

Abstract
Theiler's murine encephalomyelitis virus (TMEV) is a highly cytolytic picornavirus that persists in the mouse central nervous system (CNS) largely in macrophages with infection maintained by macrophage-to-macrophage spread. Infected macrophages in the CNS undergo apoptosis. We recently showed that M1-D macrophages infected with the low-neurovirulence TMEV BeAn virus became apoptotic through the mitochondrial pathway that is Bax mediated. Our present analyses of the molecular events and signaling pathway(s) culminating in the mitochondrial outer membrane permeabilization that initiates the caspase cascade and apoptosis of BeAn virus-infected M1-D macrophages revealed activation of p38 mitogen-activated protein kinase by 2 to 3 h postinfection (p.i.), followed by phosphorylation of tumor suppressor protein p53 Ser 15 at 3 to 6 h p.i., stabilizing p53 levels until 6 h p.i. Activated p53 upregulated the transcription of proapoptotic puma and noxa genes at 2 to 4 h p.i. and their BH3-only protein expression, followed by the loss of detectable prosurvival Mcl-1 and A1 proteins at 4 to 10 h p.i. Degradation of the prosurvival proteins is known to release Bax, which forms homo-oligomers and translocates into and permeabilizes the mitochondrial outer membrane. Inhibition of phospho-p38 by two specific inhibitors, SB203580 and BIRB796, led to a significant decrease in apoptosis at 10 h p.i., with no effect on virus titers (only SB203580 tested). Together, these data indicate that p53 activation is required for the induction of apoptosis in infected M1-D cells.
Mice inoculated intracerebrally with Theiler's murine encephalomyelitis virus (TMEV) develop persistent central nervous system infection and chronic inflammatory demyelinating disease, providing an experimental animal analog for multiple sclerosis. TMEV persists primarily in macrophages, the most prominent cellular component of demyelinating lesions. Since TMEV is a highly cytolytic picornavirus, persistence is presumably maintained by cell-to-cell spread of the virus, with infection detected in only a small percentage of macrophages at any time point, which is consistent with the paradigm of persistent picornavirus infections in cell cultures (43). In the mouse central nervous system, macrophages, including those that are infected, undergo apoptosis (24, 34, 39). As a part of our ongoing efforts to elucidate the virus-cell interactions of TMEV-infected macrophages in culture, we recently showed that M1-D macrophages infected with the low-neurovirulence TMEV strain BeAn undergo Bax-mediated apoptosis through the mitochondrial pathway (38). Apoptotic M1-D cells were first detected 8 to 10 h postinfection (p.i.), and cell death from apoptosis progressed linearly from 8 to 16 h p.i. Immunoblotting revealed that capase-9 was cleaved to its 37-kDa active form 8 h p.i., with permeabilization of the mitochondrial outer membrane leading to release of cytochrome c, followed by caspase-3 cleavage to its 17-kDa active form. Thus, apoptosis in this infection largely evolves between 8 and 16 h p.i. Interestingly, overexpression of Bcl-2 provided minimal although significant protection, and overexpression of Bcl-xL provided no protection against apoptosis (38).
Programmed cell death through the mitochondrial pathway is regulated by the Bcl-2 family of proteins, which contains three structurally and functionally distinct subgroups: (i) Bcl-2-like prosurvival proteins, which share up to four Bcl-2 homology (BH) domains; (ii) proapoptotic Bax/Bak proteins, which contain the BH1, BH2, and BH3 domains; and (iii) the proapoptotic BH3-only proteins which share only short BH3 domains. Apoptosis is initiated when prosurvival Bcl-2 family proteins are engaged by proapoptotic BH3-only proteins through the interaction between an α-helix formed by the BH3-only proteins and the hydrophobic groove formed by the BH1, BH2, and BH3 domains of the prosurvival Bcl-2 members (28, 44). Chen et al. (2) found that these interactions were not promiscuous but vary over 10,000-fold in affinity, indicating that only certain pairs associate inside the cell. Moreover, various cellular stresses appear to play a role in selectively activating the proapoptotic BH3-only proteins.
To identify the principal proapoptotic BH3-only and prosurvival multi-BH domain Bcl-2 family members engaged in BeAn virus-induced apoptosis in M1-D cells and to obtain insight into the cellular stress(es) involved, we examined the sequence of molecular events leading to apoptosis in BeAn virus-infected M1-D cells. Our analyses revealed activation of the p38 mitogen-activated protein kinase (MAPK) by 3 h p.i., phosphorylation of p53 Ser 15 and stabilization of p53 expression at 3 and 6 h p.i., transcriptional upregulation of proapoptotic Puma and Noxa at 2 to 4 h p.i. and their subsequent expression, and loss of prosurvival Mcl-1 and A1 protein expression, 4 to 10 h p.i. This sequence of events results in the release of the downstream multi-BH domain Bcl-2 member Bax (38) that permeabilizes the mitochondrial outer membrane to initiate the caspase cascade and apoptosis.
MATERIALS AND METHODS
Cells and viruses.
BHK-21 cells were grown in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 7.5% tryptose phosphate, 2 mM l-glutamine, 100 U of penicillin/ml, and 100 μg of streptomycin/ml at 37°C in 5% CO2. Cells of the immature myelomonocytic cell line M1, derived from the SL mouse strain, were induced to differentiate into M1-D macrophages with supernatants from L929 and P388D1 cells, as described previously (14). The origin and passage history of the BeAn virus stock has been described (33). Virus titers of clarified lysates of infected cells were determined by standard plaque assay in BHK-21 cells (33).
Virus infections.
After virus adsorption at a multiplicity of infection (MOI) of 10 for 45 min at 24°C, the cell monolayers were washed twice with phosphate-buffered saline (PBS) containing 1 mM CaCl2 and 0.5 mM MgCl2, followed by incubation in Dulbecco modified Eagle medium containing 1% FBS at 37°C for the times indicated. The end of the adsorption period was designated as time zero.
Apoptosis assay.
The number of apoptotic cells was determined by using DAPI (4′,6′-diamidino-2-phenylindole) staining. Briefly, M1-D cells grown and infected on glass coverslips (Fisher Scientific Co., Pittsburgh, PA) were fixed in 4% paraformaldehyde for 15 min at room temperature, washed twice in PBS (pH 7.2), and incubated with DAPI at a final concentration of 0.5 μg/ml for 5 min. Coverslips were washed with 0.5% Tween 20 in PBS and distilled H2O and viewed with a Zeiss digital confocal microscope. At least three randomly chosen fields, each field containing ca. 75 to 100 cells, from a coverslip were photographed at ×400 magnification, and the percentage of cells with condensed chromatin and fragmented nuclei was determined by a blinded observer; each experiment was repeated twice. The results of this assay based on nuclear changes characteristic of apoptosis (condensed chromatin and smaller and fragmented nuclei) correlated with those obtained by TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) staining and fluorescence-activated cell sorting of annexin V- and propidium iodide-stained cells in previous reports on BeAn virus infection in M1-D cells (10, 38).
Reagents.
The following reagents and antibodies were purchased commercially: rabbit anti-actin, rabbit anti-A1, rabbit anti-Bcl-w, rabbit anti-Bim, rabbit anti-caspase-3, rabbit anti-Flag M2, rabbit anti-phospho-p53 (Ser 15), rabbit anti-Puma, and mouse anti-caspase-9 antibody from Cell Signaling Technology (Beverly, MA); mouse anti-Bax, rabbit anti-Noxa, rabbit anti-Mcl-1, rabbit anti-p53, rabbit anti-p38, rabbit anti-Bak, and rabbit anti-phospho-JNK from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); goat anti-mouse immunoglobulin G (IgG)-horseradish peroxidase, goat anti-rabbit IgG-horseradish peroxidase rabbit anti-Bcl-xL antibody from BD Pharmingen (San Diego, CA); rabbit anti-phospho-ERK1/2 and phospho-p38 from R&D Systems, Inc. (Minneapolis, MN); anti-rabbit-IgG Alexa Fluor 568, puromycin, and MitoTracker Red CMXRos from Invitrogen (Carlsbad, CA); p38 MAPK inhibitors SB203580 from Calbiochem (Darmstadt, Germany); BIRB796 from Axion Medchem (Groningen, The Netherlands); and enhanced chemiluminescence solution from Amersham (Piscataway, NJ). The construct pBABE-puro retroviral vector for p53 GSE56 expression was provided by Nissam Hay at UIC (Chicago, IL), and pcDNA3-human Mcl-1 was provided by W. Douglas Cress at the H Lee Moffitt Cancer Center (Tampa, FL).
Subcellular fractionation.
Cytosol, heavy membranes/organelles, and nuclear fractions of M1-D cells were isolated by using a subcellular proteome extraction kit (Calbiochem). Briefly, 106 cells in a 35-mm well were washed with PBS for 10 min at 4°C amd incubated in 400 μl of ice-cold extraction buffer I containing protease inhibitors, with removal of the supernatant as the cytosolic fraction. Monolayers were then incubated for 30 min at 4°C in 400 μl of extraction buffer II containing protease inhibitors, with removal of the supernatant as the heavy membranes/organelle fraction. Finally, monolayers were incubated for 10 min at 4°C in 200 μl of ice-cold extraction buffer III containing protease inhibitors and benzonase nuclease, which served as the nuclear fraction.
RNA isolation and real-time quantitative RT-PCR.
RNA was isolated from mock-infected and virus-infected cells by using a PerfectPure RNA isolation kit from 5 PRIME (Boulder, CO). Puma, Noxa, Mcl-1, and A1 mRNA levels were determined by real-time quantitative reverse transcription-PCR (RT-PCR) using TaqMan probes. Sequences of forward and reverse primers and TaqMan probes (Table 1) were designed by Primer Express from Applied Biosystems (Foster City, CA). A standard curve for each target was generated by using a synthesized amplicon equivalent to the PCR product. PCRs were monitored in real time in an ABI Prism 7700 sequence detector (Applied Biosystems, Inc.). Target gene mRNA levels expressed in atograms were normalized to pg of GAPDH (glyceraldehyde-3-phosphate dehydrogenase).
TABLE 1.
Primers and probes for Puma, Noxa, Mcl-1, and A1 in real-time RT-PCR analysis
| Gene | Sequence (5′-3′)
|
||
|---|---|---|---|
| Forward primer | Reverse primer | Probe | |
| Puma | ACGACCTCAACGCGCAGT | GTGAGGGTCGGTGTCGATG | 6FAM-CGAGCGGCGGAGACAAGAAGAGC-TAMRA |
| Noxa | GTCGCAAAAGAGCAGGATGAG | CGTCCTTCAAGTCTGCTGGC | 6FAM-AGCCCAAGCCCAACCCGGG-TAMRA |
| Mcl-1 | AGCGCAACCACGAGACG | AGATTTAACATCGCCTTCGTTTTT | 6FAM-CCAGGGCATGCTCCGGAAACTG-TAMRA |
| A1 | TGACTTTCACGTGGAATCCATAGA | CCCCAGTTAATGATGCCATCTT | 6FAM-CCGCCAGAATAATATTCAACCAAGTGATGGA-TAMRA |
Transient transfection.
Subconfluent monolayers of M1-D cells in 35-mm six-well plates were transfected with pcDNA3-human-Mcl-1 (6), pcDNA3 empty vector or enhanced green fluorescent protein (EGFP) and Lipofectamine 2000 (Invitrogen) at a 2.5:1 ratio of reagent to DNA. Complexes incubated for 20 min at 24°C were added to cell monolayers for 4 h at 37°C, the growth medium was replaced, and incubation was continued for 24 to 48 h at 37°C in 5% CO2.
Statistical analysis.
Paired Student t test was used to compare groups, and differences were considered significant at P < 0.05.
RESULTS
Role of prosurvival Bcl-2 family members in BeAn virus-infected M1-D cells.
The antiapoptotic Bcl-2 family members Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1 play a central role in cell survival, and Mcl-1 and A1 in particular are expressed in hematopoietic cell lineages and promote viability during proliferation, differentiation, or in response to stress (25). We previously found that overexpression of Bcl-2, but not Bcl-xL, in BeAn-infected M1-D cells delayed the cleavage of caspases-9 and -3 and provided modest but significant protection from cell death (38). To determine whether another prosurvival family member might provide still greater protection from apoptosis than Bcl-2, we analyzed the expression profiles of the five antiapoptotic Bcl-2 proteins by immunoblotting them in both undifferentiated M1 promyelomonocytes and differentiated M1-D macrophages based on the demonstrated regulation of Bcl-2 and Bcl-xL mRNA levels as a function of differentiation of M1 into M1-D cells (13). Bcl-xL expression increased, Bcl-2, Bcl-w, and Mcl-1 decreased, and A1 did not change in M1-D cells compared to undifferentiated M1 cells (Fig. 1A). After infection of M1-D cells, the expression of Bcl-2 and Bcl-w was barely detectable and that of Bcl-xL, which was more robust, did not change between 1 and10 h p.i. (Fig. 1B). In contrast, expression of Mcl-1 and to a lesser extent A1, was initially upregulated but then decreased to low levels at 5 to 10 h p.i. (Fig. 1B) (38), suggesting that Mcl-1 and A1 were degraded, thereby releasing Bax to initiate the caspase cascade and apoptosis.
FIG. 1.

Expression of prosurvival (antiapoptotic) Bcl-2 family members. (A) Difference in expression in uninfected promyelomonocytic M1 cells and in M1 cells differentiated M1-D macrophages. (B) Expression in BeAn-infected (MOI = 10) M1-D cells, showing decreased levels of Mcl-1 and A1 after 4 h p.i. but no change in the other prosurvival proteins. (C and D) Densitometric analysis of the immunoblot data for Mcl-1 and A1, respectively, in panel B. β-Actin was the loading control in panels A and B.
Upregulation of Puma and Noxa proapoptotic BH3-only proteins after infection.
Of the proapoptotic BH3-only proteins known to interact with Mcl-1 and A1, Bim and Puma have the highest affinity, whereas the interaction affinity of Bik and Hrk with A1 is ∼10-fold lower and that of Noxa with Mcl-1 is ∼5-fold lower and with A1 ∼30-fold lower than Bim and Puma (2). Based on these selective interactions and because Bik, which is prominently expressed in epithelial tissues, and Hrk, which is expressed in neurons, are both dispensable for hematopoietic cell apoptosis (4, 5), we examined only the expression of Bim, Puma, and Noxa in BeAn virus-infected M1-D cells. Immunoblotting analysis revealed no difference in the level of Bim after infection from that in mock-infected cells, whereas expression of Puma increased twofold at 2 to 4 h p.i., and Noxa, which was undetectable in uninfected cells and early after infection, was substantially upregulated at 6 to 10 h p.i. (Fig. 2). The temporal upregulation of Noxa paralleled the initiation and progression of apoptosis in these infected cells.
FIG. 2.
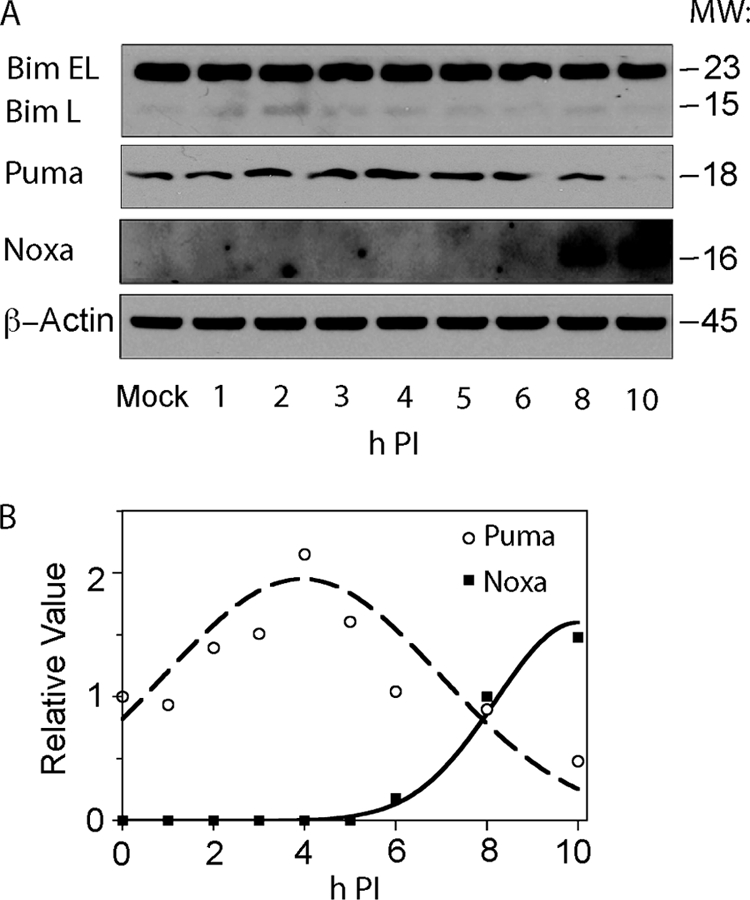
Expression of proapoptotic BH3-only family members in BeAn-infected (MOI = 10) M1-D cells. (A) Immunoblot analysis of Bim EL and L, Puma, and Noxa. (B) Densitometric values for the immunoblot data showing that Puma and Noxa were upregulated at different times p.i. β-Actin served as a loading control in panel A.
Transcriptional upregulation of relevant proapoptotic and prosurvival Bcl-2 family members after infection.
Quantitation of Puma, Noxa, Mcl-1, and A1 mRNAs by real-time RT-PCR revealed that only modest upregulation of Mcl-1 and A1 (∼30%) was seen by 4 h p.i., with declining levels thereafter (Fig. 3A and B, respectively); however, little is known about the transcriptional control of Mcl-1 and A1 in nontumor cells (42). In contrast, upregulation of Puma and Noxa mRNAs showed ∼4- and ∼3-fold increases, respectively, with peak levels at 4 h p.i. and with a subsequent decline of levels to approximately those of mock-infected cells (Fig. 3C and D, respectively). The results for Puma are consistent with the protein expression results, whereas the kinetics of Noxa expression were delayed by several hours.
FIG. 3.

Quantitation of mRNAs by real-time RT-PCR reflecting transcriptional upregulation of puma, noxa, mcl-1, and A1 genes in BeAn virus-infected (MOI = 10) M1-D cells. Peak levels of Puma and Noxa RNAs were seen at ∼4 h p.i. with three- to fourfold increases, respectively, but there was only modest upregulation of Mcl-1 and A1 RNAs.
Overexpression of Mcl-1 protects against apoptosis.
Because of the known role of Mcl-1 in macrophage survival and because Mcl-1 degradation p.i. corresponded temporally with initiation of apoptosis, M1-D cells were transiently transfected to overexpress Mcl-1 (Fig. 4A), infected with virus 16 h later, and compared to cells transfected with EGFP and virus infected for Mcl-1, Bcl-2, and caspase-3 and -9 protein expression (Fig. 4B to E). In Mcl-1-overexpressing cells infected at MOIs of 10 and 20, cleavage of caspase-9 and -3 to their active forms at 10 h p.i. was reduced compared to levels in their transfected counterparts (Fig. 4B; Fig. 4C shows the relative densitometric cleavage values of the immunoblot data). The effect of Mcl-1 overexpression was also compared to that of Bcl-2 overexpression in virus-infected M1-D cells. Mcl-1 overexpression significantly reduced apoptosis in infected M1-D cells (MOI = 10) at 8 (P < 0.01) and 10 (P = 0.02) h p.i. (Fig. 4D), which is especially notable considering that the efficiency of transient transfection was ∼40%, whereas overexpression of Bcl-2 did not protect against apoptosis (Fig. 4E). These results indicate that Mcl-1 plays a greater prosurvival role in BeAn virus-infected M1-D cells than either Bcl-2, or as previously reported, Bcl-xL (38).
FIG. 4.

Effect of transient overexpression of Mcl-1 in M1-D cells infected with BeAn virus 16 h after transfection. (A) Overexpression of Mcl-1 in uninfected M1-D cells compared to nontransfected, uninfected M1-D cells, the latter lane requiring an ∼50-fold longer exposure. The Mcl-1-overexpressing cells showed a 40- to 42-kDa doublet (7) and a 34-kDa truncated form. (B) Immunoblot showing a reduced cleavage of caspases-9 and -3 to their active forms in Mcl-1-transfected versus EGFP-transfected cells infected at MOIs 10 and 20. (C) Densitometric analysis of the immunoblot data in panel B. Relative cleavage of caspase-9 and -3 to their active forms was based on normalizing the percent cleavage (cleaved product/proform plus cleaved product) in the EGFP-transfectants (MOI = 10 data). (D and E) Protection of M1-D cells from apoptosis at 8 h (P < 0.01) and 10 h (P = 0.02) p.i. by Mcl-1 overexpression (D) but not by Bcl-2 (P < 0.05) (E).
Early activation of p53 in infected M1-D cells.
The BH3-only proapoptotic proteins Noxa and Puma are critical for p53-mediated apoptosis, and the noxa and puma genes are both direct transcriptional targets of p53 (23, 36, 41), although apoptosis can also be induced by p53-independent pathways. Because Puma and Noxa protein expression was upregulated in BeAn virus-infecied M1-D cells (see Fig. 2), we examined the expression of the p53 tumor suppressor protein and its modification by Ser-15 phosphorylation, which leads to stabilization of p53 by reducing its interaction with MDM2. Immunoblot analyses showed that p53 levels increased at 1 to 2 h p.i., remained elevated until 5 to 6 h p.i., and decreased thereafter, whereas the temporal kinetics of Ser-15 phosphorylation were slightly delayed but otherwise similar to the p53 expression pattern (Fig. 5A and B), a finding consistent with an important role for Ser-15 phosphorylation. The possible phosphorylation of other p53 amino acids was not assessed.
FIG. 5.

Role of p53 activation in BeAn virus-induced apoptosis. (A) Immunoblot demonstrating increased levels of total p53 beginning at 1 to 2 h p.i. and of phospho-p53 Ser 15 at 3 to 6 h p.i. (B) Relative densitometric values ± the standard deviation of three immunoblots (a representative immunoblot is shown in panel A). (C) Double-immunofluorescence staining showing p53 (green) in the nucleus and MitoTracker (red) staining in the cytoplasm of mock-infected cells, whereas p53 staining is seen in the cytoplasm but does not colocalize with mitochondria of infected cells at 3 h p.i. (D) Immunoblotting after cell fractionation showing p53 primarily in the nuclear (PARP-positive) fraction of mock-infected cells (M), whereas p53 was observed in both the nuclear and cytoplasmic fractions (β-actin positive) but not in the mitochondrial fraction (Cox IV positive) at 3 h p.i. The faint band in lanes 3 and 4 migrated faster than p53.
Since p53 translocates from the nucleus to the cytoplasm, transcriptionally activates Bax (3, 21), and targets the mitochondrial membranous compartment (20), we assessed the localization of p53 within the cell at 2 to 4 h p.i. Immunofluorescence staining with rabbit polyclonal antibody to p53 revealed translocation of p53 from the nucleus to the cytoplasm of infected but not mock-infected cells as early as 2 h p.i.; however, colocalization of p53 with MitoTracker was not detected (Fig. 5C shows the results at 3 h p.i.). Fractionation of infected cells and immunoblotting detected no signal in the mitochondrial fraction of infected cells at 3 h p.i. (Fig. 5D; the faint bands lanes 3 and 4 migrated faster than those of p53). Together, these results indicate that p53 is activated within 1 to 2 h p.i., transactivates two of its target genes (puma and noxa), and translocates to the cytoplasm without localization to mitochondria in these experiments.
p53 transcriptional activity is required for apoptosis in BeAn-infected M1-D cells.
To examine the role of p53 transcriptional activation of Noxa in BeAn virus-induced apoptosis, M1-D cell clones constitutively expressing a p53 genetic suppressor element (GSE56) (26, 32), which acts as a dominant-negative (dn) peptide, were selected by antibiotic resistance (Fig. 6A). Expression of GSE56, a C-terminal region of p53 (amino acids 275 to 368), results in the accumulation of a transcriptionally inactive form of p53. In a representative experiment, the amount of Noxa at 8 to 10 h p.i. was reduced by >50%, and the levels of Mcl-1 increased at least twofold even in mock-infected cells at 4 to 10 h p.i., as detected by immunoblotting (Fig. 6B-D), indicating that p53 expression in infected M1-D cells was responsible for the transactivation of Puma and Noxa and, in turn, for the degradation of Mcl-1. In dn GSE56-expressing cells, there was also a 1.5-fold increase in Bax p.i. (results not shown). Moreover, the constitutive expression of the dn p53 peptide GSE56 in M1-D cells significantly inhibited apoptosis (Fig. 6E), indicating the role of p53 transcriptional activity in mediating the death of these infected cells.
FIG. 6.

Transcriptionally activated p53 is required for BeAn virus-induced apoptosis in M1-D cells. (A) p53 expression in M1-D cells constitutive expressing the dn GSE56 element (11-kDa GSE56 peptide) compared to wild-type (Wt) cells. (B) Immunoblot analysis of M1-D cells expressing GSE56 revealed reduction of Noxa at 8 to 10 p.i. and increased Mcl-1 in mock-infected and infected cells at 4 to 10 h p.i. (C and D) Respective densitometric analysis of the immunoblot data in panel B. (E) Significantly reduced apoptosis at 8 (P = 0.02) and 10 h (P = 0.03) h p.i. in M1-D cells expressing the dn GSE56 construct.
p38 kinase is activated by BeAn virus infection.
Because extracellular signal-regulated kinase (ERK) and p38 MAPK induce phosphorylation of p53 serine 15 (35), we examined the phosphorylated forms of MAPKs by immunoblotting. Experiments revealed robust phospho-p38 levels after overnight growth of M1-D cells in medium containing 10% FBS but not after reducing FBS to 1% 6 h prior to virus adsorption (Fig. 7A, lane 11, shows the results at 1 h before virus adsorption). Virus adsorption at 24°C transiently stimulated phospho-p38 (Fig. 7A, lanes 1 and 2; 0 h p.i. represents the beginning of incubation at 37°C after virus adsorption). The amount of phospho-p38 kinase began increasing by 3 p.i., the amount of phospho-SAPK-JNK1/3 increased only at 6 h p.i., and that of phospho-ERK remained constant over time p.i. (Fig. 7A and B), suggesting that activation of p38 kinase accounts for phosphorylation of p53 Ser-15 in BeAn virus infection. Indeed, addition of the p38 kinase inhibitor SB203580 (10 μM) to the medium 2 h before and during infection of M1-D cells led to a >50% decrease in the amounts of phospho-p38 at 3 and 5 h p.i. and a 25% decrease at 6 h p.i.; phospho-p53 Ser-15 levels were below the level of detection during this period (Fig. 7C). Inhibition of phospho-p38 by SB203580 and by a more potent and specific inhibitor, BIRB796 (1), also resulted in a significant decrease in apoptosis at 10 h p.i. (Fig. 7D); BIRB796 also inhibited phosphorylation of p38 and p53 Ser15 (data not shown). The effect of the p38 MAPK inhibitor SB203580 on apoptosis was not due to a reduction in BeAn virus replication, since virus titers at 12 h p.i., the peak of virus multiplication in M1-D cells incubated with 10 μM SB203580 2 h before and during infection with BeAn virus (MOI = 10), revealed no significant reduction (P > 0.05) in mean titers (± the standard error; n = 3; 35.2 ± 6.0 PFU/cell mock-treated versus 23.8 ± 4.3 PFU/cell SB203580-treated cells). The same concentration of SB203580 was reported to have no effect on yields of other picornaviruses (12, 37).
FIG. 7.

Role of MAPKs in BeAn virus-induced apoptosis in M1-D cells. (A) Immunoblot of phospho-p38, phospho-ERK1/2, and phospho-SAPK/JNK1/3, showing that phospho-p38 levels increased early and phospho-SAPK/JNK1/3 levels increased late p.i. Lane 1 shows the levels of phospho-MAPKs at the beginning of the incubation period at 37°C after virus adsorption at 24°C, and lane 11 shows the levels of these proteins 1 h before virus adsorption. β-Actin served as a loading control. (B) Densitometric values of the immunoblot in panel A. (C) Incubation of cells in medium containing the p38 inhibitor SB203580 (10 μM) during infection led to decreased levels of total p38 and phospho-p38 at 3 to 6 h p.i. compared to medium containing dimethyl sulfoxide. (D) Incubation of infected M1-D cells in the presence of SB203580 and BIRB796 led to a significant reduction in apoptosis: *, P < 0.05; and **, P < 0.001.
DISCUSSION
p38 MAPK is known to be proapoptotic and to both transcriptionally activate p53 and phosphorylate Ser-15, preventing Mdm2-mediated ubiquitination of p53 and its subsequent proteasomal degradation (11, 16). In BeAn virus-infected M1-D cells, p38 MAPK was activated by 2 to 3 h p.i. and followed by phosphorylation of the p53 tumor suppressor protein Ser-15, stabilizing the levels of p53 between 3 and 6 h p.i. Treatment of infected cells with the p38 MAPK inhibitors SB203580 and BIRB796 prevented p53 activation and apoptosis. Based on our experiments using the dn p53 genetic suppresser element (GSE56), p53 transcriptionally upregulated the proapoptotic puma and noxa genes at 2 to 4 h p.i., with increased expression of Puma and Noxa occurring later. The kinetics of Noxa expression appeared to be delayed compared to that of Puma; however, since resting and initial Noxa expression levels p.i. were undetectable, it is conceivable that Noxa expression was actually upregulated before 6 h p.i. Thus, Puma and Noxa appear to play a predominant role in apoptosis in infected M1-D cells because their expression coincided with a decrease in levels of Mcl-1 and A1 and with the onset of apoptosis at 8 to 10 h p.i. Puma is usually more important than Noxa for cell killing because it binds with high affinity to all prosurvival Bcl-2 family members, whereas Noxa binds only to Mcl-1 and A1 (2). After the binding of proapoptotic BH3-only proteins to prosurvival Bcl-2 family members, degradation of the prosurvival proteins releases Bak/Bax, which form homo-oligomers and permeabilize the mitochondrial outer membrane, initiating the caspase cascade and apoptosis. Together, our data indicate that p53 activation plays a central role in the induction of apoptosis in infected M1-D cells.
The basis of macrophage resistance to apoptosis includes expression of prosurvival Bcl-2 family members, among which Mcl-1 predominates in differentiated human macrophages to regulate the macrophage life span (19). We were unable to generate an M1-D cell line expressing high levels of Mcl-1 using a murine Mcl-1 construct assembled in M1-D cells in our lab; other investigators have also not succeeded in generating highly expressing Mcl-1 cell lines for reasons that are unclear. In contrast, human Mcl-1 (pcDNA3-Mcl-1) was transiently expressed at high levels. Overexpression of human Mcl-1 in virus-infected M1-D cells inhibited the cleavage of caspase-9 and -3 and protected the cells from apoptosis at 8 and 10 h p.i., whereas neither Bcl-xL (38) nor Bcl-2 overexpression protected against apoptosis. A1, originally identified as a novel murine granulocyte/macrophage colony-stimulating factor-inducible gene, is expressed in hematopoietic cell lineages, including T-helper cells, macrophages, and neutrophils (17). Because of the predominant role of Mcl-1 in macrophage survival and because Noxa binds to Mcl-1 with eightfold higher affinity than to A1 (Puma binds these prosurvival proteins with similar affinity), we focused on Mcl-1. However, a prosurvival role for A1 in virus-infected M1-D cells cannot be excluded.
Recently, we showed that the BeAn virus L protein induces apoptosis in transiently transfected BHK-21 and M1-D cells (10). Cardioviruses regulate nuclear trafficking of cellular proteins and RNA through the L protein, which contains a CHCC zinc finger motif near the N terminus, an acidic domain with potential Thr and Tyr phosphorylation sites and, only in TMEV, a C-terminal Ser/Thr domain (8, 47). The cardiovirus L binds Ran-GTPase and phosphorylates nucleoporins, leading to disruption of the nuclear pore complex that spans the nuclear envelope and the Ran gradient (29-31). Phosphorylation of nucleoporins by encephalomyocarditis virus is dependent on phosphorylation of p38 and ERK early in infection, since specific MAPK inhibitors prevent phosphorylation (Ann Palmenberg, unpublished data). It is possible that BeAn L is responsible for phosphorylation of p38, leading to transcriptional activation of p53 and apoptosis. Finally, the expression of JAZ, a novel C2H2 class of zinc finger protein, was reported to be upregulated in hematopoietic cells undergoing cell death from the stress of interleukin-3 growth factor withdrawal and to associate with p53 to stimulate its transcriptional activity (45). Although JAZ and the L protein belong to different classes of zinc fingers, it will be important to investigate whether BeAn L can directly activate p53 or activate it by stimulating phosphorylation of p38 MAPK.
Viruses from five different RNA virus families have now been shown to induce apoptosis by activating p53. Both severe acute respiratory syndrome coronavirus virus and avian reovirus activate p53 through the MAPK pathway (18, 27). For severe acute respiratory syndrome virus, transfection of Huh7 cells with protein 3a, a triple-membrane-spanning protein that accumulates in the endoplasmic reticulum and Golgi bodies, mediated the upregulation of p53 by inhibiting the nuclear translocation of STAT 3, a negative regulator of p53 (27), whereas no avian reovirus gene product was implicated in p53 activation (18). Infection of respiratory epithelial cells by respiratory syncytial virus-induced apoptosis, which was inhibited by antibodies against the F protein, and expression of the F protein caused phosphorylation of p53 Ser-15 and activated p53 transcriptional activity (9). It remains unclear how respiratory syncytial virus fusion protein activates p53. Nasirudeen et al. (22) reported that dengue virus infection of Huh7, Vero, and BHK-21 cells induced apoptosis that was p53 dependent, but neither a virus gene product nor a mechanism of p53 was investigated. Finally, the capsid protein of West Nile virus was found to bind directly to and sequester Hdm2 in the nucleolus, perturbing the interaction of Hdm2 with p53 and leading to apoptosis (46).
We found that inhibition of p38 MAPK by SB203508 and of p53 transcriptional activity by the dn GSE56 and overexpression of Mcl-1 proved insufficient to maintain infected M1-D cells alive after 10 h p.i. (not shown). The death of activated macrophages is known to occur by both caspase-dependent and -independent mechanisms, although the latter is not well studied. Apoptosis elicited by tumor necrosis factor alpha through death receptors indicates that necrotic cell death may function as a back-up cell death pathway when caspase-dependent pathways cannot be activated (15, 40). Whether necrotic cell death commences in BeAn virus-infected M1-D cells when caspase-mediated cell death is inhibited remains to be addressed.
Acknowledgments
We thank Patricia Kallio for excellent technical assistance.
This study was supported by National Institutes of Health (NIH) grant NS23349, the Modestus Bauer Foundation, and the Wershkoff Multiple Sclerosis Research Fund.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
Published ahead of print on 5 August 2009.
REFERENCES
- 1.Bain, J., L. Plater, M. Elliott, N. Shpiro, C. J. Hastie, H. McLauchlan, I. Klevernic, J. S. C. Arthur, D. R. Alessi, and P. Cohen. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408:297-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen, L., S. N. Willis, A. Wie, B. J. Smith, J. I. Fletcher, M. G. Hinds, P. M. Colman, and C. L. Day. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. Biol. 17:393-403. [DOI] [PubMed] [Google Scholar]
- 3.Chipuk, J. E., T. Kuwana, L. B. Bouchier-Hayers, N. M. Droin, D. D. Newmeyer, M. Schuler, and D. R. Green. 2004. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010-1014. [DOI] [PubMed] [Google Scholar]
- 4.Coultas, L., P. Bouillet, E. G. Stanley, T. C. Brodnicki, J. M. Adams, and A. Strasser. 2004. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol. Cell. Biol. 24:1570-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coultas, L., S. Terzano, T. Thomas, A. Voss, K. Reid, E. G. Stanley, C. L. Scott, P. Bouillet, P. Bartlett, J. Ham, J. M. Adams, and A. Strasser. 2007. Hrk/DP5 contributes to the apoptosis of select neuronal populations but is dispensable for hematopoietic cell apoptosis. J. Cell Sci. 120:2044-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croxton, R., Y. Ma, L. Song, E. B. Haura, and W. D. Cress. 2002. Direct repression of the Mcl-1 promoter by E2F1. Oncogene 21:1359-1369. [DOI] [PubMed] [Google Scholar]
- 7.De Biasio, A., J. A. Vrana, P. Zhou, L. Qian, C. K. Bieszczad, K. E. Braley, A. M. Domina, S. J. Weintraub, J. M. Neveu, W. S. Lane, and R. W. Craig. 2007. N-terminal truncation of antiapoptotic MCL1, but not G2/M-induced phosphorylation, is associated with stabilization and abundant expression in tumor cells. J. Biol. Chem. 282:23919-23936. [DOI] [PubMed] [Google Scholar]
- 8.Dvorak, C. M., D. J. Hall, M. Riddle, A. Pranter, J. Dillman, M. Deibel, and A. C. Palmenberg. 2001. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection, and affects the efficiency of genome translation. Virology 290:261-271. [DOI] [PubMed] [Google Scholar]
- 9.Eckardt-Michel, J., M. Lorek, D. Baxmann, T. Grunwald, G. M. Keil, and G. Zimmer. 2008. The fusion protein of respiratory syncytial virus triggers p53-dependent apoptosis. J. Virol. 82:3236-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan, J.-L., K.-N. Son, S. Y. Arslan, Z. Liang, and H. L. Lipton. 2009. The Theiler's murine encephalomyelitis virus leader protein is the only nonstructural protein tested that induces apoptosis when transfected into mammalian cells. J. Virol. 83:6546-6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haupt, Y., R. Maya, A. Kazaz, and M. Oren. 1997. Mdm2 promotes the rapid degradation of p53. Nature 387:296-299. [DOI] [PubMed] [Google Scholar]
- 12.Hirasawa, K., A. Kim, H. S. Han, J. Han, H. S. Jun, and J. W. Yoon. 2003. Effect of p38 mitogen-activated protein kinase on the replication of encephalomyocarditis virus. J. Virol. 77:5649-5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jelachich, M. L., C. Brumlage, and H. L. Lipton. 1999. Differentiation of M1 myeloid precursor cells into macrophages results in binding and infection by Theiler's murine encephalomyelitis virus (TMEV) and apoptosis. J. Virol. 73:3227-3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jelachich, M. L., and H. L. Lipton. 1996. Theiler's murine encephalomyelitis virus kills restrictive but not permissive cells by apoptosis. J. Virol. 70:6856-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kroemer, G., and S. J. Martin. 2005. Caspase-independent cell death. Nat. Med. 11:725-730. [DOI] [PubMed] [Google Scholar]
- 16.Kubbutat, M. H., S. N. Jones, and K. H. Vousden. 1997. Regulation of p53 stability by Mdm2. Nature 387:299-303. [DOI] [PubMed] [Google Scholar]
- 17.Lin, E. Y., A. Orlofsky, M. S. Berger, and M. B. Prystowsky. 1993. Characterization of A1, a novel hemopoietic-specific early response gene with sequence similarity to bcl-2. J. Immunol. 151:1979-1988. [PubMed] [Google Scholar]
- 18.Lin, P.-Y., J.-W. Lee, M.-H. Liao, H.-Y. Hsu, S.-J. Chiu, H.-J. Liu, and W.-L. Shih. 2009. Modulation of p53 by mitogen-activated protein kinase pathways and protein kinase Cd during avian reovirus S1133-induced apoptosis. Virology 385:323-334. [DOI] [PubMed] [Google Scholar]
- 19.Liu, H., H. Perlman, L. J. Pagliari, and R. M. Pope. 2001. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages: role of Mcl-1, independent of nuclear factor (NF)-κB, Bad, or caspase activation. J. Exp. Med. 194:113-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchenko, N. D., A. Zaika, and U. M. Moll. 2000. Death signal-induced localization of p53 protein to mitochondria. J. Biol. Chem. 275:16202-16212. [DOI] [PubMed] [Google Scholar]
- 21.Miyashita, T., and J. C. Reed. 1995. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293-299. [DOI] [PubMed] [Google Scholar]
- 22.Nasirudeen, A. M. A., L. Wang, and D.-X. Liu. 2008. Induction of p53-dependent and mitochondria-mediated cell death pathway by dengue virus infection of human and animal cells. Microbes Infect. 10:1124-1132. [DOI] [PubMed] [Google Scholar]
- 23.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053-1058. [DOI] [PubMed] [Google Scholar]
- 24.Oleszak, E. L., B. E. Hoffman, J. R. Chang, E. Zaczynska, J. Gaughan, C. D. Katsetos, C. D. Platsoucas, and N. Harvey. 2003. Apoptosis of infiltrating T cells in the central nervous system of mice infected with Theiler's murine encephalomyelitis virus (TMEV). Virology 315:110-123. [DOI] [PubMed] [Google Scholar]
- 25.Opfermann, J. T., H. Iwasaki, C. C. Ong, H. Suh, S.-i. Mizuno, K. Akashi, and S. J. Korsmeyer. 2005. Obligate role of antiapoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307:1101-1104. [DOI] [PubMed] [Google Scholar]
- 26.Ossovskaya, V. S., I. A. Mazo, M. V. Chernov, O. B. Chernova, Z. Strezoska, R. Kondratov, G. R. Stark, P. M. Chumakov, and A. V. Gudkov. 1996. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc. Natl. Acad. Sci. USA 93:10309-10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padhan, K., R. Minakshi, M. A. B. Towheed, and S. Jameel. 2008. Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J. Gen. Virol. 89:1960-1969. [DOI] [PubMed] [Google Scholar]
- 28.Petros, A. M., E. T. Olejniczak, and S. W. Fesik. 2004. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 1644:83-94. [DOI] [PubMed] [Google Scholar]
- 29.Porter, F. W., Y. A. Bochkov, A. J. Albee, C. Wiese, and A. C. Palmenberg. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 103:12417-12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porter, F. W., and A. C. Palmenberg. 2009. Leader-induced phosphorylation of nucleoporins correlated with nuclear trafficking by cardioviruses. J. Virol. 83:1941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ricour, C., S. Delhaye, S. V. Hato, T. Olenyik, B. Michel, F. J. M. van Kuppeveld, K. E. Gustin, and T. Michiels. 2009. Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler's virus leader protein. J. Gen. Virol. 90:177-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rokhlin, O. W., A. V. Gudkov, S. Kwek, R. A. Glover, A. S. Gewies, and M. B. Cohen. 2000. p53 is involved in tumor necrosis factor-alpha-induced apoptosis in the human prostatic carcinoma cell line LNCaP. Oncogene 19:1959-1968. [DOI] [PubMed] [Google Scholar]
- 33.Rozhon, E. J., J. D. Kratochvil, and H. L. Lipton. 1983. Analysis of genetic variation in Theiler's virus during persistent infection in the mouse central nervous system. Virology 128:16-32. [DOI] [PubMed] [Google Scholar]
- 34.Schlitt, B. P., M. Felrice, M. L. Jelachich, and H. L. Lipton. 2003. Apoptotic cells, including macrophages, are prominent in Theiler's virus-induced inflammatory, demyelinating lesions. J. Virol. 77:4383-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.She, Q.-B., N. Chen, and Z. Dong. 2000. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 275:20444-20449. [DOI] [PubMed] [Google Scholar]
- 36.Shibue, T., K. Takeda, E. Oda, H. Tanaka, H. Murasawa, A. Takaoka, Y. Morishita, S. Akira, T. Taniguchi, and N. Tanaka. 2003. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17:2233-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Si, X., A. Morgan, J. Zhang, J. Wong, J. Yuan, M. Esfandiarei, G. Gao, C. Cheung, and B. M. McManus. 2005. Stress-activated protein kinases are involved in coxsackievirus B3 viral progeny release. J. Virol. 79:13875-13881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Son, K.-N., R. P. Becker, P. Kallio, and H. L. Lipton. 2008. Theiler's virus-induced apoptosis in M1-D macrophages is Bax-mediated through the mitochondrial pathway, resulting in loss of infectious virus: a model for persistence in the mouse central nervous system. J. Virol. 82:4502-4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsunoda, I., C. I. B. Kurtz, and R. S. Fujinami. 1997. Apoptosis in acute and chronic central nervous system disease induced by Theiler's murine encephalomyelitis virus. Virology 228:388-393. [DOI] [PubMed] [Google Scholar]
- 40.Vandenabeele, P., T. Vanden Berghe, and N. Festjens. 2006. Caspase inhibitors promote alternative cell death pathways. Sci. STKE pe44. [DOI] [PubMed]
- 41.Villunger, A., E. M. Michalak, L. Coultas, F. Mullauer, G. Bock, M. J. Ausserlechner, J. M. Adams, and A. Strasser. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science 302:1036-1038. [DOI] [PubMed] [Google Scholar]
- 42.Vrana, J. A., E. S. Cleaveland, A. Eastman, and R. W. Craig. 2006. Inducer- and cell type-specific regulation of antiapoptotic MCL1 in myeloid leukemia and multple myeloma cells exposed to differentiation-inducing or microtubule-disrupting agents. Apoptosis 11:1275-1288. [DOI] [PubMed] [Google Scholar]
- 43.Walker, D. L. 1964. The viral carrier state in animal cell cultures. Prog. Med. Virol. 6:111-148. [PubMed] [Google Scholar]
- 44.Willis, S. N., J. I. Fletcher, T. Kaufmann, M. F. van Delft, L. Chen, P. E. Czabotar, H. Ierino, E. F. Lee, W. G. Fairlie, P. Bouillet, A. Strasser, R. M. Kluck, J. M. Adams, and D. C. S. Huang. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315:856-859. [DOI] [PubMed] [Google Scholar]
- 45.Yang, M., S. Wu, X. Su, and S. May. 2006. JAZ mediates G1 cell-cycle arrest and apoptosis by positively regulating p53 transcriptional activity. Blood 108:4136-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang, M.-R., S. R. Lee, W. Oh, E.-W. Lee, J.-Y. Yeh, J.-J. Nah, Y.-S. Joo, J. Shin, H.-W. Lee, S. Pyo, and J. Song. 2008. West Nile virus capsid protein induces p53-mediated apoptosis via the sequestration of HDM2 to the nucleolus. Cell. Microbiol. 10:165-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoll, J., W. J. G. Melchers, J. M. D. Galama, and F. J. M. van Kuppevold. 2002. The Mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NF-κB. J. Virol. 76:9664-9672. [DOI] [PMC free article] [PubMed] [Google Scholar]