, J. Y., S. Li, G. C. Sen, and R. M. Krug. 2007. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 363:236-243. [DOI] [PubMed] [Google Scholar]
- 35.Nemeroff, M. E., S. M. Barabino, Y. Li, W. Keller, and R. M. Krug. 1998. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell 1:991-1000. [DOI] [PubMed] [Google Scholar]
- 36.Noah, D. L., K. Y. Twu, and R. M. Krug. 2003. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAs. Virology 307:386-395. [DOI] [PubMed] [Google Scholar]
- 37.Obenauer, J. C., J. Denson, P. K. Mehta, X. Su, S. Mukatira, D. B. Finkelstein, X. Xu, J. Wang, J. Ma, Y. Fan, K. M. Rakestraw, R. G. Webster, E. Hoffmann, S. Krauss, J. Zheng, Z. Zhang, and C. W. Naeve. 2006. Large-scale sequence analysis of avian influenza isolates. Science 311:1576-1580. [DOI] [PubMed] [Google Scholar]
- 38.Opitz, B., A. Rejaibi, B. Dauber, J. Eckhard, M. Vinzing, B. Schmeck, S. Hippenstiel, N. Suttorp, and T. Wolff. 2007. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 9:930-938. [DOI] [PubMed] [Google Scholar]
- 39.Pappas, C., P. V. Aguilar, C. F. Basler, A. Solorzano, H. Zeng, L. A. Perrone, P. Palese, A. Garcia-Sastre, J. M. Katz, and T. M. Tumpey. 2008. Single gene reassortants identify a critical role for PB1, HA, and NA in the high virulence of the 1918 pandemic influenza virus. Proc. Natl. Acad. Sci. USA 105:3064-3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Randolph, G. J., S. Beaulieu, M. Pope, I. Sugawara, L. Hoffman, R. M. Steinman, and W. A. Muller. 1998. A physiologic function for p-glycoprotein (MDR-1) during the migration of dendritic cells from skin via afferent lymphatic vessels. Proc. Natl. Acad. Sci. USA 95:6924-6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salvatore, M., C. F. Basler, J. P. Parisien, C. M. Horvath, S. Bourmakina, H. Zheng, T. Muster, P. Palese, and A. García-Sastre. 2002. Effects of influenza A virus NS1 protein on protein expression: the NS1 protein enhances translation and is not required for shutoff of host protein synthesis. J. Virol. 76:1206-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seo, S. H., E. Hoffmann, and R. G. Webster. 2004. The NS1 gene of H5N1 influenza viruses circumvents the host anti-viral cytokine responses. Virus Res. 103:107-113. [DOI] [PubMed] [Google Scholar]
- 43.Shin, Y. K., Y. Li, Q. Liu, D. H. Anderson, L. A. Babiuk, and Y. Zhou. 2007. SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J. Virol. 81:12730-12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin, Y. K., Q. Liu, S. K. Tikoo, L. A. Babiuk, and Y. Zhou. 2007. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 88:13-18. [DOI] [PubMed] [Google Scholar]
- 45.Solórzano, A., R. J. Webby, K. M. Lager, B. H. Janke, A. García-Sastre, and J. A. Richt. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J. Virol. 79:7535-7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45a.Stoughton, R., and H. Dai. February 2002. Statistical combining of cell expression profiles. U.S. patent 6,351,712.
- 46.Tan, S.-L., and M. G. Katze. 1998. Biochemical and genetic evidence for complex formation between the influenza A virus NS1 protein and the interferon-induced PKR protein kinase. J. Interferon Cytokine Res. 18:757-766. [DOI] [PubMed] [Google Scholar]
- 47.Taubenberger, J. K., A. H. Reid, R. M. Lourens, R. Wang, G. Jin, and T. G. Fanning. 2005. Characterization of the 1918 influenza virus polymerase genes. Nature 437:889-893. [DOI] [PubMed] [Google Scholar]
- 48.Tumpey, T. M., C. F. Basler, P. V. Aguilar, H. Zeng, A. Solorzano, D. E. Swayne, N. J. Cox, J. M. Katz, J. K. Taubenberger, P. Palese, and A. Garcia-Sastre. 2005. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310:77-80. [DOI] [PubMed] [Google Scholar]
- 49.Tumpey, T. M., A. García-Sastre, J. K. Taubenberger, P. Palese, D. E. Swayne, and C. F. Basler. 2004. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. USA 101:3166-3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tumpey, T. M., A. Garcia-Sastre, J. K. Taubenberger, P. Palese, D. E. Swayne, M. J. Pantin-Jackwood, S. Schultz-Cherry, A. Solorzano, N. Van Rooijen, J. M. Katz, and C. F. Basler. 2005. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79:14933-14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van't Wout, A. B. 2005. Gene expression profiling of HIV-1 infection using cDNA microarrays. Methods Mol. Biol. 304:455-459. [DOI] [PubMed] [Google Scholar]
- 52.van't Wout, A. B., J. V. Swain, M. Schindler, U. Rao, M. S. Pathmajeyan, J. I. Mullins, and F. Kirchhoff. 2005. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J. Virol. 79:10053-10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, X., M. Li, H. Zheng, T. Muster, P. Palese, A. A. Beg, and A. Garcia-Sastre. 2000. Influenza A virus NS1 protein prevents activation of NF-κB and induction of alpha/beta interferon. J. Virol. 74:11566-11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhirnov, O. P., and H. D. Klenk. 2007. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis 12:1419-1432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.